
Abstract
Curcuma kwangsiensis S. K. Lee & C. F. Liang, a traditional herb in southern China, accumulates substantial amounts of active components, curcuminoids and volatile oil. However, the limited hereditary information and SSR markers have hindered its breeding program and genetic analysis. Here, we examined the transcriptome of C. kwangsiensis using next-generation sequencing (NGS) technology. A dataset with 8.17 Gb of raw reads was generated and assembled into 77,976 unigenes. Moreover, 11,678 EST-SSR markers were screened from transcriptome data. Of the 800 synthesized primer pairs, 486 (60.8%) exhibited successful amplification and 115 (23.7%) were polymorphic. A set of 24 selected markers showed high cross-species transferabilities among 13 Curcuma species. In total, 277 alleles (6–19 alleles per locus) were discovered, and the polymorphic information content (PIC) ranged from 0.496 to 0.905. Based on the results of cluster and structure analyses, the 75 accessions were classified into four major groups with some admixtures. Finally, a core collection (22 accessions) was identified, exhibiting Na, Ne, I, and PIC values with retention rates of 87.7%, 107.8%, 102.5% and 102%, respectively. All these unigenes and EST-SSRs will be useful for germplasm resource evaluation and for diversifying the potential of product derivatives from Curcuma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Curcuma is a rhizomatous perennial herb comprises of more than 110 species in the family Zingiberaceae, and is commonly distributed in tropical and subtropical regions (Dosoky and Setzer 2018). Curcuma kwangsiensis S. K. Lee & C. F. Liang, also known as Guangxiezhu, is a native species in South China since ancient times. It has been broadly cultivated as cut flower and pot plant for the purposes of unique foliage and colorful bract. As a traditional Chinese medicinal material, it can be used for treating injuries, abdominal coldness and hypochondria (Zeng et al. 2008). The bioactivities of curcuminoids and volatile oil are widely applied in medicine, food and cosmetics (Xu et al. 2018). Moreover, essential oils are considered as one of the most important active constituents of C. kwangsiensis, and have been reported to elicit diverse biological effects such as antioxidant, anti-inflammatory, antiviral and antitumor activities (Sacchetti et al. 2005). In addition to C. kwangsiensis, most Curcuma species are edible, ornamental and medicinal plants, including C. alismatifolia Gagnep. (Theanphong and Mingvanish 2017), C. amada Roxb. (Banerjee et al. 2012) and C. longa L. (Zhang et al. 2017). At present, the greatest challenge is the selection of elite genotypes and development of superior parental lines from these germplasm resources. Therefore, assessment of the genetic relationship between C. kwangsiensis and related species is of prime importance.
Although there is an increasing demand for the cultivation of C. kwangsiensis due to its immense economic and pharmaceutical values, little genomic information is available regarding its evolution process and genetic variation (Sun et al. 2018). For sustainable and effective utilization of this plant resource, the analysis of genetic diversity is of foremost importance. In previous genetic studies, various DNA markers have been implemented for the detection of genetic diversity in Curcuma spp. To date, molecular marker development and genetic diversity investigations have been mainly focused on C. alismatifolia and C. longa (Ismail et al. 2016). However, informative markers for elucidating the population genetics of C. kwangsiensis are still lacking.
Compared to other genetic markers, simple sequence repeats (SSRs) are always preferable due to their codominant inheritance, good transferability, multiple alleles and high reproducibility (Zhou et al. 2019). Traditional techniques (e.g., biotin-streptavidin capture method) are usually based on the sequencing of SSR colonies and double-enriched microsatellite library (Barboza et al. 2018). With no availability of genomic information, these technologies are costly, laborious and time consuming. However, the rapid identification of expressed sequence tag (EST)-SSR markers from expressed sequences can be performed at a much lower cost (Pan et al. 2018). Through the use of next-generation sequencing, massive amounts of EST-SSRs can be obtained from transcriptome data at a high-throughput scale. In recent years, many EST-SSR markers were discovered by high-throughput sequencing in different plant species, which have been proven to be reliable and cost effective (Zhang et al. 2019).
Managing the genetic resources of precious germplasm collections is important for conserving their genetic diversity (Duan et al. 2017). However, the heterogeneous structure, redundant genotypes, and lacking of trait information can influence the usefulness of genetic variability. Considering that it is difficult to fully characterize all the individual plants due to lack of time, space and labor, the construction of a core collection with smaller accessions for obtaining the genetic information of the initial collections is urgently needed (Di Guardo et al. 2019). Previous genetic diversity analysis has revealed that a representative genotype-based core subset is able to capture the maximum amount of genetic diversity in a plant population (Frankel 1984; Frankel and Brown 1984). At present, several core collections have been successfully developed in economically important and fruit trees, including Vitis vinifera L. (Le Cunf et al. 2008), Olea europaea L. (Belaj et al. 2012), and Ziziphus jujuba Mill. (Xu et al. 2016). However, the core collection of Curcuma has yet to be constructed.
