
Abstract
Intestinal ischemia reperfusion (IR) causes injury of distant critical organs. Remote intestinal ischemic preconditioning (IP) may confer the cytoprotection in critical organs including lung. The authors hypothesized that intestinal IP would be a prophylactic factor in the prevention of distant lung injury induced by IR. Rats were randomly divided into IR, IP, and Sham (S) group. Compared with IR group in the serum and lung tissue, MPO, MDA, TNF-α, and IL-1 levels were significantly decreased in the IP group. Following the same pattern, NO level in the serum and lung tissue was significantly increased in the IP group. And intestinal IP markedly abolished lung injury scores in contrast to IR group. Moreover, intestinal IP significantly attenuated caspase-3 expression, leading to the low expression of Bax and the high expression of Bcl-2. The present study showed that intestinal IP ameliorates the capacity of anti-oxygen free radical, inhibits the release of pro-inflammatory cytokines and alleviates apoptosis in IR-induced lung injury in rats. Intestinal IP may provide a novel prophylactic strategy for treatment of IR-induced lung injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Intestinal ischemia reperfusion (IR), a common pathophysiologic process, occurs usually in clinical situations, such as acute mesenteric ischemia, hemorrhagic, traumatic, septic shock, severe burns, and intestine transplantation, playing an important role in the pathogenesis of systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndromes (MODS) [1, 2]. More severely, intestinal IR also causes injury of distant organs including lung. Fundamentally, lung injury, caused by SIRS, is a common cause of complications and death after intestinal IR [3]. These processes resulted from intestinal IR frequently contribute to acute respiratory distress syndrome (ARDS) [4] and acute lung injury [5]. Lung injury after intestinal IR has been well characterized as an acute inflammation with sequestration of leukocytes and their enzymatic products in lung tissue, increased microvascular permeability, histologic, and ultrastructural evidence of pulmonary capillary endothelial cell injury [6].
Intestinal IR injury is a complicated process which associated various mechanisms including apoptosis and proinflammatory cytokines. Apoptosis, as one of the most important mechanisms associated with IR injury, plays a vital role in the initiation and progression of intestinal IR injury [7]. Apoptosis is positively and negatively regulated by the Bcl-2 protein family, including proapoptotic proteins such as Bax and antiapoptotic proteins such as Bcl-2 [8]. Intestinal IR contribute to the release of destructive proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and oxygen free radicals into the circulation, subsequently resulting in SIRS with a high morbidity and mortality during critical illness [9]. Additionally, proinflammatory cytokines such as TNF-α and IL-1 have emerged as significant contributors to the process of neutrophil activation induced by intestinal IR-caused lung injury [10, 11].
Ischemic preconditioning (IP) refers to a phenomenon in which consists of one or more short cycles of reperfusion followed by one or more short cycles of ischemia [12]. Therefore, intestinal IP, repetitive brief periods of intestinal ischemia, protects the intestine from a subsequent longer ischemic insult.
Recently, remote IP can be applied easily and safely in the clinical setting, can attenuate SIRS, and can increase systemic tolerance to IR, imparting the cytoprotection in critical organs including lung [13, 14]. Accumulating studies have suggested that intestinal IP can reduce remote organ injuries such as lung [5], liver [15] kidney [16], and wound healing in small bowel anastomoses [17]. Accordingly, it is indisputable that IP is an important concept during organ transplantation, general surgery and others. Although intestinal IP has been shown to be beneficial in IR, prospectively controlled studies involving intestinal IP of the remote organ including lung are lacking. Then, the authors hypothesized that intestinal IP would be a prophylactic factor in the prevention of distant lung injury induced by IR. Therefore, the purposes of this study were to (1) determine the prophylactic role of intestinal IP against IR-induced lung injury in rats; (2) further elucidate several important molecules involved in intestinal IP lung protective effect; and (3) ascertain some potential mechanisms associated with the protective effect of intestinal IP decreasing lung injury.
MATERIALS AND METHODS
Animals
All procedures described herein were approved by the Animal Care Committee of Xi’an Jiaotong University, China and were performed in accordance with National Institutes of Health guidelines for the use of experimental animals. Eighteen Sprague-Dawley rats weighing 250–300 g were used in this study. Animals were fasting diet 24 h and drunk water freely prior to operation. Food was removed 12 h prior to the operation, but all animals had free access to water. All surgical interventions were performed under intraperitoneal injection pentobarbital anesthesia (35 mg/kg, 2 %) following isolated superior mesenteric artery (SMA) via median abdominal incision.
