
Abstract
Germline mutations in the DNA mismatch repair (MMR) genes cause Lynch syndrome. Classification and interpretation of intronic variants, especially those outside the consensus ± 1 ~ 2 splice sites are challenging as it is uncertain whether such variants would affect splicing accuracy and efficiency. The assessment of the pathogenicity of splice site variants in MLH1 is further complicated by the various isoforms due to alternative splicing. In this report, we describe a 42-year-old female with Lynch syndrome who carries a germline variant, MLH1 c.678-3T>A, in the splice acceptor site of intron 8. Functional studies and semiquantitative analysis demonstrated that this variant causes a significant increase in the transcripts with exon 9 or exon 9 and 10 deletions, which presumably leads to premature protein truncation or abnormal protein. In addition, we also observed MSI-H and loss of MLH1 by IHC in patient’s tumor tissue. This variant also segregated with Lynch Syndrome related cancers in three affected family members. Based on these evidence, the MLH1 c.678-3T>A variant is considered pathogenic.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lynch syndrome (LS), also called Hereditary Non-Polyposis Colorectal Cancer (HNPCC) [1], is an autosomal dominant inherited disease accounting for approximately 1–5% of all diagnosed colorectal cancers (CRC) [2]. It is characterized by an increased risk of early onset cancers including colorectum, endometrium, ovarian, small bowel, stomach, hepatobiliary, urinary, small bowel, brain or central nervous system, as well as sebaceous tumors [3]. Lynch syndrome is caused by a defect in the DNA mismatch repair (MMR) pathway due to the presence of germline pathogenic variants in the MMR genes [4] with mutations in MLH1 and MSH2 identified in almost 90% of LS patients [5]. Germline mutations in MMR genes lead to tumors with characteristic mutational signature, microsatellite instability (MSI), and loss of expression of one or more MMR proteins [6].
Many MMR gene mutations identified to date are truncating variants (nonsense and small insertions/deletions), large deletions/duplications and splice site variants affecting the highly conserved intronic dinucleotides 5′ GT and 3′ AG [7]. These variants are considered clinically significant as they clearly disrupt the normal function of the protein. However, the biological consequences and clinical implications of missense, silent, small in-frame deletions/insertions and variants outside the ± 1 ~ 2 consensus splicing site (SS) are often classified as variants of unknown significance (VUS). The clinical ambiguity of the VUSs is problematic because it is uncertain whether these subtle changes alter function sufficiently to predispose cells to cancer development. As a result, carriers of VUSs and their families cannot take advantages of the risk assessment, prevention, and therapeutic measures that are available to carriers of known pathogenic mutations. Classification and interpretation of MLH1 variants are further complicated by the presence of naturally occurring alternative splicing isoforms, which may create transcripts that encode proteins with abrogated function due to protein truncation, dominant negative effects or concomitant decrease in full length transcript levels. A total of thirty MLH1 alternatively spliced transcripts were reported [8], which complicates the interpretation of RNA data derived from patients carrying MLH1 variants. Quantitative studies can be utilized to assess transcript levels to distinguish natural expression fluctuation and mutation-induced aberrant splicing.
The MLH1 c.678-3T>A variant was previously reported in a 41-year old female tested for LS [9]. However, no functional studies were performed and its pathogenicity was uncertain. In our study, our proband diagnosed with colon cancer at age 42 was identified to have the same variant. We investigated the effects of this intronic variant by semi-quantification of transcript levels. This substitution results in transcripts with complete skipping of exon 9 or exons 9 and 10, which presumably leads to premature protein truncation or abnormal protein. This variant is associated with loss of MLH1 expression and MSI-H in the tumor. It also segregated with LS related cancers in three family members. Taken together, our data indicate that the MLH1 c.678-3T>A variant is considered pathogenic.