In the present study, we aimed to (1) characterize EST-SSR markers based on transcriptome data at a high-throughput scale; (2) assess the genetic diversity of 75 Curcuma accessions through the use of these markers; and (3) construct a core subset for the entire population.
Material and methods
Total RNA and genomic DNA extraction
Fresh leaves of C. kwangsiensis were collected and immediately frozen in liquid nitrogen at − 80˚C for RNA isolation. Total RNA was extracted from each sample using the RNAprep pure plant kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. The quality and quantity of RNA were examined using an Agilent 2100 Bioanalyzer and a Nanodrop 2000 spectrophotometer, respectively. For SSR marker analysis, 75 Curcuma accessions collected from different areas were employed as a test population. The information of the collection process are summarized in Supplementary Table S1. Meanwhile, genomic DNA was extracted from fresh leaf tissue by following the method of Ye et al. (2019). The integrity and quantity of DNA were assessed by 1% TAE agarose gel electrophoresis and a Nanodrop 2000 spectrophotometer, respectively.
RNA sequencing and functional analysis
cDNA library construction was performed with the RNA samples of C. kwangsiensis, and the sequence length was set to 200 bp. For paired-end RNA sequencing, Illumina HiSeq 2500 platform (Illumina, San Diego, CA) was used to sequence both ends of the cDNA fragments. The quality of raw reads was evaluated with SeqQC-V2.2 program, and high-quality reads (Q < 20) were assembled using the Trinity software with default parameters. Only the high-quality, filtered reads were utilized for subsequent analyses. The assembled sequences were then compared against the non-redundant protein database (nr) by running BLASTX with a threshold E-value of 10–6. The unigene annotations and mapping routines were conducted with gene ontology (GO) and kyoto encyclopedia of genes genomes (KEGG) analyses by searching and comparing public databases. Finally, the assembled unigenes were deposited to the figshare database archived at: https://doi.org/10.6084/m9.figshare.11955270.v4.
EST-SSR validation and polymorphism examination
Micro Satellite (MISA, http://www.pgrc.ipkgatersleben.de/misa) was employed to identify SSR markers based on the unigene sequences. The search principle and primer design of each EST-SSR were described previously (Wu et al. 2014). PCR amplification for 6 randomly selected samples was assessed with 1.5% agarose gel electrophoresis. The successfully amplified polymorphic markers was further examined with 8% polyacrylamide gel electrophoresis (PAGE). To determine genetic relationships, the forward primer of SSR markers was elongated from the M13 primer appended to the 5′-end (Schuelke 2000). Approximately 0.5 μL of PCR products with different fluorescent labels and sizes were pooled and detected using a DNA Analyzer.
Genetical and statistical analyses
Allele sizes were corrected with FlexiBin ver. 2.0 and GeneMarker ver. 2.20 (SoftGenetics LLC., State College, PA, USA). The allele number and polymorphic information content (PIC) of each polymorphic SSR loci were determined using PowerMarker ver. 3.25 program (Liu and Muse 2005). The basic genetic statistics, principal coordinate analysis (PCoA) and analysis of molecular variance (AMOVA) were conducted using GenAlEx ver. 6.0 software (Peakall and Smouse 2006) and POPGENE ver. 1.32 software (Yeh et al. 1999).
The population structure of 75 Curcuma accessions was estimated using STRUCTURE ver. 2.3 program (Pritchard et al. 2000). Delta K Evanno's method (Evanno et al. 2005) was used to calculate the optimal K value in STRUCTURE HARVEST (Earl and vonHoldt 2012). The bar plots for the probability of membership obtained from STRUCTURE data were visualized using DISTRUCT (Rosenberg 2004) and CLUMPAK (Kopelman et al. 2015). Genetic distance matrix was constructed by PowerMarker using the shared allele distance. Cluster analysis was performed to explore the relationships among 73 Curcuma accessions by using an unweighted pair-group with arithmetic mean (UPGMA) based on Nei’s unbiased genetic distances (Backeljau et al. 1996).
Construction of core collection
According to the number of accessions, a progressive sampling strategy was used to develop the core subset, by establishing 12 core collections to verify the optimum size. To ensure the reliability of the core subset establishment, 5 repetitive runs were performed with two different approaches (i.e., random search and simulated annealing) by using PowerMarker ver. 3.25 program. PowerCore software (Kim et al. 2007) was employed for subsequent data screening. The analysis was repeated 1000 times until meeting the requirement of representativeness or achieving the appropriate number of accessions. Lastly, genetic parameters-based T-test was used to determine the association between the initial collection and the core subset. The statistical analysis was carried out using SPSS ver. 18.0 package.
