
Abstract
Heart failure (HF) can be caused by a variety of causes characterized by abnormal myocardial systole and diastole. Ca2+ current through the L-type calcium channel (LTCC) on the membrane is the initial trigger signal for a cardiac cycle. Declined systole and diastole in HF are associated with dysfunction of myocardial Ca2+ function. This disorder can be correlated with unbalanced levels of phosphorylation / dephosphorylation of LTCC, endoplasmic reticulum (ER), and myofilament. Kinase and phosphatase activity changes along with HF progress, resulting in phased changes in the degree of phosphorylation / dephosphorylation. It is important to realize the phosphorylation / dephosphorylation differences between a normal and a failing heart. This review focuses on phosphorylation / dephosphorylation changes in the progression of HF and summarizes the effects of phosphorylation / dephosphorylation of LTCC, ER function, and myofilament function in normal conditions and HF based on previous experiments and clinical research. Also, we summarize current therapeutic methods based on abnormal phosphorylation / dephosphorylation and clarify potential therapeutic directions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) remains an unsolved public health problem. A failing heart is unable to efficiently supply oxygenated blood to the body, resulting in insufficient oxygen supply and nutrients to the body. Cardiovascular-related diseases, including chronic cardiac overload or injury (e.g., high blood pressure, valvular heart disease), myocardial infarction or ischemia, cardiac remodelling, functional abnormalities, and genetic disorders, can eventually lead to HF [1]. Response of the heart can initially be compensatory to additional load or heart damage by increasing its size and contractility [2]. The increase in heart size and mass is thought to be accompanied by biochemical, molecular, structural, and metabolic changes that maintain the function of an enlarged heart. However, chronic stress or disease can lead to dilation of the ventricles and decline of the systolic function, eventually progressing to HF [3]. The most obvious functional change in HF is the decline of diastolic and systolic function, an important cause of which is Ca2+ dysfunction in the myocardium.
Ca2+ dysfunction is associated with abnormal activation or inactivation of key kinases and phosphatases, which can cause phosphorylation and dephosphorylation imbalance in HF (Fig. 1). Protein kinase A (PKA) is a central regulator of cardiac function and morphology [4]. The typical PKA signaling pathway is essential for cardiac activity, especially catecholamines, including norepinephrine secreted from cardiac sympathetic nerve terminals in the heart and epinephrine released from the adrenal medulla [5]. Acute PKA activation improves cardiac performance and is associated with enhanced myocardial contractility, but chronic PKA activation or inhibition can lead to HF. Constitutive PKA activation induces hyperphosphorylation of phospholamban (PLN) and ryanodine (RyR2) of the sarcoplasmic reticulum (SR), leading to reduced contractility and dilated cardiomyopathy [6]. Protein kinase C (PKC) is another member of the serine-threonine kinase family. The increased expression and activity of PKC in HF are closely related to the activation of PKC-α [7, 8]. PKC-α is a fundamental regulator of cardiac contractility and Ca2+ processing in cardiomyocytes. Regulation of PKC-α activity affects dephosphorylation of the SR Ca2+ ATPase-2 pump (SERCA-2) inhibiting PLN and altering the SERCA-2 Ca2+ load and transients. Other kinases, such as protein kinase D (PKD), calmodulin kinase 2 (CaMKII), etc., have also been shown to be activated in HF, while protein kinase G (PKG) activity is reduced [9, 10]. The activation mechanism of PKD involves PKC-mediated PKD phosphorylation, which can be attenuated by PKC inhibition [11]. And neurohormonal stimulation of PKD activity may be enhanced under conditions where PKA activity is down-regulated [12], perhaps allowing PKD-mediated pathways to assume greater significance in the acute regulation of contractile function in HF [13]. PKD-mediated myofilament phosphorylation may have physiological significance in the neurohormonal regulation of myocardial contractile function. PKG can be activated by PKA, and play a role in inhibiting adrenaline, which is related to anti-myocardial hypertrophy [14]. PKG also phosphorylates many PKA-related sites, including the L-type Ca2+ channel (LTCC), PLN, troponin I, myosin-binding protein C (cMyBP-C), and titin. CaMKII is a highly validated signal associated with a variety of diseases, especially those of the cardiovascular system [15].

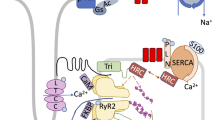
Calcium function in normal myocardium and failing myocardium. In normal hearts, appropriate phosphorylation and dephosphorylation of LTCC, ER, and myofilament are associated with faster Ca2+ release and recovery and lower Ca2+-sensitivity; while in failing hearts, abnormal phosphorylation and dephosphorylation of LTCC, ER, and myofilament is associated with faster Ca2+ release but weakened recovery ability and higher myofilament Ca2+-sensitivity
Dephosphorylation in the heart related to systolic and diastolic function is mainly mediated by protein phosphatase 1(PP1), protein phosphatase 2A (PP2A), and phosphodiesterase (PDE). PP1 is a serine-threaminophosphatase phosphatase that primarily targets the PLN of SR [16]. Activity of PP1 has also been shown to increase in failing hearts and is associated with reduced Ca2+ recovery in ER [17, 18]. PP2A is another major phosphatase in the heart that regulates Ca2+ processing [19]. PP2A coordinates the excitation and contraction of the heart. The importance of PP2A in the heart lies in its ability to antagonize the effects of β adrenergic receptor (β-AR) stimulation, reducing Ca2+ transient amplitude while increasing the Ca2+ sensitivity of myofilaments in force development. PP2A is the main phosphatase of LTCC. PP1 and PP2A form complexes on the RyR and have regulatory effects on the RyR. In HF, PP2A expression and activity are dysregulated [19]. PDE superfamily consists of several distinct subtypes that regulate the strength and duration of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) signaling in discrete compartments of cardiomyocytes [14], with the PDE4 and PDE3 subtypes controlling Ca2+ release and reuptake in the sarcoplasmic reticulum via RyR2 and SERCA-2 [20,21,22,23]. In pathological hypertrophy and HF, levels of PDE1, PDE2, PDE5, PDE9, and PDE10 are elevated. Overall, decreased activity of PDE3 and PDE4 amplified catecholamine toxicity [14].
Changes in the expression and phosphorylation / dephosphorylation imbalance are associated with changes in Ca2+ activity in cardiomyocytes. Studies on changes in phosphorylation levels have been quite mature. It has been recognized that three main amino acids can be phosphorylated: serine (Ser), threonine (Thr), and tyrosine (Tyr), which are characterized by active hydroxyl groups that can be negatively charged by binding to phosphate groups [24,25,26,27,28,29,30]. Animal experiments were conducted to study the effects of phosphorylation in different regions of cardiomyocytes on Ca2+ function by replacing amino acid targets with negatively charged aspartic acid to simulate continuous phosphorylation and positively charged alanine to simulate continuous dephosphorylation. In this review, we summarize the current research and findings of the relationship between intracellular Ca2+ dysfunction and phosphorylation/dephosphorylation imbalance in cardiomyocytes and clarify potential therapeutic directions.
Receptor activation and related-phosphorylation
One of the most prominent features of the progression of cardiac hypertrophy is the persistent activation of β-AR. The rapid positive inotropic action of β-AR activation is dependent on the activation of PKA and its downstream target phosphorylation. Epinephrine signaling directly contributes to PKA activation and stimulates downstream phosphorylation by activating β-AR. In cardiomyocytes, the major targets are LTCC, RyR2, and PLN [31]. This phosphorylation is thought to be beneficial during early hypertrophy. The phosphorylation of LTCC facilitates external Ca2+ uptake, thereby activating more ER Ca2+ release. The phosphorylation of RyR2 is beneficial to the release of Ca2+, the increase of cytosolic Ca2+ concentration, and the increase of contractility during systole. The phosphorylation of PLN enhances the function of the Ca2+ pump, which is conducive to the rapid reduction of intracellular Ca2+ concentration, the rapid completion of the relaxation and contraction cycle, and the improvement of myocardial function.
In addition to PKA activation, the downstream activator of β-AR is PKCε, which is not activated through the cAMP pathway. Studies have shown that epinephrine can activate PKCε via β-AR, independent of PKA activation [32]. The activation was not mediated by the cAMP signaling pathway but by the classical PLC/PKC pathway. In cardiomyocytes, the major phosphorylation targets of PKC are located in myofilaments and are associated with decreased myofilament contractile function. In addition to this, PKC also phosphorylated Thr286 of CaMKII [33], which was related to the decrease of CaMKII activity. Loss of PKCε promotes the occurrence and development of HF [8] indicating that PKCε plays an important role in preventing the occurrence of HF.
Epinephrine signaling activates not only β-AR but also α adrenergic receptor (α-AR). α-AR also shows positive inotropic effects. During the development of physiological hypertrophy or a stress state, epinephrine mainly activates β-AR, triggering a strong positive inotropic effect. However, the α-AR mediated positive inotropic response is predominant in HF rats, which is related to the inhibition of β-AR phosphorylation [34]. Activation of α-AR can promote the phosphorylation of the myosin light chain (MLC) by promoting the activation and expression of Ras homolog gene family (Rho) kinase, which is related to the increase of myofilament Ca2+-sensitivity [35].
In normal conditions, neurotransmitters can inhibit the β effect of epinephrine by activating muscarinic 2 receptors (M2-R). In the progression of cardiac hypertrophy, the activation of epinephrine inhibits the M2-R. When β-AR is continuously activated, parasympathetic nerves are activated in a feedback manner. Undergoing hyperphosphorylation leads to the inhibition of β-AR, and activation of M2-R can be manifested. In cardiomyocytes with suppressed β-AR, M2-R activation promotes the activation and expression of MIC kinase and Rho kinase, leading to MLC-2 phosphorylation and increased myofilament Ca2+-sensitivity [36], suggesting that the activation of these two kinases is independent of cAMP. This would act as a compensatory effect for the loss of β-AR-related inotropic effects. In addition to the partial positive inotropic effect caused by M2-R activation, the overall performance of myocardial contractility is still reduced due to the inhibition of β-AR phosphorylation and the activation of the dephosphorylation signal.
Abnormal Ca2+ and phosphorylation/dephosphorylation
LTCC and abnormal phosphorylation/dephosphorylation
LTCC, commonly referred to as dihydropyridine receptor (DHPR), is sensitive to various 1,4-dihydropyridines [37]. LTCCs in the cardiomyocyte are composed of four different polypeptide subunits (a1, b, a2, d), and the pore-forming subunit a1 is the most important part of the channel, which forms the channel pore for ion flow.
LTCC is the main entry point for Ca2+ influx (ICa2+) into cardiomyocytes and determines the activity of the entire heart [38,39,40]. The main pathway of Ca2+ channel activation is through PKA-mediated phosphorylation (Fig. 2A), which is activated by the second messenger cAMP. This process is also regulated by phosphorylation of LTCC and intracellular Ca2+ concentration [38, 41,42,43]. However, the inhibition of adenylyl cyclase (AC) activity is one of the most common pathways to interrupt PKA-dependent LTCC stimulation [44]. Another way to reduce PKA-dependent channel phosphorylation is the activation of PDEs, which hydrolyze cAMP and cGMP and reduce their intracellular concentrations [42, 45] and play an important role as PKC in LTCC regulation. PKC has a biphasic effect on ICa2+. PKCβ isoform stimulates ICa2+, whereas PKCε inhibits ICa2+ [46, 47].