Material and methods
Subject
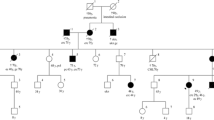
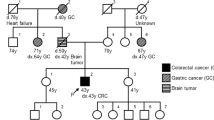
Our proband is a 69-year-old woman who was diagnosed with endometrial cancers (EC) at 49. A three-generation pedigree (Fig. 1) indicated that another eight family members on the paternal side were affected with early onset colon cancer and one died of bile duct cancer in her early 30 s. The proband’s paternal grandmother was also affected with endometrial cancer in her 30 s. The proband’s sister was diagnosed with bladder cancer at 55. The proband was tested via a commercially-available hereditary cancer multi-gene panel in a reference lab (sequencing and large rearrangement analysis of MLH1, MSH2, MSH6, PMS2 and EPCAM) and was identified to carry the MLH1 c.678-3T>A which is classified with VUS. No other mutations or variants of uncertain clinical significance were identified in the remaining four genes analyzed. Given the unknown clinical significance of this variant, the patient was assigned onto an IRB protocol and agreed to provide additional blood samples for further characterization of this variant at Memorial Sloan Kettering Cancer Center (MSKCC). Peripheral blood samples were collected and submitted to the Diagnostics Molecular Genetics Laboratory at MSKCC. Control RNAs were from unrelated individuals seen at MSKCC who do not carry the MLH1 variant.

Patient pedigree. The patient described here is a 69-year-old female who was diagnosed with colon and endometrial cancers at age of 42 and 49, respectively. Another eight family members on the paternal side were affected with early onset colon cancer and one died by bladder cancer in her early 30 s
In silico analysis
Sequence data spanning the MLH1 locus for Homo sapiens [Chromosome 3: 36,993,35037,050,846] was obtained from the Ensembl Genome Browser (https://www.ensembl.org/index.html). Primers were designed using the Primer 3 software (https://bioinfo.ut.ee/primer3-0.4.0/). In silico evaluation of the variants was performed through Alamut (Interactive Biosoftwar), which includes SSF, MaxEnt, NNSPLICE, GeneSplicer and HSF tools.
cDNA analysis
The MLH1 c.678-3T>A variant identified through commercial testing was confirmed prior to transcript analysis. Total RNA was extracted from the patient using the PAXgene BloodRNA Kit (PreAnalytiX, Qiagen, Valencia, CA) and was subsequently utilized for cDNA synthesis (Superscript III First-Strand Synthesis SuperMix, Invitrogen Life Technologies, Carlsbad, CA). Control RNA was extracted from other individuals who did not carry the MLH1 variant. RT-PCR was performed using SuperScript™ III First-Strand Synthesis SuperMix (Invitrogen) for RT and then the JumpStart REDTaq Ready Mix (Sigma) for PCR, with control cDNA or the patient’s cDNA in the presence of M13-tagged forward and reverse primers (Forward, E7F: 5′-GTA AAA CGA CGG CCA GT TGCAGGCATTAGTTTCTCAG-3′; Reverse, E11R: 5′-CAG GAA ACA GCT ATG AC CACATTCTGGGGACTGATTT-3′). Each PCR included 12.5 µl 2 × JumpStart REDTaq ready mix, 2 µl 10 µM primers (1 µl for each primer), 2 µl cDNA and water to make a final volume of 25 µl. PCR
s were performed under the following cycling conditions: 96 °C for 5 min, 94 °C for 30 s (35 ×), 64 °C for 45 s (35 ×) and 72 °C for 60 s (35 ×) with a final extension at 72 °C for 5 min (1 ×).
Cloning
To test whether the mutant allele is able to generate any normal transcript, the full-length RT-PCR product with a SNP (c.655A>G) in exon 8 from the patient was cloned into pCR4 TOPO vectors (Invitrogen, Carlsbad, CA), following procedures of the pCR4 TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA). DNA from colonies was amplified and subjected to direct DNA sequencing analysis using the forward PCR primer (BigDye Terminator v3.1 Cycle Sequencing kit and 3730 DNA Analyzer, Applied Biosystems, Foster City, CA).