Rats were randomly divided into (1) ischemia reperfusion (IR) group (n = 6), which was performed by clamping SMA 40 min and closed the abdomen following 4 h reperfusion; (2) ischemia preconditioning (IP) group (n = 6), which was induced by three cycles of 5 min ischemic and 5 min reperfusion following the same IR group as above; and (3) sham (S) group (n = 6), which only was submitted to isolate SMA 40 min then closed the abdomen.
Measurement of MPO Activity
At the end of reperfusion, the ischemic/reperfused lung tissues and serum were frozen immediately and stored at −80 °C until assessment. MPO activity, an essential enzyme existing in neutrophils an enzyme, was determined by a commercial assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Briefly, the lung tissues were homogenized in 50 mmol/L potassium phosphate buffer, pH 6, containing 0.5 % hexadecyltrimethyl ammonium bromide. The homogenates were centrifuged for 10 min at 12,500×g at 4 °C. The supernatants were collected and reacted with 0.167 g/L o-dianisodine dihydrochloride and 0.0005 % H2O2 in 50 mmol/L phosphate buffer, and absorbance was determined by spectrophotometrically at 460 nm.
Measurement of NO Level
NO level in the lung tissue and serum was determined using a NO kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Briefly, the method involved measuring the amount of NO metabolites (nitrite and nitrate), which were more stable than NO. NO level was estimated by determining total nitrate and nitrite concentration in the sample. Nitrate in the tissues and serum was reduced first to nitrite by the action of nitrate reductase. Then, the reaction was initiated by the addition of Griess reagent, and absorbance of the mixture at 550 nm was determined. The NO level in the lung tissue and serum was expressed as μmol/L of serum.
Assay of MDA Level
Lung samples were frozen immediately and stored at −80 °C until assessment. A homogenate of 10 % (w/v) was prepared. MDA level in lung tissue and serum was determined by a MDA detection kit (Nanjing JianCheng Bioengineering Institute, Nanjing, China) following the colorimetric method provided by the manufacturer. The optical densities were read in above spectrophotometer at 532 nm. Each sample was analyzed in duplicate, and the results were averaged.
Determination of TNF-α and IL-1
At the end of reperfusion, the ischemic/reperfused lung tissues and serum were frozen immediately and stored at −80 °C until assessment. TNF-α and IL-1 levels were determined using ELISA kits (R&D, Minneapolis, Minn, USA) according to manufacturer’s procedure. The results were expressed as pg/mL.
Examination of Lung Injury
Representative lung tissue blocks from all lung lobes were embedded in paraffin.
Each slide was evaluated by two expert investigators blinded to the experiment groups. Sections stained with hematoxylin-eosin (H&E) were tested by light microscopy for evidence of lung injury, and the lung injury was scored for edema, neutrophil infiltration, hemorrhage, bronchiole epithelial desquamation, and hyaline membrane formation. A score scaled at 0 to 4 represents the severity: 0 for no or very minor, 1 for modest and limited, 2 for intermediate, 3 for widespread or prominent, and 4 for widespread and most prominent. Then, the total lung injury scores were calculated by adding the individual scores for each category. And ultrastructure was used to observe injurious situation.
Electron Microscope
Approximate, a 1 mm × 2 mm × 1 mm piece of the lung tissue from each group fixed with 2.5 % glutaraldehyde for 3 h and 1 % osmium tetroxide for 2 h was dehydrated with gradient alcohol and embedded in Epoxy Resin 812. After ultrathin slicing and staining with uranyl acetate and lead citrate, the sections were measured under transmission electron microscopy (HitachiH-600).
Immunohistochemical Procedure
Bcl-2 and Bax was detected by immunohistochemical method. Histological sections (5 μm) were deparaffinized and rehydrated. Endogenous peroxidase activity was blocked with methanol/0.3 % H2O2 for 10 min and blocked with non-specific staining blocking reagent. After overnight incubation at 4 °C with anti-Bcl-2 and Bax, and antibody (diluted 1:100, 1:100 respectively), sections were treated according to standard immunoperoxidase methods using a streptavidinbiotin peroxidise complex kit.