Phosphorylation and dephosphorylation in normal myocardium and failing myocardium. A In normal conditions, PKA is the main kinase enhancing myocardial contractility when receiving adrenaline stimulation through the β-AR. PDE mainly coordinates with the function of PKA. PP2A and PP1 are responsible for LTCC and ER dephosphorylation, facilitating myocardial dilation. B But in HF, with a weakened β-AR signal and increased PDE function, the M2-R and α-AR remain functional. Phosphorylation caused by PKC, CaMKII, Rho, and MLCK is associated with Ca2+ leakage and enhanced myofilament Ca2+ sensitivity. Dephosphorylation caused by PP1 can reduce Ca2+ recovery into ER
Previous studies have found that it may not be the number and activity of LTCC that are reduced in HF, but mainly the ability of LTCC to stimulate the release of Ca2+ in ER, which is related to the excitation-contraction coupling defect [48]. Basal ICa2+ of LTCC was also found to be weakened in HF (Fig. 1), which may be related to the Ca2+ concentration in the cytoplasm, in which the Ca2+ binding protein calmodulin (CaM) plays a key role [49]. The balance of phosphorylation and dephosphorylation of LTCC is related to their activity, and the phosphorylated form of LTCC may be related to maintaining channel activity and enhancing the ability to stimulate ER Ca2+ release. It was previously thought that PKA and PP2A regulate the phosphorylation level of Ser1928 and thereby determine channel activity [50]. However, later experiments found that the phosphorylation level of Ser1928 is not related to LTCC activity, but the distal carboxyl terminus of α1C is the required factor for the β-AR stimulation of LTCC in cardiomyocytes [51]. Another interesting phosphorylation site is the Ser1700 site of the a-subunit. Although one study demonstrated that PKA-mediated phosphorylation at Ser1700 did not have a major effect on the enhancement of ICa2+ [52], other experiments found that ICa2+ was reduced and cardiac hypertrophy developed when phosphorylation at Ser1700 was absent [53], while Ser1700 and Thr1704 double mutations accelerate cardiac hypertrophy and HF [54]. Another experiment showed that these conserved consensus PKA phosphorylation sites (in addition to those mentioned above), including Ser1512 and Ser1570 (CaMKII-mediated phosphorylation) in α1, Ser459, Ser478, and Ser479 in β2, were not responsible for elevated LTCC activity when phosphorylated [55]. In addition, the phosphorylation of LTCC by PKA is also associated with A kinase anchoring protein (AKAP). PKA without AKAP15 was ineffective in regulating LTCC in cardiomyocytes when the corresponding β-AR pathway was stimulated [56]. Genetic disruption of AKAP150 in mice significantly reduces the co-immunoprecipitation of PKA with LTCC and prevents phosphorylation of Ser1928 upon β-AR stimulation in vivo [57]. Although different experimental results have emerged, it is suggested that PKA is important for the maintenance of LTCC activity. It has been observed that the dysfunction of LTCC is consistent with β-AR depression, and the normal function of β-AR may be the basis for the maintenance of LTCC activity.
SR and abnormal phosphorylation/dephosphorylation
SR is a Ca2+ store in cardiomyocytes, which is divided into longitudinal SR (LSR) parallel to myofibrils and junctional SR (JSR) in contact with the transverse tube. The coupling reaction of JSR and LTCC on the transverse tube is the trigger mechanism of myocardial contraction. The major phosphorylation sites of the SR are located at RyR2 and PLNs (located at the Ca2+ pump/SERCA-2), and their phosphorylation status is related to intracellular Ca2+ release and recycling [58].
RyR2 and abnormal phosphorylation/dephosphorylation
RyR2, a calcium channel in the SR in cardiomyocytes, is the most important component of myofilament contraction triggered by Ca2+ release during contraction [59]. Local regulation of RyR2 channels by PKA phosphorylation is an effective mechanism for regulating SR Ca2+ release. RyR2 is a tetramer composed of four 565,000 Dalton RyR2 peptides and four 12,000 Dalton FK-506 binding proteins (FKBP12.6). FKBP12s, which stabilizes RyR channel function [60] and facilitates coupled gating between adjacent RyR channels [61], are packaged into dense arrays of special regions of the SR that release intracellular stores of Ca2+ to trigger muscle contraction. One FKBP12 molecule binds to each RyR subunit, and dissociation of FKBP12 significantly changes the biophysical properties of the channel, resulting in the appearance of subconductance states and an increase in P0 due to increased sensitivity to Ca2+-dependent activation [60, 62], while dissociation of FKBP12 from RyR channels inhibits coupling gating, resulting in random gating of the channel rather than fusion [61].
Phosphorylation of RyR2 enhances Ca2+ release, and CaMKII-mediated hyperphosphorylation promotes the occurrence of HF. Previous studies have found that hyperphosphorylation of RyR2, specifically mediated by PKA, is present in HF accompanied by decreased phosphatase activity, resulting in the increasing activity of LTCC in the diastolic period and Ca2+ leakage through inhibition of FKBP12.6 binding [59]. Moreover, it was found that not only PKA but also CaMKII was involved in the hyperphosphorylation of RyR2 (Fig. 2A, B), leading to the enhancement of RyR2 activity. The phosphorylation sites included Ser2808, Ser2814, and Ser2815 [33, 63, 64]. However, ablation of the PKA-mediated phosphorylation at Ser2808 failed to prevent the progression of cardiac dysfunction [65, 66]. CaMKII-mediated ablation at Ser2814 prevented the progression of HF [63]. In animal experiments and human hearts, both PKA and CaMKII are involved in RyR2 phosphorylation in hypertrophic hearts. However, in failing hearts, RyR2 phosphorylation is mainly mediated by CaMKII and is accompanied by a higher level of Ca2+ leakage [63, 67]. Meanwhile, lower activity of PDE4D3 has been detected in HF cardiomyocytes, which also contributes to RyR2 hyperphosphorylation and HF progression [63]. It can be speculated that the progression of HF is mainly related to late CaMKII-mediated hyperphosphorylation and reduced PDE4D3 activity. The early PKA-mediated phosphorylation of RyR2 enhances the activity of LTCC and facilitates the release of more Ca2+ to adapt to the higher strength of muscle contraction, but this requires the cooperation of the enhanced function of SERCA-2. The CaMKII-mediated hyperphosphorylation of RyR2 results in Ca2+ overload and decreases Ca2+ transients.
SERCA-2 and phosphorylation/dephosphorylation signals
SERCA-2, located in the SR, is the most important component of Ca2+ recycling during diastole. SERCA-2a is the most important subtype in adult cardiomyocytes [68]. SERCA-2a can transport cytosolic Ca2+ into the SR through ATPase activity, which keeps the cardiomyocyte’s low Ca2+ concentration in the diastolic state and provides the necessary conditions for myocardial contraction [69]. PLN, a small, reversibly phosphorylated transmembrane protein located in the cardiac SR [68], was identified as a major substrate of cAMP-dependent kinases and a regulator of the SERCA-2 [69,70,71,72,73,74,75,76], which can be phosphorylated by PKA and CaMKII (Fig. 2A, B). The main effect of dephosphorylated PLN association with SERCA-2a is to reduce the apparent affinity of SERCA-2a for Ca2+. Alleviation of SERCA-2a inhibition by PLN is a major contributor to the positive inotropic and exotropic effects of β-agonists [77,78,79,80,81].
PLN phosphorylation enhances SERCA-2a activity, and CaMKII-mediated PLN hyperphosphorylation promotes the progression of HF. The phosphorylation of PLN at Ser16 and Thr17 mediated by PKA and CaMKII has been shown to inhibit PLN activity and enhance SERCA-2 activity, which is conducive to the rapid recycling of cytosolic Ca2+, shortening the relaxation [12] and contraction cycles to adjust to the cytosolic hypercalcemia environment. The dephosphorylated form of the PLN inhibits SERCA-2a activity [12, 69, 70]. Although some animal studies support that PLN phosphorylation is reduced in HF [82], others have found that PLN phosphorylation is increased [83, 84], which may be related to the type of animal studied and the stage of HF. The major site of increased phosphorylation in animal models of HF is located at Thr17, and the increase of Thr17 phosphorylation in the HF group is negatively correlated with myocardial contractility. It was also found that 24-hour continuous induction of Ca2+ transient also promoted Thr17 phosphorylation and decreased Ser16 phosphorylation and inotropic drug response [85]. Consistent with findings in human HF samples [68, 86], decreased phosphorylation was found at Ser16 in animal models of HF [82]. This suggests that CaMKII-mediated PLN phosphorylation, but not PKA, is associated with HF progression. Meanwhile, PP1β knockdown could increase the phosphorylation of PLN [87], and increase PP1 activity in HF (Fig. 2B) [17, 18] probably reducing PLN phosphorylation and impaired Ca2+ pump function.
Myofilament and abnormal phosphorylation/dephosphorylation
The sarcomere is the basic unit of myofibril. The sarcomere consists of three different myofilament systems. The components of the thick filament system are myosin and cMyBP-C. The thin myofilament system is composed of monomers of myosin (attached to myosin), tropomyosin, and troponin. Concomitant myofibrin and titin maintain the physical structure of sarcomeres. According to the sliding filament theory, the combination of troponin and Ca2+ affects tropomyosin, exposing the binding sites of actin and myosin, and then actin slides along the myosin. Myofilament is also an important part susceptible to phosphorylation in HF.
Myosin and abnormal phosphorylation/dephosphorylation
Myosin is the main component of the thick filaments, consisting of two heavy chains and four light chains, which have ATPase activity. In failing human hearts, there was no difference in MLC-1 phosphorylation levels compared to normal hearts, while MLC-2 phosphorylation levels were significantly reduced, which is associated with increased Ca2+-sensitivity (Fig. 1) [88]. The maximum tension does not alter, although with increased Ca2+-sensitivity [88]. It was discovered that phosphorylation of myosin regulatory light chain (RLC) and phosphorylation of cRLC enhanced the Ca2+-sensitivity [89, 90], which is related to increased contractility [91].
Located between myosin and actin, cMyBP-C can respond to PKC, PKA, and CaMKII (Fig. 2A, B). Phosphorylation of the CaMKII signal is attributed to enhanced filament contractility. Previous studies have shown that healthy human hearts have higher levels of cMyBP-C phosphorylation, whereas failing human hearts have lower levels of phosphorylation [92]. Independent of cTnI, PKA phosphorylation of cMyBP-C accelerated cross-bridge kinetics [93]. The cMyBP-C can be dephosphorylated in response to cholinergic signaling in HF, which is related to calcineurin overexpression [94]. The rate of force development and filament activation were found to be inhibited by the dephosphorylation form of the cMyBP-C phosphorylation sites Ser273, Ser282, and Ser302 [95], with Ser282 possibly having the greatest impact [96].
