
Abstract
Loss-of-function germline variants of MLH1 cause Lynch syndrome. Here, we present the case of a 43-year-old male patient diagnosed with cecal and transverse colon adenocarcinomas. The characteristics of the case met the revised Bethesda guidelines, and the tumors demonstrated a high frequency of microsatellite instability. Genetic testing for mismatch repair genes (indicative of Lynch syndrome) revealed a novel heterozygous germline pathogenic variant, NM_000249.4:c.856A>T/NP_000240.1:p.(Lys286Ter), in MLH1.
Similar content being viewed by others

Lynch syndrome (LS, OMIM#120435) is an autosomal dominant cancer predisposition syndrome that accounts for approximately 1–3% of all colorectal cancers (CRCs) and is associated with an increased risk of extracolonic malignancies, such as endometrial, ovarian, stomach, small bowel, hepatobiliary, and urothelial cancers1. LS is caused either by germline loss-of-function (LoF) pathogenic (P) and likely pathogenic (LP) variants in one of the four DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) or by germline deletions in the epithelial cell adhesion molecule (EPCAM) gene leading to epigenetic silencing of the adjacent MSH21,2. The microsatellite instability (MSI) phenotype is a hallmark of LS-associated tumors caused by MMR system deficiency (dMMR)2. For the definitive diagnosis of LS, genetic testing of these MMR genes is currently used in clinical practice. Therefore, accumulated knowledge regarding germline variants in MMR genes is necessary for the accurate diagnosis of LS. Genetic identification of LS patients not only alerts the probands to their own life and health risks but also warns their relatives of their own cancer risk and enables subsequent genetic testing, with significant benefits in terms of the timing, cost, and effectiveness of surveillance, early detection, and reduced cancer mortality.
MLH1 and MSH2 are the major pathogenic genes for LS2. Additionally, the majority of variants of MLH1 and MSH2 reported in one of the disease-related databases (InSiGHT variant database, https://www.insight-group.org/variants/databases/) are truncated (predominantly nonsense or frameshift variants)2, frequently leading to the LoF of these genes. Here, we report a novel MLH1 nonsense variant, NM_000249.4:c.856A>T/NP_000240.1:p.(Lys286Ter), associated with LS and classified as LP according to the joint consensus guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP)3.
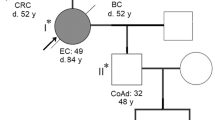
A 43-year-old Japanese male (III-2, Fig. 1) was admitted with sudden abdominal pain. The patient had no significant medical history. Through imaging evaluation via abdominal computed tomography (CT), the patient was diagnosed with perforation of the cecal and transverse colon and panperitonitis. On the same day, the patient underwent an emergency right hemicolectomy. Pathological examination revealed pT3N3 adenocarcinoma on the basis of the TNM classification4 for both cecal and transverse colon cancers. Detailed imaging examinations via contrast-enhanced CT and magnetic resonance imaging before postoperative chemotherapy revealed multiple metastases in the liver. The results of MSI testing of the resected CRC tumors demonstrated a high frequency of MSI (MSI-H); however, the tumors were negative for the BRAF V600E variant.