Results
Transcriptome assembly and annotation
In total, 8.17 Gb of raw reads were obtained and assembled into 77,976 unigenes with a mean length of 1433 bp (N50 = 1969 bp; Fig. 1a). The length of the unigenes ranged from 201 to 13,396 bp with a total of 87,071,461 nucleotides. The unigenes with a length of 1000–2000 bp (25,414, 32.6%) were the most abundant types. Besides, species classification results found that 78% of the unigenes were successfully annotated in the Musa acuminata Colla database (Fig. 1b). Other species displayed sequence similarities of less than 5%, including Elaeis guineensis Jacq. (4.6%), Phoenix dactylifera L. (3.6%), Ananas comosus (L.) Merr. (2.4%), etc.

Characterization and functional classification of C. kwangsiensis unigenes. a Unigene length distribution; b Species classification of the annotated unigenes; c GO annotation; and d KEGG pathway mapping
For the functional analysis of unigenes in C. kwangsiensis, 24,321 unigenes were classified into biological processes (11,651, 47.9%), cellular components (7023, 28.9%) and molecular functions (5647, 23.2%) by using Blast2GO software (Fig. 1c). Within the biological processes category, cell process (22.7%) and metabolic process (21%) were the dominant terms. Of the 18 cellular component terms, cell (19.9%) and cell part (19.9%) were the most abundant components. Among the 10 molecular function terms, the binding (47.9%) component was most highly represented. KEGG analysis showed that 27,010 unigenes were classified into 158 pathways and categorized into 5 clades (Fig. 1d). Of the 19 subgroups, translation (2577, 9.5%) was the most represented pathway, followed by carbohydrate metabolism (2286, 8.5%), folding, sorting and degradation (1842, 6.8%), and overview pathways (1645, 6.1%).
Identification of EST-SSR markers
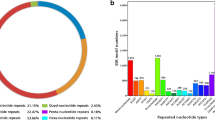
In total, 11,678 EST-SSR markers were obtained from the 77,976 identified EST sequences (Table 1). Trinucleotide repeats were found to be the most common (6955, 59.6%), while di- (4048, 34.7%), tetra- (371, 3.2%), hexa- (203, 1.7%) and penta-nucleotides (101, 0.8%) were the next most frequent in consecutive order (Fig. 2a). The number of SSR repeat motifs ranged from 5 to 36, and trinucleotides with 5–8 repeat motifs (55.9%) were the most dominant, followed by dinucleotides with 5–8 (22.9%) and 13–16 (7.7%) repeat motifs. In addition, the frequency distributions of major di- and tri-nucleotide repeat units were also evaluated. Among the dinucleotide motifs, AG/CT with 6 repeats (18.3%) was the most frequent repeat type (Fig. 2b). Moreover, the richest repeat type of trinucleotide motifs was GAA/TTC with5 repeats (12.4%). All the primer sequences were deposited to the figshare database archived at: https://doi.org/10.6084/m9.figshare.11948541.v1.

Basic characteristics of the EST-SSR markers. a The frequencies of different repeat classes. b The distributions of di- and trinucleotide motifs
Polymorphism detection and transferability of EST-SSRs
In total, 800 EST-SSR primers were ultimately synthesized according to the program criteria, including 268 (33.5%) dinucleotide, 420 (59.5%) trinucleotide and 112 (12.9%) other motif types. Of these primer pairs, 486 (60.8%) of them exhibited successful amplification with the correct sizes. However, 30 PCR products demonstrated larger sizes than the expected ones, suggesting the existence of an intron in the amplified regions. Considering that the remaining SSRs may not produce any PCR bands, they were excluded from subsequent analyses. Of the successfully amplified primers, 115 (23.7%) of them were found to be polymorphic in 8 Curcuma accessions.
Next, 24 EST-SSR markers were employed to determine the genetic relationship among 75 Curcuma accessions (Table 2). Altogether, 277 polymorphic alleles were screened with an average of 11.542 alleles for each primer pair. The parameters, such as Ne, I, Ho and He are shown in Table 3. It is worth noting that PIC can serve as a valuable index for assessing the performance of EST-SSR markers. In this study, the values of PIC ranged from 0.496 (JHH28) to 0.905 (JHH54) with an average of 0.754, implicating that the polymorphic EST-SSR markers can be used to analyze the genetic diversity of Curcuma species. Consequently, the established EST-SSR markers were employed to measure cross-species transferability and evolutionary conservation, and a high transfer rate of 73.1% was detected among 13 Curcuma species (Supplementary Table S2).