Western Blot Analysis
Protein samples (20 μg) were separated on 12 % SDS-PAGE gels and transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5 % nonfat dry milk in Tris-buffered saline containing 0.1 % Tween 20, and incubated with specific antibodies against caspase-3 (1:200), Bax (1:400), Bcl-2 (1:400), and β-actin (1:400) (Santa Cruz, CA, USA). The expression of β-actin was used as a loading control. Reagents (Pierce Corp., Rockford, IL, USA) for the enhanced chemiluminescence were applied to the blots, and the light signals were detected by X-ray film. Optical densities of the bands were scanned and quantified with the Syngene Gene Tools (Syngene Corp., Cambridge, UK). Three independent experiments were carried out to study protein expressions.
Statistical Analysis
Data were expressed as mean ± standard deviation (SD). Biochemical assays for MDA, MPO, NO, TNF-α, and IL-1 and were performed in triplicate for each specific sample. Significance was evaluated using ANOVA (GraphPad Prism 5, San Diego, CA) followed by Tukey’s post hoc test. P value < 0.05 was considered statistically significant.
RESULTS
Serum NO, MDA, MPO, TNF-α, and IL-1 Levels in Rats
As shown in Fig. 1a, serum NO level in the IR group was significantly decreased than that in the S group (14.55 ± 5.14 versus 89.09 ± 3.64, P < 0.01). IP markedly increased serum NO level comparable to the IR group (52.72 ± 7.71 versus 14.55 ± 5.14, P < 0.01). As shown in Fig. 1b, serum MDA level in the IR group was significantly increased than that in the S group (8.66 ± 0.40 versus 2.60 ± 0.47, P < 0.01). IP remarkably decreased serum MDA level comparable to the IR group (6.15 ± 0.54 versus 2.60 ± 0.47, P < 0.01). And as shown in Fig. 1c, serum MPO level in the IR group was significantly increased than that in the S group (18.86 ± 0.73 versus 3.40 ± 0.21, P < 0.01). IP evidently decreased serum MPO level comparable to the IR group (15.18 ± 0.27 versus 18.86 ± 0.73, P < 0.01). As described in Fig. 1d, e, serum TNF-α and IL-1 levels in the IR group were significantly increased than that in the S group. IP remarkably decreased serum TNF-α and IL-1 levels comparable to the IR group (P < 0.01).

Serum NO (a), MDA (b), MPO (c),TNF-α (d), and IL-1(e) level in rats. S sham group, IP ischemic preconditioning group, IR ischemia reperfusion group. Data are presented as the mean ± SD (n = 6). **P < 0.01 compared with S group; ++ P < 0.01 compared with IR group.
Lung Tissue NO, MDA, MPO, TNF-α, and IL-1 Levels in Rats
Similarly, as described in Fig. 2a, lung tissue NO level in the IR group was significantly decreased than that in the S group (0.51 ± 0.13 versus 5.46 ± 0.38, P < 0.01). IP obviously increased lung tissue NO level comparable to the IR group (0.93 ± 0.14 versus 0.51 ± 0.13, P < 0.01). As shown in Fig. 2b, lung tissue MDA level in the IR group was significantly increased than that in the S group (7.04 ± 0.45 versus 1.42 ± 0.15, P < 0.01). IP markedly decreased lung tissue MDA level comparable to the IR group (5.09 ± 0.55 versus 7.04 ± 0.45, P < 0.01). And as shown in Fig. 2c, lung tissue MPO level in the IR group was significantly increased than that in the S group (0.319 ± 0.021 versus 0.077 ± 0.005, P < 0.01). IP apparently decreased serum MPO level comparable to the IR group (0.224 ± 0.015 versus 0.319 ± 0.021, P < 0.01). As described in Fig. 2d, e, lung tissue TNF-α and IL-1 levels in the IR group were significantly increased than that in the S group. IP obviously decreased serum TNF-α and IL-1 levels comparable to the IR group (P < 0.01).

Lung tissue NO (a), MDA (b), MPO (c), TNF-α (d), and IL-1(e) level in rats. Data are presented as the mean ± SD (n = 6). S sham group, IP ischemic preconditioning group, IR ischemia reperfusion group. **P < 0.01 compared with S group; ++ P < 0.01 compared with IR group.