Troponin and abnormal phosphorylation/dephosphorylation
Troponin includes three subunits: troponin T (cTnT), troponin C (cTnC), and troponin I (cTnI), of which cTnT is a tropomyosin-binding subunit and cTnC is a Ca2+-binding subunit. The role of cTnI is to prevent myosin and actin from binding to one another. Experiments showed that cTnT was phosphorylated at the same level in cardiomyocytes of human failing and normal hearts, while cTnI was phosphorylated at a lower level in the failing hearts [88, 97].
Human cTnI contains 209 amino acids, including 12 Ser residues, 8 Thr residues, and 3 Tyr residues, and the phosphorylation of cTnI is mediated by PKA and PKC (Fig. 2A, B). The PKA-mediated phosphorylation of cTnI is associated with increased length-dependence and decreased Ca2+-sensitivity [98], while the PKC-mediated phosphorylation may be associated with cardiac disorder [99]. The percentage of cTnI dephosphorylated states was found to be elevated in human failing hearts compared to normal hearts [88]. A study found that in human failing hearts, phosphorylation decreased in some sites of cTnI including Ser5, Ser6, Ser5/Ser6 duplex, Ser23, Ser24, Ser23 /Ser24, Tyr26, while increased in Ser42, Ser44, Thr51, Ser77, Thr78, Ser77/Thr78, Thr143, Ser166, Thr181, and Ser199 [100]. It was also found that pseudo-phosphorylation at both Ser42/44 and Ser23/24 reduced myofilament Ca2+-sensitivity [101]. Double phosphorylation at Ser23/24 is essential for reducing Ca2+-sensitivity, whereas phosphorylation at a single Ser23 or 24 is not [102]. However, a later experiment found that a single Ser23 or 24 phosphorylation was sufficient to reduce Ca2+-sensitivity [103]. Pseudo-phosphorylation of Ser42/44, the PKC phosphorylation sites, is linked to a greater decrease in myofilament Ca2+-sensitivity [101]. Meanwhile, the pseudo-phosphorylation of Ser42/44 weakened the length dependence and blunted the length dependence mediated by PKA, while the pseudo-phosphorylation at Ser23/24 enhanced this length dependence and could be reinforced by PKA [101]. In another experiment, PKCα and phosphorylation levels at Ser44 were found to increase in human and rat failing hearts, which decreased following the implantation of a ventricular assist device [104]. Increased phosphorylation of Ser23/24 and Ser150 was found in the ischemic myocardium [105]. At neutral PH (PH = 7), cardiomyocytes with phosphorylation at Ser150 showed higher Ca2+-sensitivity, whereas phosphorylation at Ser23/24 showed lower Ca2+-sensitivity. However, co-phosphorylation of Ser23/24/150 alleviated the low Ca2+-sensitivity caused by Ser23/24 at neutral PH (PH = 7) [105, 106]. The presence of a phosphorylated acidic environment was shown to attenuate myofilament Ca2+-sensitivity. At acidic PH (PH = 6.5), a greater decrease of Ca2+-sensitivity was found when Ser23/24 and Ser23/24/150 were phosphorylated than single phosphorylation of Ser150, indicating that phosphorylation at Ser150 enhanced the tolerance of cardiomyocytes to an acidic environment [105]. It was also found that phosphorylation of Ser23/24 accelerated the rate of Ca2+ dissociation from troponin, whereas phosphorylation of Ser150 blunted this increase. Independent of the acidic environment, the presence of Ser150 phosphorylation slowed the speed of Ca2+ dissociation from troponin [105]. Another study found that, similar to phosphorylation at Ser23/24, phosphorylation at Tyr26 reduced Ca2+-sensitivity while accelerating the rate of dissociation of Ca2+ from troponin. It’s also has been found that co-phosphorylation of Ser23/24 and Tyr26 did not further reduce Ca2+-sensitivity but further accelerated the rate of dissociation of Ca2+ from troponin [27]. Compared with non-failing myocardium, Ser199 phosphorylation was increased in end-stage HF [100]. Ser199 was found to be phosphorylated mainly mediated by PKC [107], and its elevated phosphorylation increased myofilament Ca2+-sensitivity without affecting its length dependence [108]. Phosphorylation at Thr143 is also mediated by PKC, and pseudo-phosphorylation of Thr143 increases Ca2+-sensitivity but does not alter length dependence [109].
Phosphorylation of cTnT can be mediated by PKC, among which PKCa has four phosphorylation sites on cTnT including Thr197, Ser201, Thr206, and Thr287, and phosphorylation of these sites is associated with decreased myofilament Ca2+ sensitivity [110, 111]. The functional consequences of the phosphorylation of Thr144 were unknown [111]. Although PKC phosphorylation can play different roles in cTnT/cTnI, it is mainly related to elevated Ca2+-sensitivity [107].
Titin and abnormal phosphorylation/dephosphorylation
The primary function of titin is to maintain the integrity and stability of myofibrils. HF is often accompanied by increases in myocardial stiffness-based titin [112, 113].
The increased phosphorylation of Ser4043 and Ser12884, which can be phosphorylated by CaMKII, was found in the failing heart [114], suggesting that excessive activation of CaMKII is an adaptive response in HF patients and HF animal models [114, 115]. Titin is also a substrate of PKG, of which PKG1α is the major isoform expressed in the myocardium and involved in the phosphorylation of several cardiac target proteins, playing a key role in the sarcomole [116]. PKG has been shown to phosphorylate the N2Bus region in titin, thereby reducing titin-based myocardial stiffness [112]. As for the phosphorylation modification of Titin, in addition to the two major kinases, other kinases are also involved in the post-translational modification of Titin. In HFrEF rats, the kinase PKCα showed increased activity in the ventricle and was shown to phosphorylate the PEVK element of titin [117]. PKCα-dependent phosphorylation at Ser11878 in the PEVK-titin fragment was found to be hyperphosphorylated in the HFpEF animal model [118], enhancing the passive tension of titin. PKD, through its mediated phosphorylation, regulates heat shock protein 27 to alleviate titin aggregation, thereby inhibiting titin-dependent cardiomyocyte stiffness [118]. PKA and extracellular regulating kinases 2 (ERK2) were shown to phosphorylate titin springs at specific sites within heart-specific N2-Bus elements, and this modification alters the molecular stiffness of N2-Bus [118].
Treatment directions focusing on phosphorylation/dephosphorylation
Restoration of the balance of key kinases and phosphatases is important to the normal Ca2+ function in cardiomyocytes, which is highly related to the phosphorylation-dephosphorylation balance. Here are some treatments based on the phosphorylation-dephosphorylation disorder to treat HF (Table 1).
The increase in PP1 activity observed in HF was associated with decreased Inhibitor-1 phosphorylation as well as increased I-2 phosphorylation [119,120,121]. Inhibitor-1 (I-1) is the first putative endogenous inhibitor of PP1 (Table 1) [122], and I-1-deficient mice exhibit increased PP1 activity, decreased cardiac function, blunted β-AR response, and reduced PLN phosphorylation (Table 1) [119]. PP1 can be phosphorylated after treatment with isoproterenol [123,124,125], resulting in reduced PP1 activity; PP1 can also be dephosphorylated by PP2A and PP2B, which allows restoration of function to basal levels by relieving PP1 inhibition [126]. When PKA is phosphorylated at Thr35, I-1 efficiently inhibits PP1 activity [122, 127, 128]. However, the phosphorylation of PKCα at Ser67 and Thr75 of I-1 was associated with increased PP1 activity and decreased contractility in vivo [129, 130]. Inhibitor-2 (I-2) is a heat-stable phosphoprotein similar to I-1 [122]. Expression of I-2 resulted in reduced PP1 activity is associated with enhanced contractile parameters and increased instantaneous kinetics of Ca2+, which shows that it manifested by increased phosphorylation at Ser16 of PLN. But not at Thr17, suggesting that the PP1c/I-2 complex may preferentially dephosphorylate the PKA sites in the PLN [131, 132]. Studies on the external application of PP1 inhibitors are also ongoing. It was found that adenovirus-mediated I-35 in the truncated form of I-1c expression which lacks Ser67 and Thr75 enhances the contractile response and Ca2+ dynamics in human failing myocytes [133]. It was also found to attenuate the progression of HF in experimental mice, which was characterized by a reduced degree of cardiac hypertrophy. Similar results were found in I-2 as a therapeutic modality [18]. These beneficial effects are mediated by the enhanced phosphorylation of PLN, while the phosphorylation level of RyR2 remains unchanged. This may be important at the therapeutic level, as increased RyR phosphorylation may potentially lead to diastolic leakage and arrhythmogenic activity [134, 135].