Genetic diversity and population structure analyses
A clustering tree using neighbor-joining (NJ) method was constructed to analyze the genetic relationship, in which the 75 accessions were clearly divided into four clades (Fig. 3). All the C. alismatifolia cultivars were classified into clade 1, clade 2 and clade 3, comprising of 26, 10 and 16 accessions, respectively. However, there were some exceptions in clade 3, such as Cros, Ccor and Cpha01. The remaining accessions were assigned into clade 4, including ten Curcuma species. Clade 4 revealed a unique evolutionary pattern of this species, which were consistent with its origins and previous findings. The genetic parameters were highest in clade 4, whereas the lowest value were found in clade 2 (Fig. 4). At the population level, 114 private alleles were found in the four populations and distributed across 24 loci, with frequencies ranging from 0.019 to 0.400. Notably, 11 private alleles were observed in C. phaeocaulis Valeton, followed by C. kwangsiensis (n = 7) and C. cordata Wall. (n = 6). In overall, 44.7% of the private alleles were detected in clade 4, suggesting that the accessions in this group possess informative genetic diversity and may share the same ancestry.

Neighbor-joining tree of 75 Curcuma accessions based on 24 EST-SSR markers

Graphical representation of the allelic patterns across different clades
A two-dimensional scatter plot illustrating the dispersion among the 75 accessions was constructed using PCoA (Supplementary Fig. S1). The four clusters had good consistency with the corresponding clades produced by the clustering tree. Coordinate 1 explained 36.99% of the variance in the genotype data and distinguished clusters 3 and 4 from clusters 1 and 2. Coordinate 2 explained 21.25% of the variance in the genotype data and separated clusters 1 and 4 from clusters 2 and 3.
In the population structure analysis, the delta K approach generated an obvious peak at K = 4, where all accessions were classified into four main groups (Supplementary Fig. S2). Overall, the structure results corroborated the dendrogram with some exceptions due to the mixed ancestry (membership value < 80%). The highest number of samples was found in group 1 (29), followed by group 4 (24), group 2 (11) and group 3 (11). The accessions in group 1–3 were referred to C. alismatifolia cultivars with some admixture among groups. In particular, group 4 contained some accessions with high importance values, and most of them were typical ornamental and medical plants.
Construction of the core subset
Twelve core collections were established, accounting for 11–40% of the total accessions. The results demonstrated that the I, Na, Ne and PIC values of a core subset with 22 genotypes were increased, with the corresponding retention rates of 102.5%, 87.7%, 107.8% and 102%, respectively (Fig. 5). Meanwhile, the 22 core individuals were divided into four clusters using the newly developed markers. Seven Curcuma species were grouped into cluster 1, while the other 15 C. alismatifolia cultivars were grouped into the remaining three clusters. The core individuals in each cluster were highly consistent with the origins of each clade based on the NJ tree above (Supplementary Fig. S3). The allele frequencies of the core subset were highly correlated (R2 = 0.923) with those of the whole collection, indicating the excellent representation of the core collection (Supplementary Fig. S4).

Comparison of the genetic parameters among different core collections, including a Number of alleles. b Number of effective alleles. c Shannon's information index. d Polymorphic information content
Discussion
Curcuma kwangsiensis, an important perennial herb in South China, has been widely applied in cosmetics, food and medicine (Zhang et al. 2011). Previous research on this species has mainly focused on plant tissue culture and medicinal component analysis (Komatsu et al. 2008). However, the exploration and decipherment of novel genomic information and genetic variation are yet to be performed. To improve its yield and industrial value, it is necessary for breeders to understanding the hereditary information of this crop at the molecular level. However, the limited number of molecular markers has severely hindered the genetic assessment of C. kwangsiensis and its breeding programs.