Semiquantitative analysis of MLH1 transcripts
cDNA products were further analyzed with the same primer sequences and PCR conditions as mentioned above in the cDNA analysis, except that the reverse primer sequence in this reaction was labeled with 5′ 56-JOEN fluorophore. The RT-PCR products amplified with the JOE fluorophore labeled primer were then subjected to fragment analysis on 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA) using the internal lane standard 600 (ILS 600) (Promega Corporation, Madison, WI) as a DNA marker.
Results
Patient’s personal and family histories and segregation studies
Our proband is a 69-year-old woman who was diagnosed with endometrial cancers at 49. Another eight family members on the paternal side were affected with early onset colon cancer and one died of bile duct cancer in her early 30 s. Although several affected family members are not available for testing, this variant co segregated with LS related cancers in three affected family members (Fig. 1).
The MLH1 c.678-3T>A variant disrupts normal splicing and presumably leads to premature protein truncation
To evaluate the potential effects of the variant on splicing, we used Alamut software, which incorporates five tools to predict the potential effects of MLH1 c.678-3T>A on normal mRNA splicing. Three out of the five tools predicted that the variant significantly weaken the 3′ acceptor splice site with two of them predicted complete loss of the natural acceptor site and another one predicted a score reduction of 59%. The other two tools predicted that this variant may not affect splicing as the score reduction a very lower proportion (4.5% and 2.1%) (Fig. 2a, b).

In silico predictions of the MLH1 c.678-3T>A variant. The Alamut software was used to evaluate the potential effects of the variant on splicing. Three out of the five tools predicted that the variant significantly weaken the 3′ acceptor splice site with two of them predicted complete loss of the natural acceptor site and another one predicted a score reduction of 59%. The other two tools predicted that this variant does not significantly affect splicing
The effect of MLH1 c.678-3T>A variant on RNA splicing was subsequently evaluated by amplifying regions of MLH1 from cDNA derived from the patient. PCR was designed to generate a fragment that spanned part of exon 7 and the entire coding region of exons 8, 9,10 and 11, which are likely affected by the variant. Two additional bands were identified in the patient, but they are absent in controls (lane 6, Fig. 3a). Further sequencing results revealed that this variant leads to loss of the entire exon 9 and skipping of both exon 9 and 10 (Fig. 3b).

RT-PCR analysis demonstrates MLH1 c.678-3T>A leads to exon 9 or exons 9 and 10 skipping. a RT-PCR products run on QIAxcel. Two extra bands were observed in the patient, but not in controls. b Electropherogram showing that the variant causes exon skipping. The boundary of exons is marked by red arrow. c Semi-quantitative fragment analysis of RNA transcripts from the patient and controls using the GeneMapper software. d Percentage of different transcripts. The percentage of the wild-type allele in the was calculated as an average of peak height × peak area of wild-type allele/sum of peak heights × peak areas of wild type and mutant alleles from at least nine independent experiments
MLH1 naturally occurring alternative transcripts have been reported in the literature and databases [8]. To assess whether the transcripts observed in the patient is due to alternative splicing, we included negative samples from patients who do not carry the MLH1 variant (n = 19) in fragment analysis. The semiquantitative analysis of RT-PCR products spanning MLH1 exons 7–11 resulted in four transcripts (Fig. 3c). One transcript of predicted length, “Wild type (WT)”, was observed in all controls tested as well as in the patient. Another transcript observed in all controls tested and the patient was 94 bp shorter than the full-length fragment, “Del10”. This fragment is consistent with alternative MLH1 transcript that skips exon 10. The third transcript in both controls and the patient was 113 bp shorter than the wild type transcript, “Del9”, which presumably leads to a truncated protein (p.Glu227Serfs*42). The fourth and the shortest transcript in all samples corresponds to transcript skipping both exons 9 and 10, “Del9 + del10”, and it is expected to produce an in frame deletion (p.Glu227_Ser295del). We then calculated the proportions of the alternative transcripts and the wild type full length transcript, comparing the amounts of these transcripts to the sum of all transcripts as an approximation for the total MLH1 transcript level. Percentages of each transcript obtained within each of the 19 control samples and the affected patient were averaged and shown in Fig. 3d. Our fragment analysis revealed that the proportion of transcripts with exon 10 skipping is comparable between the patient and negative controls (Fig. 3d) which is less than 1% of the total transcripts (Fig. 3d). However, the patient sample showed a dramatically elevated level of the exon-9 skipped transcript (approximately 5%), compared to the negative controls (< 0.1%) (Fig. 3d). Therefore, the MLH1 c.678-3T>A variant can substantially alter the level of transcript lacking exon 9, which would give rise to a frameshift resulting in premature protein truncation. The transcript lacking both exons 9 and 10 accounts for about 40% of the total transcripts in the patient sample, which is significantly increased than that in the controls (< 1%) (Fig. 3d).