Lung Histology
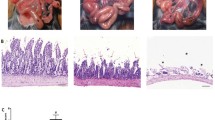
Average lung injury worsened in the intestinal IR group and improved in the IP group, as shown representative of morphological lung tissues in Fig. 3a. Lung injury scores in the IR group were significantly increased than that in the S group (3.8 ± 0.76 versus 0.20 ± 0.45, P < 0.01). And IP considerably decreased lung injury scores comparable to the IR group (2.6 ± 0.55 versus 3.8 ± 0.76, P < 0.05), as demonstrated in Fig. 3b. With respect to ultrastructural lung, as shown in Fig. 3c, lung tissue was observed under electron microscope. Mitochondria, air-blood barrier, and surfactant in the S group were closing normal, and apoptosis bodies were not found. In the IR group, however, mitochondria and air-blood barrier were obvious swelling, and surfactant lost and apoptosis bodies appeared. In the IP group, mitochondria and air-blood barrier were mild swelling, and majority of surfactant existed, but apoptosis bodies were few found, suggesting that preconditioning potently decreased lung injuries.

Morphological changes of lung tissue in rats. a Representative of morphological pictures in lung tissue of rats (HE stain, original magnification ×400). b Lung injury scores in rats, S sham group, IP ischemic preconditioning group, IR ischemia reperfusion group. c Representative of ultrastructural pictures in lung tissue of rats (original magnification ×20,000). Data are presented as the mean ± SD (n = 6). **P < 0.01 compared with S group; + P < 0.05 compared with IR group.
Lung Tissue Bcl-2 and Bax Expression in Rats
In the present study, Bcl-2 and Bax were expressed in alveolar, bronchus, and epithelial cells. Immunohistochemical stainings for lung tissue Bcl-2 and Bax expression were negative in S group. As shown in Fig. 4, lung tissue Bcl-2 expression in the IR group was significantly increased than that in the S group (3.3 ± 0.25 versus 0.79 ± 0.2, P < 0.01). Moreover, IP considerably increased lung tissue Bcl-2 expression comparable to the IR group (6.02 ± 0.86 versus 3.3 ± 0.25, P < 0.01). In contrast, as shown in Fig. 5, lung tissue Bax expression in the IR group was significantly increased than that in the S group (8.58 ± 0.74 versus 0.82 ± 0.23, P < 0.01). Furthermore, IP significantly decreased lung tissue Bax expression comparable to the IR group (5.48 ± 0.63 versus 8.58 ± 0.74, P < 0.01). In accord, the Bcl-2/Bax ratio notably decreased in IR group compared with IP group (0.48 ± 0.14 versus 0.76 ± 0.16, P < 0.01), suggesting increased lung tissue apoptosis in the IR group.

Bcl-2 expression in lung tissue of rats (SP immunohistochemical, original magnification ×400). a Representative of Bcl-2 expression in lung tissue of rats. b Quantitation of Bcl-2 expression in lung tissue of rats. S sham group, IP ischemic preconditioning group, IR ischemia reperfusion group. Data are presented as the mean ± SD (n = 6). **P < 0.01 compared with S group; ++ P < 0.01 compared with IR group.

Bax expression in lung tissue of rats (SP immunohistochemical, original magnification ×400). a Representative of Bax expression in lung tissue of rats. b Quantitation of Bax expression in lung tissue of rats. c Bcl-2/Bax ratio in lung tissue of rats. S sham group, IP ischemic preconditioning group, IR ischemia reperfusion group. Data are presented as the mean ± SD (n = 6). **P < 0.01 compared with S group; ++ P < 0.01 compared with IR group.
Lung Tissue Caspase-3, Bax, and Bcl-2 Protein Expression in Rats
As shown in Fig. 6, Western blot analysis revealed that caspase-3, Bax, and Bcl-2 protein expression in the IR group were higher compared with that in the S group (P < 0.01). IP attenuated the increase in caspase-3 and Bax protein expression compared with that in the IR group (P < 0.05). Conversely, IP significantly increased lung tissue Bcl-2 protein expression comparable to the IR group (P < 0.05).

Protein expression of Caspase-3, Bax and Bcl-2 in lung tissue of rats. Protein expression of Bax, Bcl-2, and Caspase-3 were analyzed by Western blot. Data are presented as the mean ± SD (n = 6). **P < 0.01 compared with S group; + P < 0.05 compared with IR group.