Next-generation sequencing has revolutionized the fields of transcriptomics and genomics, making it possible to identify large numbers of functional genes and novel molecular markers (Ellegren 2014; Taheri et al. 2018). This technology was used to generate large volumes of data for non-model species, due to its high throughput, low cost and high accuracy (Shukla et al. 2015). By taking advantage of this technology, a large fraction of the transcriptome was obtained for C. kwangsiensis. In total, 8.17 Gb of raw data were generated after sequencing. The N50 and mean length of the assembled unigenes were 1969 bp and 1433 bp, respectively, which was relatively comparable to those obtained from the de novo transcriptome in other species, including C. alismatifolia (1501 bp, 994 bp) (Taheri et al. 2019), C. longa (424 bp, 367 bp) and C. aromatica Salisb. (410 bp, 359 bp) (Sheeja et al. 2015). Notably, the higher N50 value and longer average length of C. kwangsiensis indicate a more accurate and effective assembly for the transcriptome. For gene annotation, 78% of the unigenes were matched with wild banana (M. acuminata), which might be attributed to the fact that they all belonged to Zingiberales and shared similar genome sequences in the process of evolution. Using the GO database, we identified 24,321 unigenes involved in molecular functions, cellular components and biological processes. In addition, 27,010 unigenes were annotated into 158 pathways and 5 subcategories according to KEGG analysis. To our knowledge, GO annotation helps us elucidate the function of unigenes at the macro and physiological levels, and KEGG analysis provides a basis for systematic gene function annotation with regard to gene product networks (Kanehisa et al. 2012). These findings may facilitate further research on the biological function-specific unigenes in C. kwangsiensis or other related species.
SSR marker is preferred for genetic analysis due to its high level of codominance, reproducibility and polymorphism (Barboza et al. 2018). Prior to this study, there are limited SSR markers available for this unique ornamental and medicinal crop. In the present work, we identified 11,678 EST-SSRs from the 77,976 EST sequences. The trinucleotide motifs were the most abundant, which was consistent with prior researches conducted on C. alismatifolia (Taheri et al. 2019), Zingiber officinale Roscoe (Awasthi et al. 2017), colored calla lily (Wei et al. 2016) and pummelo (Liang et al. 2015). However, Ye et al. (2019) found that dinucleotide motifs were the most abundant types in the crape myrtle genome, instead of trinucleotide motifs. Other studies also demonstrated that dinucleotide motifs was the most frequent type (Rowland et al. 2012; Li et al. 2018). All these findings imply that the dominant repetition motifs are highly variable among different plant species.
In addition, the 800 SSR primer pairs exhibited a higher amplification efficiency (60.8%) compared to other reported species, such as Taxodium (51.1%) (Cheng et al. 2015) and tree peony (47.3%) (Wu et al. 2014). However, 30 PCR products were larger than expected, which might be attributed to the occurrence of large insertion fragments, long intervening introns, large repeat numbers, or sequence assembly errors (Wei et al. 2011). Generally, the PIC value more than 0.5 reflects a high degree of molecular marker polymorphism (Bostein et al. 1980). Herein, a mean PIC value of 0.754 implicated that these primer pairs could be used to assess the genetic diversity and relationships among Curcuma species. The high abundance of polymorphisms may be attributed to the complex genetic architecture of the tested germplasm or the contingent selection of polymorphic EST-SSR markers.
Furthermore, the EST-SSR markers were chosen to evaluate the cross-species transferability among 13 Curcuma species. Notably, a high transfer rate of 73.1% was identified in most Curcuma species. Such excellent transferability may becaused by the mild conservation of the SSR-flanking sequences among the four species (Liu et al. 2019). However, only 6 out of 24 markers amplified the expected bands in C. rubescens Roxb., suggesting that it may differ from other species evolutionarily. Two markers (JHH2 and JHH12) exhibited strong cross-species transferability in all the Curcuma species. Hence, these reliable EST-SSR markers can be applied for genome mapping, ancestry tracing and evolutionary genetic analysisin the near future.
The population structure and genetic diversity of the whole collection were evaluated using 24 polymorphic EST-SSR makers. Our findings indicated that the grouping in STRUCTURE was in good agreement with the cluster analysis. Although few exceptions were observed in the four populations, the data of cluster analysis was convincing. The results of dendrogram analysis revealed that all the individual plants were primarily clustered according to their geographical origins and growth habits, which were partially consistent with those of Syamkumar and Sasikumar (2007) as well as Jan et al. (2011). Nearly all the C. alismatifolia cultivars were divided into 3 clades, while the other Curcuma species were clustered together. This indicates that genetic clustering can be used to assess whether the four populations share the same pedigrees or breeding strategies. However, few exceptions still exist, for example, Cros, Ccor and Cpha01 in clade 3. Given that the four Curcuma species were procured, transported and bred concurrently, the mislabeling may have happened because of the nearly similar crop types and indiscernible flowers.
Additionally, we found that clade 4 exhibited the highest value of genetic diversity, suggesting that the individuals in this clade can capture abundant genetic information. Previous studies also showed that the accessions of this clade were rich in the main active components, curcuminoids and volatile oil (Dosoky and Setzer 2018; Jain and Parihar 2019). In the present work, 11 private alleles were identified in C. phaeocaulis, followed by C. kwangsiensis (n = 7) and C. cordata (n = 6). The majority of private alleles (44.7%) were observed in clade 4, and it is strongly believed that these Curcuma species share a unique ancestor. Therefore, novel strategies are needed to sharply focus on the conservation of rare private alleles and utilization of precious germplasm resources.