The variant c.678-3T>A completely disrupts normal splicing in the mutant allele
Our fragment analysis showed that the wild type transcript is approximately 55% of total transcript. To determine whether the mutant allele can generate any MLH1 wild type full length transcripts, we searched for heterozygous variant in the RT-PCR region. A heterozygous variant, c.655A>G present in exon 8 allowed us to perform this assessment. We extracted and sequenced the full-length RT-PCR product from the patient. As shown in Fig. 4, the sequencing result revealed that the full-length transcript contains only the A allele at the c.655 nucleotide position, indicating that c.655A is in cis with the wild type allele and that c.655G is in cis with the mutant allele in the patient.

SNP tagging demonstrates that the mutant allele does not produce any wild type transcript. The sequencing result revealed that c.655A is in cis with the wild type allele and that c.655C is in cis with the mutant allele in the patient. a All clones (n = 58) with the full length transcript contained the normal A; b all clones with the exon 9 deletion (n = 18) contained the mutant G allele; c All clones with both exons 9 and 10 deletion (n = 15) contained the mutant G allele
Given the low sensitivity of Sanger sequencing, to exclude the possibility of low frequency mutant alleles being undetected by this approach, we used cloning approach to determine whether the c.655G was present in the wildtype transcript. We cloned the RT-PCR products into the TOPO sequencing vector and then sequenced 93 colonies. All of the fifty-eight clones from the patient containing the full-length transcript had the normal A at the c.655 position, indicating that the mutant allele was unable to generate any normal transcript (Fig. 4a). However, all clones with the exon 9 deletion (n = 18) or with both exons 9 and 10 deletion (n = 15) contained the mutant G allele (Fig. 4b, c). These results indicate that the aberrant splicing caused by this mutation is quite efficient as the mutant G allele completely abolishes normal splicing. It is worth noting that two clones containing transcripts lacking exon 10 had the normal A indicating exon 10 skipping was generated from the normal allele instead of the mutant one. This is consistent with the observation that the proportion of exon 10 exclusion transcript is extremely low (< 1%) and, more importantly, comparable in the patient and negative controls (Fig. 3c, d).
Discussion
VUSs in MMR genes are commonly seen in patients with suspected LS and continue to lead to considerable clinical challenge. In most of the cases, there is not too much information on segregation and cases-control studies. Functional studies have been proven to be very useful in these situations and this is the reason we pursued cDNA studies to evaluate the functional consequences of this splice variant.
Multiple in silico tools have been developed to predict splicing as it relates to creation or loss of splice sites at exonic or intronic level. In general, they have higher sensitivity (around 90–100%) relative to specificity (60–80%) in predicting the effects on splicing. It has been recommended to use different in silico tools for variant interpretation [10]. These predictions can be considered as one piece of evidence to help classify a VUS but it is not recommended to use as the sole source of evidence to make a final classification. In our case, we used five different in silico tools to have more robustness, two of them predicted loss of the natural acceptor site and another one predicted a score reduction of 59%. The other two tools also predicted score reduction but in a very lower proportion (4.5% and 2.1%). In summary, these five tools predicted the variant to be likely pathogenic at RNA level for loss of the natural acceptor site of the exon 9. Therefore, it was very important to do RNA in vitro analysis to prove the in silico predictions.