DISCUSSION
The major findings in the present study are that intestinal IP protects lung injury induced by IR as the following pathways: (1) IP might work as an anti-oxygen free-radical agent by elevating NO activity and decreasing MDA content. (2) IP could alleviate acute inflammatory response by attenuating aggregation of neutrophils and release of pro-inflammatory cytokines TNF-α and IL-1. (3) IP may suppress IR-induced apoptosis by upregulation of Bcl-2 and downregulation of Caspase-3 and Bax.
In 1996, although Hotter et al. first reported the protection of IP from intestinal IR injury [18], the precise mechanism responsible for acute lung injury afforded by intestinal IP remains to be poorly understood so far. Intestinal IP is a multifactorial process requiring the interaction of anti-oxidation and anti-apoptosis, intracellular enzymes, and cytokines [19, 20]. Previously, the findings reported that plasma MDA level was significantly increased after intestinal IR, indicating that lipid peroxidation plays an important role in the pathogenesis of superior arterial occlusion shock [21]. As to anti-oxidation, lipid peroxidation inhibition is among the well-documented effects of IP. In our study, we measured serum and lung MDA as an index of oxidative burst and demonstrated that intestinal IP ameliorated IR generated a systemic oxidative stress. NO performs critical functions in modulating the physiological role of the gastrointestinal tract, maintaining the intestinal mucosal integrity in patients with various intestinal disorders [22]. Several studies have shown that NO has protective effects on injuries resulting from intestinal IR [23, 24]. In the present study, we quantified serum and lung NO as an index of protective ability and showed that intestinal IP decreased IR resulted in lung injuries effects. Previous study has indicated that IR injury is accompanied by recruitment of neutrophils into the intestine [25]. Intestine IR induces an acute inflammatory response in which increased adherence and emigration of neutrophils occurs, significantly contributing to intestinal IR injury [26]. Activated neutrophils are considered to be an important factor in tissue injury [25, 26]. MPO represents aggregation and adhesion of neutrophils in serum and lung, one cell types which interact with both physical and metabolic responses during acute inflammation. In our study, we tested serum and lung MPO as acute inflammation marker and showed that intestinal IP ameliorated acute inflammatory response with lung injuries induced by IR.
In ischemia/reperfusion injury, a large of cytokines may be responsible for priming neutrophils. These pro-inflammatory molecules such as TNF-α and IL-1 can induce direct tissue damage and are also potent activators of neutrophils [27]. Moreover, TNF-α and IL-1 have emerged as significant contributors to lung injury [28, 29]. The neutrophils and their enzymatic products are sequestrated in lung tissue, resulting in increased microvascular permeability, perivascular and interstitial edema, and pulmonary edema [30]. In this study, we examined serum and lung TNF-α and IL-1 as pro-inflammatory molecules and displayed that intestinal IP decreased acute inflammatory response with lung injuries induced by IR in rats.
Importantly, a similar pattern was noticed for ultrastructural change and lung injuries scores. In the present study, both average ultrastructural change and lung injuries scores worsened in the IR group and improved in the IP group. It is proposed that intestinal IP inhibits IR-caused lung injuries in histologic and cytologic features.
Apoptosis has been implicated in physiological and pathological conditions including intestinal IR and acute lung injury [31]. And this process is controlled by a number of regulatory molecules mediated by apoptotic signaling. Among them, the Bcl-2 family of proteins plays a central role, which consists of both cell death promoters and cell death preventers [32]. Bax is one of pro-apoptotic proteins, whereas Bcl-2 is one of anti-apoptotic proteins. Moreover, the ratio of Bcl-2 to Bax has also been reported to be essential in determining whether a cell survives or dies the ratio of anti- to pro-apoptotic molecules such as Bcl-2/Bax contributes to the response to an apoptotic signaling [32, 33]. In the present study, Bax expression was downregulated, whereas Bcl-2 expression was upregulated in IP group compared with IR group. In accord, Bcl-2/Bax ratio increased in the IP group, suggesting decreased lung tissue apoptosis. These results suggest that intestinal IP may suppress IR-induced apoptosis by concomitant stimulation of the anti-apoptotic protein Bcl-2 and inhibition of the pro-apoptotic protein Bax.