Indeed, it is difficult and expensive to characterize the genetic diversity of the entire germplasm collection. A core subset with minimum repetitiveness and maximum genetic diversity should be constructed to represent the entire collection of a plant species (Anoumaa et al. 2017). Previous research has suggested that an appropriate sampling ratio should be employed according to the genetic features of various germplasm collections (Xu et al. 2016). Typically, the sample size of 10–30% is required to cover the high levels of genetic variability in a plant population. Several important indices of I, Na, Ne and PIC are commonly used to estimate the genetic diversity of a core collection. Herein, we established a core subset with 29.3% sampling ratio, with the highest values of I, Na, Ne, and PIC (retention rates = 102.5%, 87.7%, 107.8% and 102%, respectively). The results are comparable to the findings of recently published core collection studies, such as O. europaea (Belaj et al. 2012), Sinojackia huangmeiensis J.W. Ge & X.H. Yao (Zhao et al. 2016), and Z. jujuba (Xu et al. 2016). No significant difference was observed between the core subset and the entire collection (P < 0.05), implying that the core subset can be used to represent the entire collection. Therefore, a core subset with the highest retention rate and lowest sample size is highly recommended.
The core subset established in this study is particularly useful to breed C. kwangsiensis, which can also serve as applied for effective sampling and gene mining of a germplasm collection. However, it should be noted that the detection and identification of genetic variants in a core subset is not sufficient to the entire genetic information of a plant population. The sample size of the entire collection and limited genotype data can affect the performance of the core subset. Thus, further research should focus on the improvement of this core collection via morphological characterization of the accessions, incorporation of new accessions and enrichment of the available genetic data.
Conclusions
In this study, we developed 11,678 EST-SSR markers from the transcriptome data of C. kwangsiensis. Of the 115 polymorphic markers, a set of 24 selected markers showed high cross-species transferabilities among 13 Curcuma species. Based on the results of genetic diversity and population structure analysis, the 75 accessions could be effectively divided into four major groups according to their geographical origins and growth habits. Finally, a core collection (22 accessions) was constructed, which was useful for effective sampling and gene mining of the germplasm resources. In order to improve the accuracy and reliability of genetic study in Curcuma, more sample size and genotype data should be incorporated for the further research.
References
Anoumaa M, Yao NK, Kouam EB, Kanmegne G, Machuka E, Osama SK, Nzuki I, Kamga YB, Fonkou T, Omokolo DN (2017) Genetic diversity and core collection for potato (Solanum tuberosum L.) cultivars from cameroon as revealed by SSR markers. Am J Potato Res 94:449–463
Awasthi P, Singh A, Sheikh G, Mahajan V, Gupta AP, Gupta S, Bedi YS, Gandhi SG (2017) Mining and characterization of EST-SSR markers for Zingiber officinale Roscoe with transferability to other species of Zingiberaceae. Physiol Mol Biol Pla 23:925–931
Backeljau T, De Bruyn L, De Wolf H, Jordaens K, Van Dongen S, Winnepennincks B (1996) Multiple UPGMA and neighbor-joining trees and the performance of some computer packages. Mol Biol Evol 13:309
Banerjee S, Singh S, Pandey H, Pandey P, Rahman LU (2012) Conservation and storage of Curcuma amada Roxb. synseeds on Luffa sponge matrix and RAPD analysis of the converted plantlets. Ind Crop Prod 36:383–388
Barboza K, Beretta V, Kozub PC, Salinas C, Morgerfeld MM, Galmarini CR, Cavagnaro PF (2018) Microsatellite analysis and marker development in garlic: distribution in EST sequence, genetic diversity analysis, and marker transferability across Alliaceae. Mol Genet Genomics 293:1091–1106
Belaj A, del Carmen D-G, Atienza SG, Urdíroz NM, De la Rosa R, Satovic Z, Martín A, Kilian A, Trujillo I, Valpuesta V, Del Río C (2012) Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet Genomes 8:365–378
Bostein D, White RL, Sholnick M, David RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am J Hum Genet 32:314–331