The RNA in vitro analysis is a well-known in vitro study that can support the damage effect of the variant. Our in vitro results showed that c.678-3T>A variant produced two different transcripts, a complete skipping of exon 9 and an exclusion of exons 9 and 10. The exon 9 skipping is predicted to produce a frameshift protein (p.Glu227Serfs*42) while skipping of exons 9 and 10 produce an in frame deletion (p.Glu227_Ser295del). These two transcripts have been reported several times in the literature among other mRNA isoforms [11,12,13]. Also, our semiquantitative analysis showed that in control individuals both exon-skipped transcripts are present with a very low expression. In c.678-3T>A carrier, the expression of skipping of exon 9–10 is around 40% while skipping of 9 is only of 5% and the total transcripts. Since we already demonstrated that the mutant allele was unable to generate any full-length mRNA transcript, one possible explanation for is that the del 9, and del9 + del10 mRNA transcripts are less stable comparing with the full-length, which alters the ratio of full-length mRNA and the shorter mRNA transcripts. The region of MLH1 protein encoded by exon 9 and 10 is located between the two-major protein–protein interaction domains of MLH1 protein, the ATPase binding domain and the PMS2 binding pocket. It has been reported that hMLH1Δ9/10 displays MMR deficiency and cannot restore MMR function in MLH1 deficient cells in vitro [14]. It also exhibits a dominant negative effect in MMR-proficient cell lines [15]. The dominant negative effect might be due to a competitive sequestration of PMS2, reducing stability of the WT MLH1 protein. As shown in the pedigree in Fig. 1, we observed unusually early onset CRC and EC in our patient family where eight (8) family members had early onset CRC: one at 25, four in their 30 s and three in their 40 s at 41, 45 and 47, while the mean age at diagnosis of CRC in LS patients is 44–61 years. One member had endometrial cancer in her 30 s (she also had CRC in her 30 s) and another one at 49 years old while the mean age at diagnosis of endometrial cancer in LS patients is 48–62 years (https://www.ncbi.nlm.nih.gov/gtr/conditions/C1333991/). The early onset CRC and EC in these nine family members clearly indicates a dominant negative effect of this splice site variant on the MLH1 protein. Our data demonstrated that the in-frame deletion of exons 9 and 10 generated by the MLH1 c.678-3T>A variant affects the normal function of MLH1 protein.
Different splice site mutations leading to exon 9 and exons 9–10 deletion have been reported. The c.790+2_+3insT mutation produced an exon 9 skipping and there are segregations in the family [16]. The c.790+1G>A produce an exon 9–10 skipping and it has been seen in a patient with presence of LOH in tumor [17, 18]. The c.790+4A>T MLH1 variant generates exon 9 and exon 9–10 skipping transcripts. They also reported that both skippings have been seeing in the controls, this agrees with our results. Interestingly, the patient also presented LOH in the tumor [19].The c.790+5G>T mutation has been study using functional assays and they detected predominantly of exon 9 skipping [20, 21]. All these variants have been classified as pathogenic, which supported the pathogenicity of the MLH1 c.678-3T>A variant.
Other important evidences are the segregation, clinical manifestation and frequency in general population. The c.678-3T>A variant has not been identified in population databases such as GnomAD or ExAC among others. Also, this family presented Amsterdam criteria with MSI-H in the tumor. Although several affected family members are not available for testing, this variant does co-segregate with LS related cancers in at least three affected family members, which provided additional evidence to support our conclusion that the variant is responsible of the LS related diseases in this family.
In summary, we show the relevance of in vitro splicing analysis in proving the pathogenicity of MLH1 variant. The information obtains from in vitro analysis has been widely utilized in the classification of MMR variants [21,22,23,24]. Now, the next generation sequencing is being routinely used in diagnostic laboratories, the detection of multiple variants in the same or distinct cancer genes is increasing, and it is necessary to have in vitro assays that can help us to classify VUS detected. The new classification of c.678-3T>A variant will lead to a better clinical management of LS patients.