It has also been showed that caspase plays a key role in the execution phase of apoptosis and contributes to amounts of the morphological features normally related to this form of cell death [34]. Usually, caspase is a common pathway associated with apoptosis signal transduction. Activated by several proapoptotic factors, caspase then activates one another and consequently can initiate specific caspase cascades and apoptosis occur finally [35]. Among them, caspase-3 is the way which must be passed through in the apoptosis cascade, and it is the central one in mammalian cell apoptosis [36]. In the present study, the expression of caspase-3 in lung tissue was detected by Western blot. The results demonstrated that intestinal IP attenuated the increase in caspase-3 protein expression compared with that in the IR group. As the executioner of apoptosis, caspase-3 plays a pivotal role in apoptosis, and thus the protective role of intestinal IP is able to be achieved by inhibiting caspase-3. Intestinal IP may suppress IR-caused apoptosis by upregulation of Bcl-2 and downregulation of caspase-3 and Bax.
In conclusion, the present study investigates that intestinal IP ameliorates the capacity of anti-oxygen free radical, inhibits the release of pro-inflammatory cytokines, and alleviates apoptosis in IR-caused lung injury in rats. Based on these evidences, intestinal IP may provide a novel prophylactic strategy for the treatment of IR-caused lung injury. However, there remains an urgent need to gain additional mechanistic insight into the molecular events involved in the protective role of intestinal IP decreasing IR-induced lung injury and that could be exploited therapeutically.
References
Liu, K.X., Y.S. Li, W.Q. Huang, C. Li, J.X. Liu, and Y. Li. 2009. Immediate but not delayed postconditioning during reperfusion attenuates acute lung injury induced by intestinal ischemia/reperfusion in rats: comparison with ischemic preconditioning. Journal of Surgical Research 157: e55–e62.
Dwivedi, A.J., R. Wu, E. Nguyen, S. Higuchi, H. Wang, K. Krishnasastry, C.P. Marini, T.S. Ravikumar, and P. Wang. 2007. Adrenomedullin and adrenomedullin binding protein-1 prevent acute lung injury after gut ischemia-reperfusion. Journal of the American College of Surgeons 205: 284–293.
Sorkine, P., O. Szold, P. Halpern, M. Gutman, M. Greemland, V. Rudick, and G. Goldman. 1997. Gut decontamination reduces bowel ischemia-induced lung injury in rats. Chest 112: 491–495.
Ware, L.B., and M.A. Matthay. 2000. The acute respiratory distress syndrome. New England Journal of Medicine 342: 1334.
Guzel, A., M. Kanter, A. Guzel, A. Pergel, and M. Erboga. 2012. Anti-inflammatory and antioxidant effects of infliximab on acute lung injury in a rat model of intestinal ischemia/reperfusion. Journal of Molecular Histology 43: 361–369.
Ito, K., H. Ozasa, and S. Horikawa. 2005. Edaravone protects against lung injury induced by intestinal ischemia/reperfusion in rat. Free Radical Biology Medicine 38: 369–374.
Sun, Q., Q.T. Meng, Y. Jiang, H.M. Liu, S.Q. Lei, W.T. Su, W.N. Duan, Y. Wu, and Z.Y. Xia. 2013. Protective effect of ginsenoside Rb1 against intestinal ischemia-reperfusion induced acute renal injury in mice. PloS One 8: e80859.
Shen, M., R.X. Wu, L. Zhao, J. Li, H.T. Guo, R. Fan, Y. Cui, Y.M. Wang, S.Q. Yue, and J.M. Pei. 2012. Resveratrol attenuates ischemia/reperfusion injury in neonatal cardiomyocytes and its underlying mechanism. PloS One 7: e51223.
Zhang, T., Y. Wang, R. Ban, L. Tong, H. Qiao, H. Lao, H. Zhao, X. Jiang, X. Sun, and F. Zhang. 2012. Oral administration of lactoferrin attenuates intestinal ischemia-reperfusion injury in rats. European Surgical Research 49: 99–106.
Caty, M.G., K.S. Guice, K.T. Oldham, D.G. Remick, and S.I. Kunkel. 1990. Evidence for tumor necrosis factor-induced pulmonary microvascular injury after intestinal ischemia-reperfusion injury. Annals of Surgery 212: 694–700.