Changes in PDE in HF are diverse, as mentioned above, levels of PDE1, PDE2, PDE5, PDE9, and PDE10 are increased in pathological hypertrophy and HF. However, the PDE3 and PDE4 changes are diverse, and their overall reduced activity is correlated with hypertrophy. Treatment options specific to PDE are also constantly emerging. It has been clinically demonstrated that inhibitors of PDE3 and PDE5 are ineffective [136]. PDE1 enzymes bind to and hydrolyze cAMP and cGMP in a mutually competitive manner. PDE1A regulates the unique cAMP and cGMP pools, predominantly in the perinuclear and nuclear regions of cardiac fibroblasts. Inhibitors of PDE1 or PDE1A gene silencing have been shown to inhibit the adrenalin-induced reduction in PKG activity [137]. PDE1A is also upregulated in cardiac fibroblasts activated by profibrotic stimuli, and inhibition or silencing of PDE1A was shown to limit myofibroblast transformation and the synthesis of extracellular matrix [138]. The pan-PDE1 inhibitor vinpocetine prevented cardiomyocyte hypertrophy and fibroblast activation, thereby blunting pathological remodeling induced by angiotensin II (Table 1) [139]. ITI214 is a drug directed against the PDE1 inhibitor, which can produce acute inotropic and lusitropic effects by promoting a cAMP pool independent of β-AR signaling and increasing ICa2+ (Table 1). However, it was less associated with Ca2+ transients and myofilament phosphorylation [140]. ITI-214 augmented cardiac inotropy, cardiac output, and heart rate (Table 1) [141]. PDE2 has a low affinity for cAMP and cGMP but a high hydrolytic capacity. Increased expression of PDE2 and the hydrolysis activation of cAMP were associated with a diminished β-AR response [142,143,144], suggesting a possible association with the progression of HF [144]. An antihypertrophic effect of PDE2 inhibition was reported in isolated cardiomyocytes [145, 146]. Inhibition of PDE2 was also shown to counteract cardiac hypertrophy and pathological remodeling (Table 1), particularly in fibrosis. In contrast, cardiomyocyte activation by PDE2 may favor ischemic HF to improve Ca2+ homeostasis, limit systolic dysfunction, and prevent arrhythmia [147, 148]. PDE2 cardiomyocytes may be beneficial to counteract the pressure overload caused by pathological remodeling [145,146,147, 149]. Isoforms of PDE4 and PDE3 were found to control Ca2+ release and reuptake in the SR by RyR2 and SERCA-2 respectively [20,21,22,23], or localized to the myofilaments [150, 151] or the nuclear envelope [23, 152, 153]. PDE3 activity is present in the cytosolic and microsomal fractions and constitutes the majority of cAMP-hydrolysing activity in the latter [154]. PDE3 regulates ICa2+ [155,156,157,158,159] and Ca2+ uptake in the SR by modulating cAMP-PKA [160, 161]. The role of its expression in HF was different in different experiments, with some experiments finding that its activity is unchanged in human failing hearts [162, 163], while others have found that its activity is reduced [164]. Some animal experiments reported reduced PDE 3 activity in HF [164,165,166,167,168,169], while others found elevated PDE 3 activity and expression [170,171,172,173]. However, PDE3 inhibition was shown to increase the incidence of arrhythmias in patients [174, 175]. PDE4D3 localizes within the RyR2 macromolecular complex, whose activity occurs mainly in the nuclear membrane. This localized pool of PDE4 also controls the integration of β-AR to AMP-PKA signaling in the nucleus. PDE4 activity can be regulated by PKA phosphorylation or by MAPK1 [176, 177], which is a major negative regulator of β-AR responses in healthy rat cardiomyocytes [156, 178], and the conversion from PDE4 to PDE3 occurs in cardiac hypertrophy and HF [166, 179, 180]. Reduced PDE4 activity in HF increased RyR2 phosphorylation and promoted Ca2+ leakage [22]. On the other hand, this promotes the phosphorylation of SERCA-2, contributes to Ca2+ uptake, and may be beneficial for HF [82, 86, 181,182,183]. PDE5 is localized at the Z-band [184], which is cGMP-activated and specifically hydrolyzes cGMP. PDE5 expression is elevated in animal and human failing hearts [185,186,187,188,189], although some experiments found it to be reduced [190]. PDE5 inhibition provided cardioprotection by promoting cGMP-PKG signaling to prevent cardiomyocyte hypertrophy and cardiac dysfunction [191]. PDE5 inhibition was also shown to attenuate diastolic dysfunction and decrease fibrosis and collagen type I deposition [192, 193]. The cGMP-PKG pathway under the control of PDE5 counteracts the effects of adverse cardiac remodeling (Table 1). PDE8 is a high-affinity, cAMP-specific enzyme [194, 195]. PDE8A was found to regulate excitation-contraction coupling by controlling a specific pool of cAMP involved in β-AR regulation of Ca2+ homeostasis [196]. PDE9 is highly specific for cGMP hydrolysis and is mainly located on the membrane, transducing the np-coupled signal. PDE9 inhibition may lead to improved diastolic function and impaired systolic function (Table 1) [197]. Animal studies have demonstrated that the inhibition of PDE 9 contributes to cardiac output [198]. PDE10 is upregulated in failing hearts in animals and humans [199], and PDE10 was shown to reduce epinephrine-induced cardiac hypertrophy [199]. However, other experiments simultaneously suggested that PDE overexpression exerted protective effects on the heart and reduced cardiac hypertrophy and cardiac hypertrophy caused by β-AR stimulation [143, 147], which can be associated with decreased phosphorylation of RyR2 and reduced ER Ca2+ leaks in early stages of HF or ischemic heart disease, preventing the occurrence of arrhythmia.
PP2A can dephosphorylate many sites on LTCC [200,201,202,203], RyR2 [204, 205], as well as TnI, TnT, and MyCP-c [206,207,208,209], which regulates the relaxation and contraction capacity of cardiomyocytes. The relationship between reduced LTCC activity and altered PP2A activity is unclear at present, and the treatment of PP2A is mainly in the cancer field. Whether PP2A inhibition is helpful in the treatment of HF still needs further experiments to be proven.
PKA is important in the progression of HF, and the restoration of its activity depends on the normal function of β-AR. However, for PKA, whether promoting the activity of PKA alone is helpful in the treatment of HF, more animal experiments are needed. PKG is another important kinase that is related to the phosphorylation of MLC [210]. PKG decrease is associated with HFpEF (with normal systolic function but impaired diastolic function, because PKG facilitates Ca2+ recovery into ER) [211]. PKG activation has broad prospects [212] (Table 1), and PKG may be the best alternative in the absence of normal β-AR function. PKC and CaMKII are activated in HF. Experiments proved that infusion of the oral PKCα/β/γ inhibitor ruboxistaurin increased contractility in wild-type and PKCβγ (- / -) mice, but not in PKCα (- / -) mice, which showed that the inhibitory effect of PKCα improved HF (Table 1) [8]. The same effects were found in other animals [213], providing a new direction for HF treatment. Hesperadin is a CaMKII inhibitor, and its application ameliorates cardiomyocyte injury and HF (Table 1) [214]. CaMKII activation is mainly associated with ER dysfunction, and its inhibitory effect may help to restore ER function. CaMKII inhibitors have broad prospects for the treatment of HF.
As shown in Fig. 3, a comprehensive treatment plan targeted abnormal kinase and phosphatase can be started by PKG activation and PP1 inhibition, for they are benefical to restoring the function of SERCA-2. CaMKII inhibition is also important because former studies indicated a relationships between activation of CaMKII and ER dysfunction and myofilament dysfunction. Although PKCα is activated in HF, it is mainly related to a rising Ca2+ sensitivity. PKCα inhibition can be considered. PDE inhibitiors should be used with caution, because they may cause arrhythmias. Future experiments are expected to prove the effect of the potential treatment plan.

Potential treatment plan targeted on abnormal kinase and phosphatase. Conventional treatments including diuresis, angiotensin inhibition, and β-AR inhibition. Potential treatment targeted abnormal kinase and phosphatase should be sequential. PKG activation and PP1 inhibition can be firstly used for they are responsible for Ca2+ recovery. CaMKII inhibition is also important for it is beneficial to normal ER function. PKC inhibition is the last for it is mainly related to Ca2+ sensitivity recovery (a higher Ca2+ sensitivity may play a compensation role with relatively low intracellular Ca2+)
Conclusions
Calcium dysfunction because of abnormal phosphorylation and dephosphorylation is directly related to impaired myocardial systolic and diastolic functions. β-AR recovery is the end point of HF. Considering the relationships between Ca2+ and CaMKII and PKC, recovery of SR (Ca2+ release and recovery) is the most important, and PKG activation and PP1 inhibition may play a great role in the absence of normal β-AR function. Other potential therapeutic directions in HF can be focused on CaMKII and PKCα inhibitions with the removal of HF-leading risks, which can facilitate a normal Ca2+ function and have benign circular effects, and also improve the function of myofilament. Also, myofilament dysfunction is related to α-AR and M2-R activation. Inhibition of α-AR and M2-R inhibition can be considered, but it can cause many side effects. Treatment plans targeted on abnormal kinase/phosphatase should be sequential, for sudden Ca2+ decrease may cause a more serious systolic disorder. Future experiments are expected to prove the combined therapeutic effect and establish an appropriate dosage and course of treatment.
References
Katz AM, Rolett EL (2016) Heart failure: when form fails to follow function. Eur Heart J 37(5):449–454. https://doi.org/10.1093/eurheartj/ehv548
Wu S, Chen L, Zhou X (2022) Circular RNAs in the regulation of cardiac hypertrophy. Mol Ther Nucleic Acids 27:484–490. https://doi.org/10.1016/j.omtn.2021.12.025
Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR (2015) Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol 89(9):1401–1438. https://doi.org/10.1007/s00204-015-1477-x
Liu Y, Chen J, Fontes SK, Bautista EN, Cheng Z (2022) Physiological and pathological roles of protein kinase A in the heart. Cardiovasc Res 118(2):386–398. https://doi.org/10.1093/cvr/cvab008
Wang J, Gareri C, Rockman HA (2018) G-Protein-coupled receptors in heart disease. Circ Res 123(6):716–735. https://doi.org/10.1161/CIRCRESAHA.118.311403
Antos CL, Frey N, Marx SO et al (2001) Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ Res 89(11):997–1004. https://doi.org/10.1161/hh2301.100003