References
Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, Kloor M, Kunstmann E, Vogelsang H, Keller G, Dietmaier W, Mangold E, Friedrichs N, Propping P, Kruger S, Gebert J, Schmiegel W, Rueschoff J, Loeffler M, Moeslein G, German HC (2006) Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC Consortium. J Clin Oncol 24(26):4285–4292. https://doi.org/10.1200/JCO.2005.03.7333
Vasen HF (2005) Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer 4(3):219–225. https://doi.org/10.1007/s10689-004-3906-5
Cohen SA, Leininger A (2014) The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. Appl Clin Genet 7:147–158. https://doi.org/10.2147/TACG.S51483
Das FC, Wernhoff P, Dominguez-Barrera C, Dominguez-Valentin M (2016) Update on hereditary colorectal cancer. Anticancer Res 36(9):4399–4405. https://doi.org/10.21873/anticanres.10983
Lynch HT, de la Chapelle A (2003) Hereditary colorectal cancer. N Engl J Med 348(10):919–932. https://doi.org/10.1056/NEJMra012242
Boland CR, Koi M, Chang DK, Carethers JM (2008) The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Fam Cancer 7(1):41–52. https://doi.org/10.1007/s10689-007-9145-9
Peltomaki P, Vasen H (2004) Mutations associated with HNPCC predisposition—update of ICG-HNPCC/INSiGHT mutation database. Dis Mark 20(4–5):269–276
Thompson BA, Martins A, Spurdle AB (2015) A review of mismatch repair gene transcripts: issues for interpretation of mRNA splicing assays. Clin Genet 87(2):100–108. https://doi.org/10.1111/cge.12450
O’Leary E, Larson E, Lin AY, Reardon S, Lee P, Russell M, Melnitchouk N, Hart S, Ghassemi K, Hecht JR, Wainberg Z, Yoo J (2014) A multi-disciplinary cancer program enhances hereditary colorectal cancer detection. Am J Digest Dis 1(1):62–66
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Kohonen-Corish M, Ross VL, Doe WF, Kool DA, Edkins E, Faragher I, Wijnen J, Khan PM, Macrae F, St John DJ (1996) RNA-based mutation screening in hereditary nonpolyposis colorectal cancer. Am J Hum Genet 59(4):818–824
Genuardi M, Viel A, Bonora D, Capozzi E, Bellacosa A, Leonardi F, Valle R, Ventura A, Pedroni M, Boiocchi M, Neri G (1998) Characterization of MLH1 and MSH2 alternative splicing and its relevance to molecular testing of colorectal cancer susceptibility. Hum Genet 102(1):15–20
Charbonnier F, Martin C, Scotte M, Sibert L, Moreau V, Frebourg T (1995) Alternative splicing of MLH1 messenger RNA in human normal cells. Cancer Res 55(9):1839–1841
Trojan J, Zeuzem S, Randolph A, Hemmerle C, Brieger A, Raedle J, Plotz G, Jiricny J, Marra G (2002) Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology 122(1):211–219
Peasland A, Matheson E, Hall A, Irving J (2010) Alternative splicing of hMLH1 in childhood acute lymphoblastic leukaemia and characterisation of the variably expressed Delta9/10 isoform as a dominant negative species. Leuk Res 34(3):322–327. https://doi.org/10.1016/j.leukres.2009.08.015
Arnold S, Buchanan DD, Barker M, Jaskowski L, Walsh MD, Birney G, Woods MO, Hopper JL, Jenkins MA, Brown MA, Tavtigian SV, Goldgar DE, Young JP, Spurdle AB (2009) Classifying MLH1 and MSH2 variants using bioinformatic prediction, splicing assays, segregation, and tumor characteristics. Hum Mutat 30(5):757–770. https://doi.org/10.1002/humu.20936
Auclair J, Busine MP, Navarro C, Ruano E, Montmain G, Desseigne F, Saurin JC, Lasset C, Bonadona V, Giraud S, Puisieux A, Wang Q (2006) Systematic mRNA analysis for the effect of MLH1 and MSH2 missense and silent mutations on aberrant splicing. Hum Mutat 27(2):145–154. https://doi.org/10.1002/humu.20280