Cheng YL, Yang Y, Wang ZY, Qi BY, Yin YL, Li HG (2015) Development and characterization of EST-SSR markers in Taxodium ‘zhongshansa.’ Plant Mol Biol Rep 33:1–11
Di Guardo M, Scollo F, Ninot A, Rovira M, Hermoso JF, Distefano G, La Malfa S, Batlle I (2019) Genetic structure analysis and selection of a core collection for carob tree germplasm conservation and management. Tree Genet Genomes 15:41
Dosoky NS, Setzer WN (2018) Chemical composition and biological activities of essential oils of Curcuma species. Nutrients 10:1196
Duan HJ, Cao S, Zheng HQ, Hu DH, Lin J, Cui BB, Lin HZ, Hu RY, Wu B, Sun YH, Li Y (2017) Genetic characterization of Chinese fir from six provinces in southern China and construction of a core collection. Sci Rep 7:13814
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellegren H (2014) Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol (Amst) 29:51–63
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the sofware STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Frankel H (1984) Genetic perspectives of germplasm conservation. In: Arber WK, Llimensee K, Peacock WJ, Starlinger P (eds) Genetic manipulation: impact on man and society. Cambridge University Press, Cambridge, pp 161–170
Frankel H, Brown AHD (1984) Current plant genetic resources-A critical appraisal. In: Chopra VL, Joshi BC, Sharma RP, Bansal HC (eds) Genetics: new frontiers. Oxford & IBH Publishing Co., New Delhi India, pp 3–13
Ismail NA, Rafii MY, Mahmud TMM, Hanafi MM, Miah G (2016) Molecular markers: a potential resource for ginger genetic diversity studies. Mol Biol Rep 43:1347–1358
Jain A, Parihar DK (2019) Molecular marker based genetic diversity study of wild, cultivated and endangered species of Curcuma from Chhattisgarh region for in situ conservation. Biocatal Agric Biotechnol 18:101033
Jan HU, Rabbani MA, Shinwari ZK (2011) Assessment of genetic diversity of indigenous turmeric (Curcuma longa L.) germplasm from Pakistan using RAPD markers. J Med Plants Res 5(5):823–830
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114
Kim KW, Chung HK, Cho GT, Ma KH, Chandrabalan D, Gwag JG, Kim TS, Cho EG, Park YJ (2007) PowerCore: a program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics 23:2155–2162
Komatsu K, Sasaki Y, Tanaka K, Kuba Y, Fushimi H, Cai SQ (2008) Morphological, genetic, and chemical polymorphism of Curcuma kwangsiensis. J Nat Med 62:413–422
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Le Cunff L, Fournier-Level A, Laucou V, Vezzulli S, Lacombe T, Adam-Blondon AF, Boursiquot JM, This P (2008) Construction of nested genetic core collections to optimize the exploitation of natural diversity in Vitis vinifera L. subsp. sativa. BMC Plant Biol 8(1):31
Li N, Zheng YQ, Ding HM, Li HP, Peng HZ, Jiang B, Li HB (2018) Development and validation of SSR markers based on transcriptome sequencing of Casuarina equisetifolia. Trees 32:41–49
Liang M, Yang XM, Li H, Su SY, Yi HL, Chai LJ, Deng XX (2015) De novo transcriptome assembly of pummelo and molecular marker development. PLoS ONE 10:e0120615
Liu FM, Hong Z, Yang ZJ, Zhang NN, Liu XJ, Xu DP (2019) De novo transcriptome analysis of Dalbergia odorifera T. Chen (Fabaceae) and transferability of SSR markers developed from the transcriptome. Forests 10(2):98
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Pan L, Huang T, Yang ZF, Tang L, Cheng YJ, Wang JP, Ma X, Zhang XQ (2018) EST-SSR marker characterization based on RNA-sequencing of Lolium multiflorum and cross transferability to related species. Mol Breeding 38:80
Peakall R, Smouse PE (2006) GenAlEx 6.5: genetic analysis in Excel. Population genetic sofware for teaching and research-an update. Bioinformatics 28:2537–2539
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, Main D (2012) Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol 12:46
Sacchetti G, Maietti S, Muzzoli M, Scaglianti M, Manfredini S, Radice M, Bruni R (2005) Comparative evaluation of 11 essential oils of different origin as functional antioxidants, antiradicals and antimicrobials in foods. Food Chem 91:621–632
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Sheeja TE, Deepa K, Santhi R, Sasikumar B (2015) Comparative transcriptome analysis of two species of Curcuma contrasting in a high-value compound curcumin: insights into genetic basis and regulation of biosynthesis. Plant Mol Biol Rep 33:1825–1836