Bowling N, Walsh RA, Song G et al (1999) Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation 99(3):384–391. https://doi.org/10.1161/01.cir.99.3.384
Liu Q, Chen X, Macdonnell SM et al (2009) Protein kinase calpha, but not PKCbeta or PKCgamma, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res 105(2):194–200. https://doi.org/10.1161/CIRCRESAHA.109.195313
Bossuyt J, Helmstadter K, Wu X et al (2008) Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res 102(6):695–702. https://doi.org/10.1161/CIRCRESAHA.107.169755
Paulus WJ, Tschope C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62(4):263–271. https://doi.org/10.1016/j.jacc.2013.02.092
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298(5600):1912–1934. https://doi.org/10.1126/science.1075762
Mattiazzi A, Mundina-Weilenmann C, Guoxiang C, Vittone L, Kranias E (2005) Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc Res 68(3):366–375. https://doi.org/10.1016/j.cardiores.2005.08.010
Haworth RS, Roberts NA, Cuello F, Avkiran M (2007) Regulation of protein kinase D activity in adult myocardium: novel counter-regulatory roles for protein kinase cepsilon and protein kinase A. J Mol Cell Cardiol 43(6):686–695. https://doi.org/10.1016/j.yjmcc.2007.09.013
Kamel R, Leroy J, Vandecasteele G, Fischmeister R (2023) Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure. Nat Rev Cardiol 20(2):90–108. https://doi.org/10.1038/s41569-022-00756-z
Reyes Gaido OE, Nkashama LJ, Schole KL et al (2023) CaMKII as a therapeutic target in cardiovascular disease. Annu Rev Pharmacol Toxicol 63:249–272. https://doi.org/10.1146/annurev-pharmtox-051421-111814
Steenaart NA, Ganim JR, Di Salvo J, Kranias EG (1992) The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch Biochem Biophys 293(1):17–24. https://doi.org/10.1016/0003-9861(92)90359-5
Gupta RC, Mishra S, Rastogi S, Imai M, Habib O, Sabbah HN (2003) Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am J Physiol Heart Circ Physiol 285(6):H2373–H2381. https://doi.org/10.1152/ajpheart.00442.2003
Yamada M, Ikeda Y, Yano M et al (2006) Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J 20(8):1197–1199. https://doi.org/10.1096/fj.05-5299fje
Lei M, Wang X, Ke Y, Solaro RJ (2015) Regulation of Ca2+ transient by PP2A in normal and failing heart. Front Physiol 6:13. https://doi.org/10.3389/fphys.2015.00013
Beca S, Ahmad F, Shen W et al (2013) Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res 112(2):289–297. https://doi.org/10.1161/CIRCRESAHA.111.300003
Beca S, Helli PB, Simpson JA et al (2011) Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res 109(9):1024–1030. https://doi.org/10.1161/CIRCRESAHA.111.250464
Lehnart SE, Wehrens XH, Reiken S et al (2005) Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123(1):25–35. https://doi.org/10.1016/j.cell.2005.07.030
Lugnier C, Keravis T, Le Bec A, Pauvert O, Proteau S, Rousseau E (1999) Characterization of cyclic nucleotide phosphodiesterase isoforms associated to isolated cardiac nuclei. Biochim Biophys Acta 1472(3):431–446. https://doi.org/10.1016/s0304-4165(99)00145-2
Kranias EG, Solaro RJ (1982) Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature 298(5870):182–184. https://doi.org/10.1038/298182a0
Layland J, Solaro RJ, Shah AM (2005) Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res 66(1):12–21. https://doi.org/10.1016/j.cardiores.2004.12.022
Pena JR, Wolska BM (2004) Troponin I phosphorylation plays an important role in the relaxant effect of beta-adrenergic stimulation in mouse hearts. Cardiovasc Res 61(4):756–763. https://doi.org/10.1016/j.cardiores.2003.12.019
Salhi HE, Walton SD, Hassel NC et al (2014) Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation. J Mol Cell Cardiol 76:257–264. https://doi.org/10.1016/j.yjmcc.2014.09.013
Noland TA Jr, Raynor RL, Kuo JF (1989) Identification of sites phosphorylated in bovine cardiac troponin I and troponin T by protein kinase C and comparative substrate activity of synthetic peptides containing the phosphorylation sites. J Biol Chem 264(34):20778–20785
Jideama NM, Noland TA Jr, Raynor RL et al (1996) Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem 271(38):23277–23283. https://doi.org/10.1074/jbc.271.38.23277
Swiderek K, Jaquet K, Meyer HE, Schachtele C, Hofmann F, Heilmeyer LM Jr (1990) Sites phosphorylated in bovine cardiac troponin T and I. characterization by 31P-NMR spectroscopy and phosphorylation by protein kinases. Eur J Biochem 190(3):575–582. https://doi.org/10.1111/j.1432-1033.1990.tb15612.x
Papa A, Kushner J, Marx SO (2022) Adrenergic regulation of calcium channels in the heart. Annu Rev Physiol 84:285–306. https://doi.org/10.1146/annurev-physiol-060121-041653
Li L, Cai H, Liu H, Guo T (2015) Beta-adrenergic stimulation activates protein kinase cepsilon and induces extracellular signal-regulated kinase phosphorylation and cardiomyocyte hypertrophy. Mol Med Rep 11(6):4373–4380. https://doi.org/10.3892/mmr.2015.3316
Oestreich EA, Malik S, Goonasekera SA et al (2009) Epac and phospholipase cepsilon regulate Ca2+ release in the heart by activation of protein kinase cepsilon and calcium-calmodulin kinase II. J Biol Chem 284(3):1514–1522. https://doi.org/10.1074/jbc.M806994200
Sjaastad I, Schiander I, Sjetnan A et al (2003) Increased contribution of alpha 1- vs. beta-adrenoceptor-mediated inotropic response in rats with congestive heart failure. Acta Physiol Scand 177(4):449–458. https://doi.org/10.1046/j.1365-201X.2003.01063.x
Suematsu N, Satoh S, Kinugawa S et al (2001) Alpha1-adrenoceptor-Gq-RhoA signaling is upregulated to increase myofibrillar Ca2+ sensitivity in failing hearts. Am J Physiol Heart Circ Physiol 281(2):H637–H646. https://doi.org/10.1152/ajpheart.2001.281.2.H637
Hussain RI, Qvigstad E, Birkeland JA et al (2009) Activation of muscarinic receptors elicits inotropic responses in ventricular muscle from rats with heart failure through myosin light chain phosphorylation. Br J Pharmacol 156(4):575–586. https://doi.org/10.1111/j.1750-3639.2009.00016.x
Yamakage M, Namiki A (2002) Calcium channels–basic aspects of their structure, function and gene encoding: anesthetic action on the channels–a review. Can J Anaesth 49(2):151–164. https://doi.org/10.1007/BF03020488
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415(6868):198–205. https://doi.org/10.1038/415198a
Richard S, Perrier E, Fauconnier J et al (2006) Ca2+-induced Ca2+ entry’ or how the L-type Ca2+ channel remodels its own signalling pathway in cardiac cells. Prog Biophys Mol Biol 90(1–3):118–135. https://doi.org/10.1016/j.pbiomolbio.2005.05.005
Bers DM, Despa S (2006) Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci 100(5):315–322. https://doi.org/10.1254/jphs.cpj06001x
Kamp TJ, Hell JW (2000) Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res 87(12):1095–1102. https://doi.org/10.1161/01.res.87.12.1095
Jurevicius J, Fischmeister R (1996) cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc Natl Acad Sci USA 93(1):295–299. https://doi.org/10.1073/pnas.93.1.295
Lindegger N, Niggli E (2005) Paradoxical SR Ca2+ release in guinea-pig cardiac myocytes after beta-adrenergic stimulation revealed by two-photon photolysis of caged Ca2+. J Physiol 565(Pt 3):801–813. https://doi.org/10.1113/jphysiol.2005.084376
Mery PF, Abi-Gerges N, Vandecasteele G, Jurevicius J, Eschenhagen T, Fischmeister R (1997) Muscarinic regulation of the L-type calcium current in isolated cardiac myocytes. Life Sci 60(13–14):1113–1120. https://doi.org/10.1016/s0024-3205(97)00055-6
Fischmeister R, Castro LR, Abi-Gerges A et al (2006) Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res 99(8):816–828. https://doi.org/10.1161/01.RES.0000246118.98832.04
Alden KJ, Goldspink PH, Ruch SW, Buttrick PM, Garcia J (2002) Enhancement of L-type Ca2+ current from neonatal mouse ventricular myocytes by constitutively active PKC-betaII. Am J Physiol Cell Physiol 282(4):C768–774. https://doi.org/10.1152/ajpcell.00494.2001
Yue Y, Qu Y, Boutjdir M (2004) Beta- and alpha-adrenergic cross-signaling for L-type ca current is impaired in transgenic mice with constitutive activation of epsilon PKC. Biochem Biophys Res Commun 314(3):749–754. https://doi.org/10.1016/j.bbrc.2003.12.155
Gomez AM, Valdivia HH, Cheng H et al (1997) Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276(5313):800–806. https://doi.org/10.1126/science.276.5313.800
Ouadid H, Albat B, Nargeot J (1995) Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol 25(2):282–291. https://doi.org/10.1097/00005344-199502000-00014
Davare MA, Horne MC, Hell JW (2000) Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem 275(50):39710–39717. https://doi.org/10.1074/jbc.M005462200
Ganesan AN, Maack C, Johns DC, Sidor A, O’Rourke B (2006) Beta-adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1C but not serine 1928. Circ Res 98(2):e11–18. https://doi.org/10.1161/01.RES.0000202692.23001.e2
Yang L, Katchman A, Samad T, Morrow J, Weinberg R, Marx SO (2013) Beta-adrenergic regulation of the L-type Ca2+ channel does not require phosphorylation of alpha1C Ser1700. Circ Res 113(7):871–880. https://doi.org/10.1161/CIRCRESAHA.113.301926
Fu Y, Westenbroek RE, Scheuer T, Catterall WA (2014) Basal and beta-adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. Proc Natl Acad Sci USA 111(46):16598–16603. https://doi.org/10.1073/pnas.1419129111
Yang L, Dai DF, Yuan C et al (2016) Loss of beta-adrenergic-stimulated phosphorylation of CaV1.2 channels on Ser1700 leads to heart failure. Proc Natl Acad Sci USA 113(49):E7976–E7985. https://doi.org/10.1073/pnas.1617116113
Katchman A, Yang L, Zakharov SI et al (2017) Proteolytic cleavage and PKA phosphorylation of alpha(1 C) subunit are not required for adrenergic regulation of ca(V)1.2 in the heart. Proc Natl Acad Sci USA 114(34):9194–9199. https://doi.org/10.1073/pnas.1706054114
Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA (2003) Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with a kinase-anchoring protein 15. Proc Natl Acad Sci USA 100(22):13093–13098. https://doi.org/10.1073/pnas.2135335100
Hall DD, Davare MA, Shi M et al (2007) Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry 46(6):1635–1646. https://doi.org/10.1021/bi062217x
Michalak M, Opas M (2009) Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell Biol 19(6):253–259. https://doi.org/10.1016/j.tcb.2009.03.006
Marx SO, Reiken S, Hisamatsu Y et al (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101(4):365–376. https://doi.org/10.1016/s0092-8674(00)80847-8
Brillantes AB, Ondrias K, Scott A et al (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77(4):513–523. https://doi.org/10.1016/0092-8674(94)90214-3
Marx SO, Ondrias K, Marks AR (1998) Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281(5378):818–821. https://doi.org/10.1126/science.281.5378.818