Casey G, Lindor NM, Papadopoulos N, Thibodeau SN, Moskow J, Steelman S, Buzin CH, Sommer SS, Collins CE, Butz M, Aronson M, Gallinger S, Barker MA, Young JP, Jass JR, Hopper JL, Diep A, Bapat B, Salem M, Seminara D, Haile R, Colon Cancer Family R (2005) Conversion analysis for mutation detection in MLH1 and MSH2 in patients with colorectal cancer. JAMA 293(7):799–809. https://doi.org/10.1001/jama.293.7.799
Bianchi F, Raponi M, Piva F, Viel A, Bearzi I, Galizia E, Bracci R, Belvederesi L, Loretelli C, Brugiati C, Corradini F, Baralle D, Cellerino R (2011) An intronic mutation in MLH1 associated with familial colon and breast cancer. Fam Cancer 10(1):27–35. https://doi.org/10.1007/s10689-010-9371-4
Takahashi M, Furukawa Y, Shimodaira H, Sakayori M, Moriya T, Moriya Y, Nakamura Y, Ishioka C (2012) Aberrant splicing caused by a MLH1 splice donor site mutation found in a young Japanese patient with Lynch syndrome. Fam Cancer 11(4):559–564. https://doi.org/10.1007/s10689-012-9547-1
Tournier I, Vezain M, Martins A, Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP, Soret J, Tazi J, Frebourg T, Tosi M (2008) A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat 29(12):1412–1424. https://doi.org/10.1002/humu.20796
Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, du Sart D, Fabre A, Farrell MP, Farrington SM, Frayling IM, Frebourg T, Goldgar DE, Heinen CD, Holinski-Feder E, Kohonen-Corish M, Robinson KL, Leung SY, Martins A, Moller P, Morak M, Nystrom M, Peltomaki P, Pineda M, Qi M, Ramesar R, Rasmussen LJ, Royer-Pokora B, Scott RJ, Sijmons R, Tavtigian SV, Tops CM, Weber T, Wijnen J, Woods MO, Macrae F, Genuardi M (2014) Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 46(2):107–115. https://doi.org/10.1038/ng.2854
Borras E, Pineda M, Brieger A, Hinrichsen I, Gomez C, Navarro M, Balmana J, Ramon y Cajal T, Torres A, Brunet J, Blanco I, Plotz G, Lazaro C, Capella G (2012) Comprehensive functional assessment of MLH1 variants of unknown significance. Hum Mutat 33(11):1576–1588. https://doi.org/10.1002/humu.22142
Tricarico R, Kasela M, Mareni C, Thompson BA, Drouet A, Staderini L, Gorelli G, Crucianelli F, Ingrosso V, Kantelinen J, Papi L, De Angioletti M, Berardi M, Gaildrat P, Soukarieh O, Turchetti D, Martins A, Spurdle AB, Nystrom M, Genuardi M, InSi GHTVIC (2017) Assessment of the InSiGHT interpretation criteria for the clinical classification of 24 MLH1 and MSH2 gene variants. Hum Mutat 38(1):64–77. https://doi.org/10.1002/humu.23117
Funding
This study was funded by Department of Pathology, Memorial Sloan Kettering Cancer Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Cadoo reports institutional support for therapeutic clinical trial, travel and expenses from Astra Zeneca, institutional support for therapeutic clinical trial from Syndax Pharmaceuticals, personal fees and other from Tessaro, personal fees from OncLive, outside the submitted work. Dr. Zhang: honoraria (future technology research LLC, BGI, Illumina); honoraria and travel and accommodation expenses (Roche Diagnostics Asia Pacific). Family member has a leadership position and ownership interest of Shanghai Genome Center. The other authors do not have any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, C., Sheehan, M., Borras, E. et al. Characterization of a germline splice site variant MLH1 c.678-3T>A in a Lynch syndrome family. Familial Cancer 19, 315–322 (2020). https://doi.org/10.1007/s10689-020-00180-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-020-00180-7