The arrow indicates the proband (P).
Additionally, the characteristics of the case met the criteria of the revised Bethesda guidelines5; therefore, the patient was referred to the Clinical Genetics Department of our hospital for hereditary tumor risk assessment, although the characteristics of the family members did not meet the Amsterdam Criteria II for LS6 (Fig. 1). Several commercially available genetic tests have been proposed by clinical geneticists for the definitive diagnosis of hereditary CRC. Following pretest genetic counseling and the acquisition of informed consent, the patient opted for and underwent MMR gene testing for the evaluation of the MLH1, MSH2, MSH6, PMS2, and EPCAM genes via very long amplicon sequencing (vLAS), an optimized long-range polymerase chain reaction (PCR)-based next-generation sequencing method (Center for Clinical Genomics, Kanazawa Medical University Hospital, Uchinada, Japan)7. Using vLAS technology, single-nucleotide variants (SNVs), small insertions or deletions (indels), large indels, and structural variants, including exon-level copy number variants (CNVs) within the regions covered by long-range PCR, can be detected. The identified variants were interpreted on the basis of ACMG/AMP Guidelines3.
The heterozygous nonsense variant NM_000249.4:c.856A>T/NP_000240.1:p.(Lys286Ter) (NC_000003.12:g.37017571A>T) in MLH1 was identified and confirmed via Sanger sequencing (Fig. 2). To our knowledge (4 June 2024 date last accessed), this SNV has never been reported in disease-related databases, including the Human Gene Mutation Database (HGMD) Professional (https://my.qiagendigitalinsights.com/bbp/view/hgmd/pro/start.php), Leiden Open Variation Database (LOVD) v3.0 (https://www.lovd.nl/), InSiGHT variant database, or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and has rarely been reported in population databases (PM2 ACMG/AMP variant criterion3), including gnomAD v4.1.0 (https://gnomad.broadinstitute.org/, allele frequency = 6.842 × e−7) and 54KJPN-SNV/INDEL (https://jmorp.megabank.tohoku.ac.jp/, allele frequency = 0). This SNV is predicted to generate a stop codon, possibly leading to a premature termination codon and causing a LoF (PVS1 ACMG/AMP variant criterion3) via nonsense-mediated mRNA decay (NMD). According to the ACMG/AMP guidelines3, this SNV was classified as LP on the basis of the PM2 and PVS1 criteria. No other variants that could be responsible for LS were detected in any of the tested genes. Therefore, the patient was diagnosed with LS due to a novel germline nonsense variant of MLH1. Following the diagnosis of the proband, MLH1 genetic testing of at-risk family members, especially unaffected first-degree relatives, was suggested for genetic counseling but has not yet been performed.

A Integrative Genomics Viewer (IGV) snapshot of NM_000249.4:c.856A>T (NC_000003.12:g.37017571A>T, filled arrow). B Sanger sequencing confirmation of NM_000249.4:c.856A>T (filled arrow) in MLH1. Direct PCR sequencing analysis was performed using genomic DNA from a patient peripheral blood sample (III-2 in Fig. 1) and a control sample.
At the same nucleotide position, NM_000249.4:c.856, two other nucleotide changes, A > C and A > G, which cause missense substitutions of amino acids at codon 286, p.(Lys286Gln) and p.(Lys286Glu), respectively, have been reported in disease-related databases as variants of uncertain significance or as benign or likely benign variants. Because nonsense variants in neighboring codons, such as codon 284, have been reported as P or LP in the ClinVar and InSiGHT databases, it is reasonable to predict that c.856A>T is a null variant causing NMD to lead to LS, although the nonsense SNV in codon 286 has not been reported previously. Identifying a pathogenic MMR variant in the proband is essential to confirm the genetic predisposition to LS in the proband and enable the presymptomatic diagnosis of variant carriers in family members. Therefore, reporting novel pathogenic variants responsible for LS will help in the accurate diagnosis of LS.
HGV datbase
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3439.
References
Lynch, H. T., Snyder, C. L., Shaw, T. G., Heinen, C. D. & Hitchins, M. P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 15, 181–194 (2015).
Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 15, 385–393 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Brierley, J. D., Gospodarowicz, M. K., Wittekind, C. eds. The TNM Classification of Malignant Tumours. 8th edn (Wiley Blackwell, Oxford, 2017).
Umar, A. et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl Cancer Inst. 96, 261–268 (2004).
Vasen, H. F., Watson, P., Mecklin, J. P. & Lynch, H. T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116, 1453–1456 (1999).
Niida, Y., Togi, S. & Ura, H. Streamlining genetic diagnosis with long-range polymerase chain reaction (PCR)-based next-generation sequencing for type I and type II collagenopathies. Cureus 15, e50482 (2023).
Acknowledgements
The authors thank all individuals who participated in this study. This work was supported in part by the Aichi Cancer Research Foundation (N. Takaiso) and the Japan Agency for Medical Research and Development (AMED); grant numbers JP22kk0305020 and JP23ck0106872; (I. Imoto).
Funding
This work was supported in part by the Aichi Cancer Research Foundation (N. Takaiso) and the Japan Agency for Medical Research and Development (AMED); grant numbers JP22kk0305020 and JP23ck0106872; (I. Imoto).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
This study was approved by the Institutional Review Board of Aichi Cancer Center (No. S06002). Informed consent was obtained from the patient for publication of the case details as well as genetic and genomic findings.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takaiso, N., Imoto, I., Matsumoto, T. et al. Novel MLH1 nonsense variant in a patient with suspected Lynch syndrome. Hum Genome Var 11, 36 (2024). https://doi.org/10.1038/s41439-024-00294-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-024-00294-9
- Springer Nature Limited