Cámara-Lemarroy, C.R., F.J. Guzmán-de la Garza, P. Cordero-Pérez, G. Alarcón-Galván, J.M. Ibarra-Hernández, L.E. Muñoz-Espinosa, and N.E. Fernández-Garza. 2013. Bupropion reduces the inflammatory response and intestinal injury due to ischemia-reperfusion. Transplantation Proceedings 45: 2502–2505.
Taha, M.O., R. Miranda-Ferreira, A.C. Chang, A.M. Rodrigues, I.S. Fonseca, L.B. Toral, M.R. Cardoso, M.J. Simões, I.S. Oliveira-Junior, H.P. Monteiro, D.J. Fagundes, N.S. Taha, and A. Caricati-Neto. 2012. Effect of ischemic preconditioning on injuries caused by ischemia and reperfusion in rat intestine. Transplantation Proceedings 44: 2304–2308.
Li, C., Y.S. Li, M. Xu, S.H. Wen, X. Yao, Y. Wu, C.Y. Huang, W.Q. Huang, and K.X. Liu. 2013. Limb remote ischemic preconditioning for intestinal and pulmonary protection during elective open infrarenal abdominal aortic aneurysm repair: a randomized controlled trial. Anesthesiology 118: 842–852.
Camara-Lemarroy, C.R. 2014. Remote ischemic preconditioning as treatment for non-ischemic gastrointestinal disorders: beyond ischemia-reperfusion injury. World Journal of Gastroenterology 20: 3572–3581.
Kanoria, S., R. Jalan, N.A. Davies, A.M. Seifalian, R. Williams, and B.R. Davidson. 2006. Remote ischaemic preconditioning of the hind limb reduces experimental liver warm ischaemia-reperfusion injury. British Journal of Surgery 93: 762–768.
Song, T., Y.F. Peng, S.Y. Guo, Y.H. Liu, and L.Y. Liul. 2007. Brief small intestinal ischemia lessens renal ischemia-reperfusion injury in rats. Comparative Medicine 57: 200–205.
Holzner, P.A., B. Kulemann, S. Kuesters, S. Timme, J. Hoeppner, U.T. Hopt, and G. Marjanovic. 2011. Impact of remote ischemic preconditioning on wound healing in small bowel anastomoses. World Journal of Gastroenterology 17: 1308–1316.
Hotter, G., D. Closa, M. Prados, L. Fernández-Cruz, N. Prats, E. Gelpí, and J. Roselló-Catafau. 1996. Intestinal preconditioning is mediated by a transient increase in nitric oxide. Biochemical and Biophysical Research Communications 222: 27–32.
Liu, K.X., C. Li, Y.S. Li, B.L. Yuan, M. Xu, Z. Xia, and W.Q. Huang. 2010. Proteomic analysis of intestinal ischemia/reperfusion injury and ischemic preconditioning in rats reveals the protective role of aldose reductase. Proteomics 10: 4463–4475.
Moore-Olufemi, S.D., S.E. Olufemi, S. Lott, N. Sato, R.A. Kozar, F.A. Moore, R.S. Radhakrishnan, S. Shah, F. Jimenez, B.C. Kone, and C.S. Cox Jr. 2010. Intestinal ischemic preconditioning after ischemia/reperfusion injury in rat intestine: profiling global gene expression patterns. Digestive Disease and Science 55: 1866–1877.
Flessas, I.I., A.E. Papalois, K. Toutouzas, F. Zagouri, and G.C. Zografos. 2011. Effects of lazaroids on intestinal ischemia and reperfusion injury in experimental models. Journal of Surgical Research 166: 265–274.
Lee, C.H., C.C. Hsiao, C.Y. Hung, Y.J. Chang, and H.C. Lo. 2012. Long-term enteral arginine supplementation in rats with intestinal ischemia and reperfusion. Journal of Surgical Research 175: 67–75.
Kostopanagiotou, G., E.D. Avgerinos, E. Markidou, P.C. Voiniadis, K. Chondros, V. Smyrniotis Theodoraki, and N. Arkadopoulos. 2011. Protective effect of NAC preconditioning against ischemia- reperfusion injury in piglet small bowel transplantation: effects on plasma TNF, IL-8, hyaluronic acid, and NO. Journal of Surgical Research 168: 301–305.