Kaftan E, Marks AR, Ehrlich BE (1996) Effects of rapamycin on ryanodine receptor/Ca2+-release channels from cardiac muscle. Circ Res 78(6):990–997. https://doi.org/10.1161/01.res.78.6.990
Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM (2005) Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 97(12):1314–1322. https://doi.org/10.1161/01.RES.0000194329.41863.89
Walweel K, Molenaar P, Imtiaz MS et al (2017) Ryanodine receptor modification and regulation by intracellular Ca2+ and Mg2+ in healthy and failing human hearts. J Mol Cell Cardiol 104:53–62. https://doi.org/10.1016/j.yjmcc.2017.01.016
MacDonnell SM, Garcia-Rivas G, Scherman JA et al (2008) Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res 102(8):e65–e72. https://doi.org/10.1161/CIRCRESAHA.108.174722
Alvarado FJ, Chen X, Valdivia HH (2017) Ablation of the cardiac ryanodine receptor phospho-site Ser2808 does not alter the adrenergic response or the progression to heart failure in mice. Elimination of the genetic background as critical variable. J Mol Cell Cardiol 103:40–47. https://doi.org/10.1016/j.yjmcc.2017.01.001
Fischer TH, Herting J, Tirilomis T et al (2013) Ca2+/calmodulin-dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 128(9):970–981. https://doi.org/10.1161/CIRCULATIONAHA.113.001746
Periasamy M, Bhupathy P, Babu GJ (2008) Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res 77(2):265–273. https://doi.org/10.1093/cvr/cvm056
MacLennan DH, Kranias EG (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 4(7):566–577. https://doi.org/10.1038/nrm1151
Simmerman HK, Jones LR (1998) Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev 78(4):921–947. https://doi.org/10.1152/physrev.1998.78.4.921
Brittsan AG, Kranias EG (2000) Phospholamban and cardiac contractile function. J Mol Cell Cardiol 32(12):2131–2139. https://doi.org/10.1006/jmcc.2000.1270
Tada M (1992) Molecular structure and function of phospholamban in regulating the calcium pump from sarcoplasmic reticulum. Ann NY Acad Sci 671:92–102 discussion 102–103. https://doi.org/10.1111/j.1749-6632.1992.tb43787.x
Tada M, Inui M (1983) Regulation of calcium transport by the ATPase-phospholamban system. J Mol Cell Cardiol 15(9):565–575. https://doi.org/10.1016/0022-2828(83)90267-5
Tada M, Ohmori F, Kinoshita N, Abe H (1978) Cyclic AMP regulation of active calcium transport across membranes of sarcoplasmic reticulum: role of the 22,000-dalton protein phospholamban. Adv Cycl Nucleotide Res 9:355–369
Kadambi VJ, Kranias EG (1997) Phospholamban: a protein coming of age. Biochem Biophys Res Commun 239(1):1–5. https://doi.org/10.1006/bbrc.1997.7340
Koss KL, Kranias EG (1996) Phospholamban: a prominent regulator of myocardial contractility. Circ Res 79(6):1059–1063. https://doi.org/10.1161/01.res.79.6.1059
Wegener AD, Simmerman HK, Lindemann JP, Jones LR (1989) Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to beta-adrenergic stimulation. J Biol Chem 264(19):11468–11474
Talosi L, Edes I, Kranias EG (1993) Intracellular mechanisms mediating reversal of beta-adrenergic stimulation in intact beating hearts. Am J Physiol 264(3 Pt 2):H791–H797. https://doi.org/10.1152/ajpheart.1993.264.3.H791
Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM (1983) Beta-adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J Biol Chem 258(1):464–471
Garvey JL, Kranias EG, Solaro RJ (1988) Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J 249(3):709–714. https://doi.org/10.1042/bj2490709
Mundina-Weilenmann C, Vittone L, Ortale M, de Cingolani GC, Mattiazzi A (1996) Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart. J Biol Chem 271(52):33561–33567. https://doi.org/10.1074/jbc.271.52.33561
Sande JB, Sjaastad I, Hoen IB et al (2002) Reduced level of serine(16) phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc Res 53(2):382–391. https://doi.org/10.1016/s0008-6363(01)00489-8
Currie S, Smith GL (1999) Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc Res 41(1):135–146. https://doi.org/10.1016/s0008-6363(98)00241-7
Boknik P, Heinroth-Hoffmann I, Kirchhefer U et al (2001) Enhanced protein phosphorylation in hypertensive hypertrophy. Cardiovasc Res 51(4):717–728. https://doi.org/10.1016/s0008-6363(01)00346-7
Mills GD, Kubo H, Harris DM, Berretta RM, Piacentino V 3rd, Houser SR (2006) Phosphorylation of phospholamban at threonine-17 reduces cardiac adrenergic contractile responsiveness in chronic pressure overload-induced hypertrophy. Am J Physiol Heart Circ Physiol 291(1):H61–H70. https://doi.org/10.1152/ajpheart.01353.2005
Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E (1999) Reduced Ca2+-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol 31(3):479–491. https://doi.org/10.1006/jmcc.1998.0897
Aoyama H, Ikeda Y, Miyazaki Y et al (2011) Isoform-specific roles of protein phosphatase 1 catalytic subunits in sarcoplasmic reticulum-mediated Ca2+ cycling. Cardiovasc Res 89(1):79–88. https://doi.org/10.1093/cvr/cvq252
van der Velden J, Papp Z, Zaremba R et al (2003) Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res 57(1):37–47. https://doi.org/10.1016/s0008-6363(02)00606-5
Pulcastro HC, Awinda PO, Breithaupt JJ, Tanner BC (2016) Effects of myosin light chain phosphorylation on length-dependent myosin kinetics in skinned rat myocardium. Arch Biochem Biophys 601:56–68. https://doi.org/10.1016/j.abb.2015.12.014
Kampourakis T, Sun YB, Irving M (2016) Myosin light chain phosphorylation enhances contraction of heart muscle via structural changes in both thick and thin filaments. Proc Natl Acad Sci USA 113(21):E3039–E3047. https://doi.org/10.1073/pnas.1602776113
Markandran K, Yu H, Song W, Lam D, Madathummal MC, Ferenczi MA (2021) Functional and molecular characterisation of heart failure progression in mice and the role of myosin regulatory light chains in the recovery of cardiac muscle function. Int J Mol Sci 23(1):88. https://doi.org/10.3390/ijms23010088
Jacques AM, Copeland O, Messer AE et al (2008) Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol 45(2):209–216. https://doi.org/10.1016/j.yjmcc.2008.05.020
Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL (2008) Acceleration of crossbridge kinetics by protein kinase a phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res 103(9):974–982. https://doi.org/10.1161/CIRCRESAHA.108.177683
Gilda JE, Gomes AV (2013) How phosphorylated can it get? Cardiac myosin binding protein C phosphorylation in heart failure. J Mol Cell Cardiol 62:108–110. https://doi.org/10.1016/j.yjmcc.2013.05.015
Gresham KS, Stelzer JE (2016) The contributions of cardiac myosin binding protein C and troponin I phosphorylation to beta-adrenergic enhancement of in vivo cardiac function. J Physiol 594(3):669–686. https://doi.org/10.1113/JP270959
McNamara JW, Singh RR, Sadayappan S (2019) Cardiac myosin binding protein-C phosphorylation regulates the super-relaxed state of myosin. Proc Natl Acad Sci USA 116(24):11731–11736. https://doi.org/10.1073/pnas.1821660116
Messer AE, Jacques AM, Marston SB (2007) Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol 42(1):247–259. https://doi.org/10.1016/j.yjmcc.2006.08.017
Konhilas JP, Irving TC, Wolska BM et al (2003) Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J Physiol 547(Pt 3):951–961. https://doi.org/10.1113/jphysiol.2002.038117
Horn K, Leontieva L, Williams JM, Furbee PM, Helmkamp JC, Manley WG 3rd (2002) Alcohol problems among young adult emergency department patients: making predictions using routine sociodemographic information. J Crit Care 17(4):212–220. https://doi.org/10.1053/jcrc.2002.37231
Zhang P, Kirk JA, Ji W et al (2012) Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 126(15):1828–1837. https://doi.org/10.1161/CIRCULATIONAHA.112.096388
Wijnker PJ, Sequeira V, Witjas-Paalberends ER et al (2014) Phosphorylation of protein kinase C sites Ser42/44 decreases Ca2+-sensitivity and blunts enhanced length-dependent activation in response to protein kinase A in human cardiomyocytes. Arch Biochem Biophys 554:11–21. https://doi.org/10.1016/j.abb.2014.04.017
Wijnker PJ, Foster DB, Tsao AL et al (2013) Impact of site-specific phosphorylation of protein kinase A sites Ser23 and Ser24 of cardiac troponin I in human cardiomyocytes. Am J Physiol Heart Circ Physiol 304(2):H260–H268. https://doi.org/10.1152/ajpheart.00498.2012
Martin-Garrido A, Biesiadecki BJ, Salhi HE et al (2018) Monophosphorylation of cardiac troponin-I at Ser-23/24 is sufficient to regulate cardiac myofibrillar Ca2+ sensitivity and calpain-induced proteolysis. J Biol Chem 293(22):8588–8599. https://doi.org/10.1074/jbc.RA117.001292
Ravichandran VS, Patel HJ, Pagani FD, Westfall MV (2019) Cardiac contractile dysfunction and protein kinase C-mediated myofilament phosphorylation in disease and aging. J Gen Physiol 151(9):1070–1080. https://doi.org/10.1085/jgp.201912353
Nixon BR, Walton SD, Zhang B et al (2014) Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca2+ sensitivity and accelerates deactivation in an acidic environment. J Mol Cell Cardiol 72:177–185. https://doi.org/10.1016/j.yjmcc.2014.03.010
Salhi HE, Hassel NC, Siddiqui JK et al (2016) Myofilament calcium sensitivity: mechanistic insight into TnI Ser-23/24 and Ser-150 phosphorylation integration. Front Physiol 7:567. https://doi.org/10.3389/fphys.2016.00567
Kooij V, Zhang P, Piersma SR et al (2013) PKCalpha-specific phosphorylation of the troponin complex in human myocardium: a functional and proteomics analysis. PLoS ONE 8(10):e74847. https://doi.org/10.1371/journal.pone.0074847
Fugger L, Morling N, Bendtzen K et al (1989) IL-6 gene polymorphism in rheumatoid arthritis, pauciarticular juvenile rheumatoid arthritis, systemic lupus erythematosus, and in healthy Danes. J Immunogenet 16(6):461–465. https://doi.org/10.1111/j.1744-313x.1989.tb00495.x
Wijnker PJ, Sequeira V, Foster DB et al (2014) Length-dependent activation is modulated by cardiac troponin I bisphosphorylation at Ser23 and Ser24 but not by Thr143 phosphorylation. Am J Physiol Heart Circ Physiol 306(8):H1171–H1181. https://doi.org/10.1152/ajpheart.00580.2013
Kooij V, Boontje N, Zaremba R et al (2010) Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol 105(2):289–300. https://doi.org/10.1007/s00395-009-0053-z
Solaro RJ, Kobayashi T (2011) Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem 286(12):9935–9940. https://doi.org/10.1074/jbc.R110.197731
Linke WA, Hamdani N (2014) Gigantic business: titin properties and function through thick and thin. Circ Res 114(6):1052–1068. https://doi.org/10.1161/CIRCRESAHA.114.301286
Leite-Moreira AM, Almeida-Coelho J, Neves JS et al (2018) Stretch-induced compliance: a novel adaptive biological mechanism following acute cardiac load. Cardiovasc Res 114(5):656–667. https://doi.org/10.1093/cvr/cvy026