Shukla A, Singh VK, Bharadwaj DR, Kumar R, Rai A, Rai AK, Mugasimangalam R, Parameswaran S, Singh M, Naik PS (2015) De novo assembly of bitter gourd transcriptomes: gene expression and sequence variations in gynoecious and monoecious lines. PLoS ONE 10:e0128331
Sun JR, Bu JL, Zhao H, Mao YP, Zeng W, Guo J, Huang LQ (2018) Multivariate data analysis of volatile metabolites in rhizomes and radixes of four medicinal plants from Curcuma L. Acta Pharm Sin 53(8):1215–1224 ((in Chinese))
Syamkumar S, Sasikumar B (2007) Molecular marker based genetic diversity analysis of Curcuma species from India. Sci Hortic 112:235–241
Taheri S, Abdullah TL, Rafii MY, Harikrishna JN, Werbrouck SPO, Teo CH, Sahebi M, Azizi P (2019) De novo assembly of transcriptomes, mining, and development of novel EST-SSR markers in Curcuma alismatifolia (Zingiberaceae family) through Illumina sequencing. Sci Rep 9:3047
Taheri S, Abdullah TL, Yusop MR, Hanafi MM, Sahebi M, Azizi P, Shamshiri RR (2018) Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 23:399
Theanphong O, Mingvanish W (2017) Chemical constituents and antioxidant activities of essential oils from roots and rhizomes of Curcuma alismatifolia Gagnap. from Thailand. J Appl Sci 16:105–111
Wei WL, Qi XQ, Wang LH, Zhang YX, Hua W, Li DH, Lv HX, Zhang XR (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12(1):451
Wei ZZ, Sun ZZ, Cui BB, Zhang QX, Xiong M, Wang X, Zhou D (2016) Transcriptome analysis of colored calla lily (Zantedeschia rehmannii Engl.) by Illumina sequencing: de novo assembly, annotation and EST-SSR marker development. PeerJ 4: e2378. https://doi.org/10.7717/peerj.2378
Wu J, Cai CF, Cheng FY, Cui HL, Zhou H (2014) Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol Breeding 34:1853–1866
Xu CQ, Gao J, Du ZF, Li K, Wang Z, Li YY, Pang XM (2016) Identifying the genetic diversity, genetic structure and a core collection of Ziziphus jujuba Mill. Var. jujuba accessions using microsatellite markers. Sci Rep 6: 31503. https://doi.org/10.1038/srep31503
Xu XY, Meng X, Li S, Gan RY, Li Y, Li HB (2018) Bioactivity, health benefits, and related molecular mechanisms of curcumin: current progress, challenges, and perspectives. Nutrients 10:1553
Ye YJ, Feng L, Liang XH, Liu TT, Cai M, Cheng TR, Wang J, Zhang QX, Pan HT (2019) Characterization, validation, and cross-species transferability of newly developed EST-SSR markers and their application for genetic evaluation in crape myrtle (Lagerstroemia spp). Mol Breeding 39:26
Yeh FC, Yang RC, Boyle T (1999) POPGENE Version 1.31. University of Alberta Canada
Zeng JH, Xu GB, Chen X (2008) Application of the chromatographic fingerprint for quality control of essential oil from GuangXi Curcuma kwangsiensis. Med Chem Res 18:158–165
Zhang LY, Yang ZW, Cheng F, Su P, Chen DK, Pan WY, Fang YX, Dong CZ, Zheng X, Du ZY (2017) Composition and bioactivity assessment of essential oils of Curcuma longa L. collected in China. Ind Crop Prod 109:60–73
Zhang SJ, Liu N, Sheng AW, Ma GH, Wu GJ (2011) In vitro plant regeneration from organogenic callus of Curcuma kwangsiensis Lindl. (Zingiberaceae). Plant Growth Regul 64:141–145
Zhang ZY, Xie WG, Zhao YQ, Zhang JC, Wang N, Ntakirutimana F, Yan JJ, Wang YR (2019) EST-SSR marker development based on RNA-sequencing of E. sibiricus and its application for phylogenetic relationships analysis of seventeen Elymus species. BMC Plant Biol 19:235
Zhao J, Tong YQ, Ge TM, Ge JW (2016) Genetic diversity estimation and core collection construction of Sinojackia huangmeiensis based on novel microsatellite markers. Biochem Syst Ecol 64:74–80
Zhou Q, Mu KM, Ni ZX, Liu XH, Li YG, Xu LA (2019) Analysis of genetic diversity of ancient Ginkgo populations using SSR markers. Ind Crop Prod 145:111942
Acknowledgements
The financial support is from Key-Area Research and Development Program of Guangdong Province (2020B020220007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The authors declare that the experiments comply with the current laws of China.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ye, Y., Xu, Y., Li, D. et al. Characterization of EST-SSR markers in Curcuma kwangsiensis S. K. Lee & C. F. Liang based on RNA sequencing and its application for phylogenetic relationship analysis and core collection construction. Genet Resour Crop Evol 68, 1503–1516 (2021). https://doi.org/10.1007/s10722-020-01079-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-020-01079-z