Moore-Olufemi, S.D., R.A. Kozar, F.A. Moore, N. Sato, H.T. Hassoun, C.S. Cox Jr., and B.C. Kone. 2005. Ischemic preconditioning protects against gut dysfunction and mucosal injury after ischemia/reperfusion injury. Shock 23: 258–263.
Sayan, H., V.H. Ozacmak, F. Sen, M. Cabuk, D.Y. Atik, A.A. Igdem, and I.D. Ozacmak. 2009. Pharmacological preconditioning with erythropoietin reduces ischemia-reperfusion injury in the small intestine of rats. Life Science 84: 364–371.
Sukhotnik, I., N. Slijper, Y. Pollak, E. Chemodanov, R. Shaoul, A.G. Coran, and J.G. Mogilner. 2011. Parenteral omega-3 fatty acids (Omegaven) modulate intestinal recovery after intestinal ischemia-reperfusion in a rat model. Journal Of Pediatric Surgery 46: 1353–1360.
Zhao, C., J. Sun, C. Fang, and F. Tang. 2014. 1,8-cineol attenuates LPS-induced acute pulmonary inflammation in mice. Inflammation 37: 566–572.
Chen, L., L. Zhao, C. Zhang, and Z. Lan. 2014. Protective effect of p-cymene on lipopolysaccharide-induced acute lung injury in mice. Inflammation 37: 358–364.
An, S., Y. Hishikawa, J. Liu, and T. Koji. 2007. Lung injury after ischemia-reperfusion of small intestine in rats involves apoptosis of type II alveolar epithelial cells mediated by TNF-alpha and activation of Bid pathway. Apoptosis 12: 1989–2001.
Guo, Q., J. Jin, J.X. Yuan, A. Zeifman, J. Chen, B. Shen, and J. Huang. 2011. VEGF, Bcl-2 and Bad regulated by angiopoietin-1 in oleic acid induced acute lung injury. Biochemical and Biophysical Research Communications 413: 630–636.
Avgerinos, E.D., G. Kostopanagiotou, C. Costopanagiotou, N. Kopanakis, I. Andreadou, M. Lekka, G. Nakos, and V. Smyrniotis. 2010. Intestinal preconditioning ameliorates ischemia-reperfusion induced acute lung injury in rats: an experimental study. Journal of Surgical Research 160: 294–301.
Chen, W., X.B. Fu, S.L. Ge, T.Z. Sun, G. Zhou, B. Han, Y.R. Du, H.H. Li, and Z.Y. Sheng. 2005. Intravenous acid fibroblast growth factor protects intestinal mucosal cells against ischemia-reperfusion injury via regulating Bcl-2/Bax expression. World Journal of Gastroenterology 11: 3419–3425.
An, S., Y. Hishikawa, and T. Koji. 2005. Induction of cell death in rat small intestine by ischemia reperfusion: differential roles of Fas/Fas ligand and Bcl-2/Bax systems depending upon cell types. Histochemistry and Cell Biology 123: 249–261.
Bratton, S.B., M. MacFarlane, K. Cain, and G.M. Cohen. 2000. Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Experimental Cell Research 256: 27–33.
Xiao, F., W. Gao, X. Wang, and T. Chen. 2012. Amplification activation loop between caspase-8 and -9 dominates artemisinin-induced apoptosis of ASTC-a-1 cells. Apoptosis 17: 600–611.
Qu, T., B. Huang, L. Zhang, L. Li, F. Xu, W. Huang, C. Li, Y. Du, and G. Zhang. 2014. Identification and functional characterization of Two executioner caspases in Crassostrea gigas. PloS One 9: e89040.
Acknowledgments
This study was supported by the Fundamental Research Funds for the Central Universities (2013JDHZ08) and Specialized Research Fund of the Second Affiliated Hospital of Xi’an Jiaotong University School of Medicine of China, No. RC (XM) 201103.
Conflict of Interests
The authors have declared that no competing interests exist.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Wang ZD and Ji YY contributed equally to this work and share first authorship.
Rights and permissions
About this article

Cite this article
Wang, Z., Ji, Y., Wang, S. et al. Protective Effect of Intestinal Ischemic Preconditioning on Ischemia Reperfusion-Caused Lung Injury in Rats. Inflammation 38, 424–432 (2015). https://doi.org/10.1007/s10753-014-0047-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-014-0047-3