Gomori K, Herwig M, Budde H et al (2022) Ca2+/calmodulin-dependent protein kinase II and protein kinase G oxidation contributes to impaired sarcomeric proteins in hypertrophy model. ESC Heart Fail 9(4):2585–2600. https://doi.org/10.1002/ehf2.13973
Mohamed BA, Schnelle M, Khadjeh S et al (2016) Molecular and structural transition mechanisms in long-term volume overload. Eur J Heart Fail 18(4):362–371. https://doi.org/10.1002/ejhf.465
Hofmann F, Wegener JW (2013) cGMP-dependent protein kinases (cGK). Methods Mol Biol 1020:17–50. https://doi.org/10.1007/978-1-62703-459-3_2
Kovacs A, Herwig M, Budde H et al (2021) Interventricular differences of signaling pathways-mediated regulation of cardiomyocyte function in response to high oxidative stress in the post-ischemic failing rat heart. Antioxid (Basel) 10(6):964. https://doi.org/10.3390/antiox10060964
Sugimoto M, Murata M, Yamaoka Y (2020) Chemoprevention of gastric cancer development after Helicobacter pylori eradication therapy in an east Asian population: Meta-analysis. World J Gastroenterol 26(15):1820–1840. https://doi.org/10.3748/wjg.v26.i15.1820
Carr AN, Schmidt AG, Suzuki Y et al (2002) Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 22(12):4124–4135. https://doi.org/10.1128/MCB.22.12.4124-4135.2002
El-Armouche A, Pamminger T, Ditz D, Zolk O, Eschenhagen T (2004) Decreased protein and phosphorylation level of the protein phosphatase inhibitor-1 in failing human hearts. Cardiovasc Res 61(1):87–93. https://doi.org/10.1016/j.cardiores.2003.11.005
Gupta RC, Mishra S, Yang XP, Sabbah HN (2005) Reduced inhibitor 1 and 2 activity is associated with increased protein phosphatase type 1 activity in left ventricular myocardium of one-kidney, one-clip hypertensive rats. Mol Cell Biochem 269(1–2):49–57. https://doi.org/10.1007/s11010-005-2538-x
Huang FL, Glinsmann WH (1976) Separation and characterization of two phosphorylase phosphatase inhibitors from rabbit skeletal muscle. Eur J Biochem 70(2):419–426. https://doi.org/10.1111/j.1432-1033.1976.tb11032.x
Neumann J, Gupta RC, Schmitz W, Scholz H, Nairn AC, Watanabe AM (1991) Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circ Res 69(6):1450–1457. https://doi.org/10.1161/01.res.69.6.1450
Iyer RB, Koritz SB, Kirchberger MA (1988) A regulation of the level of phosphorylated phospholamban by inhibitor-1 in rat heart preparations in vitro. Mol Cell Endocrinol 55(1):1–6. https://doi.org/10.1016/0303-7207(88)90084-6
Gupta RC, Neumann J, Watanabe AM, Lesch M, Sabbah HN (1996) Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am J Physiol 270(Pt 2):H1159–1164. https://doi.org/10.1152/ajpheart.1996.270.4.H1159
El-Armouche A, Bednorz A, Pamminger T et al (2006) Role of calcineurin and protein phosphatase-2A in the regulation of phosphatase inhibitor-1 in cardiac myocytes. Biochem Biophys Res Commun 346(3):700–706. https://doi.org/10.1016/j.bbrc.2006.05.182
Foulkes JG, Strada SJ, Henderson PJ, Cohen P (1983) A kinetic analysis of the effects of inhibitor-1 and inhibitor-2 on the activity of protein phosphatase-1. Eur J Biochem 132(2):309–313. https://doi.org/10.1111/j.1432-1033.1983.tb07363.x
Huang KX, Paudel HK (2000) Ser67-phosphorylated inhibitor 1 is a potent protein phosphatase 1 inhibitor. Proc Natl Acad Sci USA 97(11):5824–5829. https://doi.org/10.1073/pnas.100460897
Braz JC, Gregory K, Pathak A et al (2004) PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 10(3):248–254. https://doi.org/10.1038/nm1000
Rodriguez P, Mitton B, Waggoner JR, Kranias EG (2006) Identification of a novel phosphorylation site in protein phosphatase inhibitor-1 as a negative regulator of cardiac function. J Biol Chem 281(50):38599–38608. https://doi.org/10.1074/jbc.M604139200
Park IK, DePaoli-Roach AA (1994) Domains of phosphatase inhibitor-2 involved in the control of the ATP-Mg-dependent protein phosphatase. J Biol Chem 269(46):28919–28928
Yang J, Hurley TD, DePaoli-Roach AA (2000) Interaction of inhibitor-2 with the catalytic subunit of type 1 protein phosphatase. Identification of a sequence analogous to the consensus type 1 protein phosphatase-binding motif. J Biol Chem 275(30):22635–22644. https://doi.org/10.1074/jbc.M003082200
Kirchhefer U, Baba HA, Boknik P et al (2005) Enhanced cardiac function in mice overexpressing protein phosphatase Inhibitor-2. Cardiovasc Res 68(1):98–108. https://doi.org/10.1016/j.cardiores.2005.05.019
Wehrens XH, Lehnart SE, Reiken SR et al (2004) Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304(5668):292–296. https://doi.org/10.1126/science.1094301
Yano M, Ono K, Ohkusa T et al (2000) Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation 102(17):2131–2136. https://doi.org/10.1161/01.cir.102.17.2131
Redfield MM, Chen HH, Borlaug BA et al (2013) Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309(12):1268–1277. https://doi.org/10.1001/jama.2013.2024
Miller CL, Oikawa M, Cai Y et al (2009) Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 105(10):956–964. https://doi.org/10.1161/CIRCRESAHA.109.198515
Miller CL, Cai Y, Oikawa M et al (2011) Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol 106(6):1023–1039. https://doi.org/10.1007/s00395-011-0228-2
Wu MP, Zhang YS, Xu X, Zhou Q, Li JD, Yan C (2017) Vinpocetine attenuates pathological cardiac remodeling by inhibiting Cardiac Hypertrophy and Fibrosis. Cardiovasc Drugs Ther 31(2):157–166. https://doi.org/10.1007/s10557-017-6719-0
Hashimoto T, Kim GE, Tunin RS et al (2018) Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation 138(18):1974–1987. https://doi.org/10.1161/CIRCULATIONAHA.117.030490
Gilotra NA, DeVore AD, Povsic TJ et al (2021) Acute hemodynamic effects and tolerability of phosphodiesterase-1 inhibition with ITI-214 in human systolic heart failure. Circ Heart Fail 14(9):e008236. https://doi.org/10.1161/CIRCHEARTFAILURE.120.008236
Aye TT, Soni S, van Veen TA et al (2012) Reorganized PKA-AKAP associations in the failing human heart. J Mol Cell Cardiol 52(2):511–518. https://doi.org/10.1016/j.yjmcc.2011.06.003
Mehel H, Emons J, Vettel C et al (2013) Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J Am Coll Cardiol 62(17):1596–1606. https://doi.org/10.1016/j.jacc.2013.05.057
Galindo-Tovar A, Vargas ML, Kaumann AJ (2018) Phosphodiesterase PDE2 activity, increased by isoprenaline, does not reduce beta-adrenoceptor-mediated chronotropic and inotropic effects in rat heart. Naunyn Schmiedebergs Arch Pharmacol 391(6):571–585. https://doi.org/10.1007/s00210-018-1480-x
Zoccarato A, Surdo NC, Aronsen JM et al (2015) Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res 117(8):707–719. https://doi.org/10.1161/CIRCRESAHA.114.305892
Baliga RS, Preedy MEJ, Dukinfield MS et al (2018) Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci USA 115(31):E7428–E7437. https://doi.org/10.1073/pnas.1800996115
Vettel C, Lindner M, Dewenter M et al (2017) Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res 120(1):120–132. https://doi.org/10.1161/CIRCRESAHA.116.310069
Wagner M, Sadek MS, Dybkova N et al (2021) Cellular mechanisms of the anti-arrhythmic effect of Cardiac PDE2 overexpression. Int J Mol Sci 22(9):4816. https://doi.org/10.3390/ijms22094816
Liu K, Li D, Hao G et al (2018) Phosphodiesterase 2A as a therapeutic target to restore cardiac neurotransmission during sympathetic hyperactivity. JCI Insight 3(9):e98694. https://doi.org/10.1172/jci.insight.98694
Barbagallo F, Xu B, Reddy GR et al (2016) Genetically encoded biosensors reveal PKA hyperphosphorylation on the myofilaments in rabbit heart failure. Circ Res 119(8):931–943. https://doi.org/10.1161/CIRCRESAHA.116.308964
Verde I, Pahlke G, Salanova M et al (2001) Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J Biol Chem 276(14):11189–11198. https://doi.org/10.1074/jbc.M006546200
Dodge-Kafka KL, Soughayer J, Pare GC et al (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437(7058):574–578. https://doi.org/10.1038/nature03966
Dodge KL, Khouangsathiene S, Kapiloff MS et al (2001) mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J 20(8):1921–1930. https://doi.org/10.1093/emboj/20.8.1921
Ahmad F, Shen W, Vandeput F et al (2015) Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2. J Biol Chem 290(11):6763–6776. https://doi.org/10.1074/jbc.M115.638585
Vandecasteele G, Verde I, Rucker-Martin C, Donzeau-Gouge P, Fischmeister R (2001) Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J Physiol 533(Pt 2):329–340. https://doi.org/10.1111/j.1469-7793.2001.0329a.x
Leroy J, Abi-Gerges A, Nikolaev VO et al (2008) Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res 102(9):1091–1100. https://doi.org/10.1161/CIRCRESAHA.107.167817
Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R (1999) Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol 127(1):65–74. https://doi.org/10.1038/sj.bjp.0702506
Ono K, Trautwein W (1991) Potentiation by cyclic GMP of beta-adrenergic effect on Ca2+ current in guinea-pig ventricular cells. J Physiol 443:387–404. https://doi.org/10.1113/jphysiol.1991.sp018839
Fischmeister R, Hartzell HC (1990) Regulation of calcium current by low-Km cyclic AMP phosphodiesterases in cardiac cells. Mol Pharmacol 38(3):426–433
Yano M, Kohno M, Ohkusa T et al (2000) Effect of milrinone on left ventricular relaxation and Ca2+ uptake function of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol 279(4):H1898–H1905. https://doi.org/10.1152/ajpheart.2000.279.4.H1898
Malecot CO, Bers DM, Katzung BG (1986) Biphasic contractions induced by milrinone at low temperature in ferret ventricular muscle: role of the sarcoplasmic reticulum and transmembrane calcium influx. Circ Res 59(2):151–162. https://doi.org/10.1161/01.res.59.2.151
Movsesian MA, Smith CJ, Krall J, Bristow MR, Manganiello VC (1991) Sarcoplasmic reticulum-associated cyclic adenosine 5’-monophosphate phosphodiesterase activity in normal and failing human hearts. J Clin Invest 88(1):15–19. https://doi.org/10.1172/JCI115272
von der Leyen H, Mende U, Meyer W et al (1991) Mechanism underlying the reduced positive inotropic effects of the phosphodiesterase III inhibitors pimobendan, adibendan and saterinone in failing as compared to nonfailing human cardiac muscle preparations. Naunyn Schmiedebergs Arch Pharmacol 344(1):90–100. https://doi.org/10.1007/BF00167387
Ding B, Abe JI, Wei H et al (2005) Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation 111(19):2469–2476. https://doi.org/10.1161/01.CIR.0000165128.39715.87
Mika D, Bobin P, Lindner M et al (2019) Synergic PDE3 and PDE4 control intracellular cAMP and cardiac excitation-contraction coupling in a porcine model. J Mol Cell Cardiol 133:57–66. https://doi.org/10.1016/j.yjmcc.2019.05.025
Abi-Gerges A, Richter W, Lefebvre F et al (2009) Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res 105(8):784–792. https://doi.org/10.1161/CIRCRESAHA.109.197947
Ding B, Abe J, Wei H et al (2005) A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci USA 102(41):14771–14776. https://doi.org/10.1073/pnas.0506489102
Smith CJ, He J, Ricketts SG, Ding JZ, Moggio RA, Hintze TH (1998) Downregulation of right ventricular phosphodiesterase PDE-3A mRNA and protein before the development of canine heart failure. Cell Biochem Biophys 29(1–2):67–88. https://doi.org/10.1007/BF02737829
Smith CJ, Huang R, Sun D et al (1997) Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation 96(9):3116–3123. https://doi.org/10.1161/01.cir.96.9.3116
Hanna R, Nour-Eldine W, Saliba Y et al (2021) Cardiac phosphodiesterases are differentially increased in diabetic cardiomyopathy. Life Sci 283:119857. https://doi.org/10.1016/j.lfs.2021.119857
Polidovitch N, Yang S, Sun H et al (2019) Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol 132:60–70. https://doi.org/10.1016/j.yjmcc.2019.04.028
Li EA, Xi W, Han YS, Brozovich FV (2019) Phosphodiesterase expression in the normal and failing heart. Arch Biochem Biophys 662:160–168. https://doi.org/10.1016/j.abb.2018.12.013
Takahashi K, Osanai T, Nakano T, Wakui M, Okumura K (2002) Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessels 16(6):249–256. https://doi.org/10.1007/s003800200032
Holmes JR, Kubo SH, Cody RJ, Kligfield P (1985) Milrinone in congestive heart failure: observations on ambulatory ventricular arrhythmias. Am Heart J 110(4):800–806. https://doi.org/10.1016/0002-8703(85)90460-0
DiBianco R, Shabetai R, Kostuk W, Moran J, Schlant RC, Wright R (1989) A comparison of oral milrinone, digoxin, and their combination in the treatment of patients with chronic heart failure. N Engl J Med 320(11):677–683. https://doi.org/10.1056/NEJM198903163201101
Beard MB, Olsen AE, Jones RE, Erdogan S, Houslay MD, Bolger GB (2000) UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase family form a discrete module via electrostatic interactions. J Biol Chem 275(14):10349–10358. https://doi.org/10.1074/jbc.275.14.10349
Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD (1999) The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J 18(4):893–903. https://doi.org/10.1093/emboj/18.4.893
Mika D, Bobin P, Pomerance M et al (2013) Differential regulation of cardiac excitation-contraction coupling by cAMP phosphodiesterase subtypes. Cardiovasc Res 100(2):336–346. https://doi.org/10.1093/cvr/cvt193
Abi-Gerges A, Castro L, Leroy J, Domergue V, Fischmeister R, Vandecasteele G (2021) Selective changes in cytosolic beta-adrenergic cAMP signals and L-type calcium channel regulation by phosphodiesterases during cardiac hypertrophy. J Mol Cell Cardiol 150:109–121. https://doi.org/10.1016/j.yjmcc.2020.10.011
Qvigstad E, Moltzau LR, Aronsen JM et al (2010) Natriuretic peptides increase beta1-adrenoceptor signalling in failing hearts through phosphodiesterase 3 inhibition. Cardiovasc Res 85(4):763–772. https://doi.org/10.1093/cvr/cvp364
Minamisawa S, Hoshijima M, Chu G et al (1999) Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99(3):313–322. https://doi.org/10.1016/s0092-8674(00)81662-1
Schwinger RH, Bohm M, Schmidt U et al (1995) Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation 92(11):3220–3228. https://doi.org/10.1161/01.cir.92.11.3220
Meyer M, Schillinger W, Pieske B et al (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92(4):778–784. https://doi.org/10.1161/01.cir.92.4.778
Takimoto E, Champion HC, Belardi D et al (2005) cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res 96(1):100–109. https://doi.org/10.1161/01.RES.0000152262.22968.72
Mokni W, Keravis T, Etienne-Selloum N et al (2010) Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PLoS ONE 5(12):e14227. https://doi.org/10.1371/journal.pone.0014227
Pokreisz P, Vandenwijngaert S, Bito V et al (2009) Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 119(3):408–416. https://doi.org/10.1161/CIRCULATIONAHA.108.822072
Nagendran J, Archer SL, Soliman D et al (2007) Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 116(3):238–248. https://doi.org/10.1161/CIRCULATIONAHA.106.655266
Shan X, Quaile MP, Monk JK, French B, Cappola TP, Margulies KB (2012) Differential expression of PDE5 in failing and nonfailing human myocardium. Circ Heart Fail 5(1):79–86. https://doi.org/10.1161/CIRCHEARTFAILURE.111.961706
Garcia AM, Nakano SJ, Karimpour-Fard A et al (2018) Phosphodiesterase-5 is elevated in failing single ventricle myocardium and affects cardiomyocyte remodeling in vitro. Circ Heart Fail 11(9):e004571. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004571
Senzaki H, Smith CJ, Juang GJ et al (2001) Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J 15(10):1718–1726. https://doi.org/10.1096/fj.00-0538com
Takimoto E, Champion HC, Li M et al (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11(2):214–222. https://doi.org/10.1038/nm1175
Nakata TM, Suzuki K, Uemura A, Shimada K, Tanaka R (2019) Contrasting effects of Inhibition of phosphodiesterase 3 and 5 on cardiac function and interstitial fibrosis in rats with isoproterenol-induced cardiac dysfunction. J Cardiovasc Pharmacol 73(3):195–205. https://doi.org/10.1097/FJC.0000000000000652
Guazzi M, Vicenzi M, Arena R, Guazzi MD (2011) PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail 4(1):8–17. https://doi.org/10.1161/CIRCHEARTFAILURE.110.944694
Soderling SH, Bayuga SJ, Beavo JA (1998) Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci USA 95(15):8991–8996. https://doi.org/10.1073/pnas.95.15.8991
Wang H, Yan Z, Yang S, Cai J, Robinson H, Ke H (2008) Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry 47(48):12760–12768. https://doi.org/10.1021/bi801487x
Patrucco E, Albergine MS, Santana LF, Beavo JA (2010) Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J Mol Cell Cardiol 49(2):330–333. https://doi.org/10.1016/j.yjmcc.2010.03.016
Methawasin M, Strom J, Borkowski T et al (2020) Phosphodiesterase 9a Inhibition in mouse models of diastolic dysfunction. Circ Heart Fail 13(5):e006609. https://doi.org/10.1161/CIRCHEARTFAILURE.119.006609
Scott NJA, Rademaker MT, Charles CJ, Espiner EA, Richards AM (2019) Hemodynamic, hormonal, and renal actions of phosphodiesterase-9 inhibition in experimental heart failure. J Am Coll Cardiol 74(7):889–901. https://doi.org/10.1016/j.jacc.2019.05.067
Chen S, Zhang Y, Lighthouse JK et al (2020) A novel role of cyclic nucleotide phosphodiesterase 10A in pathological cardiac remodeling and dysfunction. Circulation 141(3):217–233. https://doi.org/10.1161/CIRCULATIONAHA.119.042178
Hall DD, Feekes JA, Arachchige Don AS et al (2006) Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45(10):3448–3459. https://doi.org/10.1021/bi051593z
El Refaey M, Musa H, Murphy NP et al (2019) Protein phosphatase 2A regulates cardiac Na(+) channels. Circ Res 124(5):737–746. https://doi.org/10.1161/CIRCRESAHA.118.314350
Lubelwana Hafver T, Wanichawan P, Manfra O et al (2017) Mapping the in vitro interactome of cardiac sodium Na+-calcium Ca2+ exchanger 1 (NCX1). Proteomics 17:17–18. https://doi.org/10.1002/pmic.201600417
Hua F, Wu Z, Yan X et al (2018) DR region of Na(+)-K(+)-ATPase is a new target to protect heart against oxidative injury. Sci Rep 8(1):13100. https://doi.org/10.1038/s41598-018-31460-z
Belevych AE, Sansom SE, Terentyeva R et al (2011) MicroRNA-1 and – 133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 6(12):e28324. https://doi.org/10.1371/journal.pone.0028324
Terentyev D, Belevych AE, Terentyeva R et al (2009) miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res 104(4):514–521. https://doi.org/10.1161/CIRCRESAHA.108.181651
Jideama NM, Crawford BH, Hussain AK, Raynor RL (2006) Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int J Biol Sci 2(1):1–9. https://doi.org/10.7150/ijbs.2.1
Yin X, Cuello F, Mayr U et al (2010) Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol Cell Proteom 9(3):497–509. https://doi.org/10.1074/mcp.M900275-MCP200
Mumby MC, Russell KL, Garrard LJ, Green DD (1987) Cardiac contractile protein phosphatases. Purification of two enzyme forms and their characterization with subunit-specific antibodies. J Biol Chem 262(13):6257–6265
Wu SC, Solaro RJ (2007) Protein kinase C zeta. A novel regulator of both phosphorylation and de-phosphorylation of cardiac sarcomeric proteins. J Biol Chem 282(42):30691–30698. https://doi.org/10.1074/jbc.M703670200
Surks HK, Mochizuki N, Kasai Y et al (1999) Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286(5444):1583–1587. https://doi.org/10.1126/science.286.5444.1583
Ravan H, Yazdanparast R (2012) Development of a new loop-mediated isothermal amplification assay for prt (rfbS) gene to improve the identification of salmonella serogroup D. World J Microbiol Biotechnol 28(5):2101–2106. https://doi.org/10.1007/s11274-012-1014-5
Cai Z, Wu C, Xu Y, Cai J, Zhao M, Zu L (2023) The NO-cGMP-PKG Axis in HFpEF: from pathological mechanisms to potential therapies. Aging Dis 14(1):46–62. https://doi.org/10.14336/AD.2022.0523
Ladage D, Tilemann L, Ishikawa K et al (2011) Inhibition of PKCalpha/beta with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circ Res 109(12):1396–1400. https://doi.org/10.1161/CIRCRESAHA.111.255687
Zhang J, Liang R, Wang K et al (2022) Novel CaMKII-delta inhibitor hesperadin exerts dual functions to ameliorate cardiac ischemia/reperfusion injury and inhibit tumor growth. Circulation 145(15):1154–1168. https://doi.org/10.1161/CIRCULATIONAHA.121.055920
Funding
The current study was supported by the Natural Science Foundation of Shandong Province (ZR2023QH190).
Author information
Authors and Affiliations
Contributions
Yan-Bing Liu conceptualized the topic and idea and prepared the first draft and all figures. Qian Wang and Yu-Ling Song were responsible for reference research and information collection. Xiao-Min Song, Yu-Chen Fan, Lin Kong, Jing-Sai Zhang, Sheng Li, Yi-Ju Lv, and Ze-Yang Li were responsible for editing the first draft and finalizing the manuscript. Jing-Yu Dai revised and polished the language of the manuscript. Jing-Yu Dai and Zhen-Kang Qiu approved the final version.
Corresponding authors
Ethics declarations
Ethical standards
The manuscript does not contain clinical studies or patient data.
Competing interests
Yan-Bing Liu, Qian Wang, Yu-Ling Song, Xiao-Min Song, Yu-Chen Fan, Lin Kong, Jing-Sai Zhang, Sheng Li, Yi-Ju Lv, Ze-Yang Li, Jing-Yu Dai, and Zhen-Kang Qiu have no conflicts of interest or financial ties to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, YB., Wang, Q., Song, YL. et al. Abnormal phosphorylation / dephosphorylation and Ca2+ dysfunction in heart failure. Heart Fail Rev 29, 751–768 (2024). https://doi.org/10.1007/s10741-024-10395-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-024-10395-w