
Abstract
Bacterial blight (BB) caused by Xanthomonas citri pv. malvacearum (Xcm), poses a significant threat to Upland cotton (Gossypium hirsutum L.) production worldwide, and Xcm race 18 is the most virulent and widespread and can cause serious yield loss. Understanding the genetic basis of resistance in diploid Asiatic cotton (G. arboreum) and successfully transferring the resistance to tetraploid Upland cotton are crucial for developing resistant cotton cultivars. This study aimed to identify chromosomal regions for BB resistance through a genome-wide association study (GWAS) using 245 G. arboreum accessions evaluated in two replicated greenhouse tests and to evaluate an introgression BC2F7 population derived from a tri-species hybrid (G. arboreum/G. aridum/G. hirsutum). In response to Xcm race 18 infections after artificial inoculation, 80% of the accessions exhibited a high level of resistance, including 151 accessions showing immunity with no visible foliar water-soaked lesions. A GWAS based on 7009 polymorphic SNP markers detected 9 major BB resistance QTLs on chromosomes A01, A02, A05, A06, A10, A12 and A13 in the Asiatic cotton. The tri-species introgression population showed segregation in BB resistance with significantly lower disease incidence of BB than the susceptible check Acala 1517-18 GLS (30.2 vs. 100%), suggesting that the resistance in the diploid species has been successfully transferred into Upland cotton. The identification of Xcm race 18 resistant diploid Asiatic cotton germplasm and specific chromosomal regions and candidate genes delineated by SNPs for resistance for the first time provides strong evidence that the Asiatic cotton is a new genetic source of resistance to Xcm race 18. The results will facilitate further genetic and genomic studies toward the eventual identification of resistance genes in Asiatic cotton and their transfer into tetraploid cotton through marker-assisted selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacterial blight (BB), caused by Xanthomonas citri pv. malvacearum (Xcm), poses a significant threat to cotton (Gossypium spp.) production worldwide, causing various symptoms such as seedling blight, angular leaf spot, vein blight, black arm lesion, or boll blight (Smith 1920; Knight 1948; Brinkerhoff 1970; Verma 1986; Hillocks 1992). This bacterium infects plants through open stomata, wounds, and bolls (Bird and Smith 1961), particularly in windy environments with blowing rain and dust events during the growing season. There are 22 races of Xcm identified in cotton-producing regions globally over time (Hunter et al. 1968; Brinkerhoff 1970; Hillocks 1992; Delannoy et al. 2005; Jalloul et al. 2015; Phillips et al. 2017), of which race 18 is highly virulent and wide spread in the United States and worldwide (El-Zik and Thaxton 1995; Allen and West 1991; Hussain 1984; Thaxton et al. 2001; Verma and Singh 1975; Zhang et al. 2020).
BB can lead to substantial yield loss and fiber quality reduction (Knight and Hutchinson 1950; Wickens 1953). Annual yield reductions in Upland cotton caused by this disease ranged from 1 to 2% under normal conditions to 5–15% in severe outbreaks (Hillocks 1992), with potential losses exceeding 50% under rainy weather and high humidity (Knight 1950; Verma 1986). Effective control measures were established in the 1970s through the development of BB-resistant cultivars and the use of acid-delinted seeds (Zhang et al. 2020). However, BB’s resurgence in the United States due to the adoption of BB-susceptible transgenic cultivars prompted renewed efforts to combat this disease through breeding (Bird 1986; Wheeler et al. 2007, 2022; Wheeler 2018). Xcm races evolve in response to the development of new cotton cultivars with resistance genes.
Historically, research efforts focused on cotton breeding for BB resistance, notably in Sudan from the 1930s to 1950s (Zhang et al. 2020). Pioneering work by Knight in transferring resistance genes from various cotton species to Egyptian cotton (G. barbadense L.) known as Sakel, which was cultivated in Sudan (Knight 1963) using traditional breeding methods, resulted in the identification of 10 major BB resistance genes. These genes, including B1–B10, were successfully introduced into commercial Upland cotton cultivars in Sudan, significantly enhancing BB resistance. Currently, 22 BB resistance genes have been reported in cotton, of which genes B1–B12 are major genes (Zhang et al. 2020). Genes B1, B2, B3, B7, B10 and B12 were identified in Upland cotton (Knight and Clouston 1939; Brinkerhoff et al. 1979; Knight 1944; Knight 1953a, b; Green and Brinkerhoff 1956; Innes and Brown 1969; Lagiere 1960; Innes 37,38,39,d; Knight 1963; Follin et al. 1988; Wallace and El-Zik 1989). Genes B4, B6, B8 and B11 were identified in diploid cottons (Knight 1948, 1954; Knight 1953a, b; Innes 1966). Gene B5 was identified in perennial G. barbadense (Knight 1950). All these genes except B8 were transferred to Sakel and Upland cotton (Zhang et al. 2020).
In the United States, studying the genetic basis for BB resistance primarily focused on Upland cotton. Multiple major B genes, including B7, B12, BIn, BN and BS, were identified, along with several polygene modifiers or complexes (Zhang et al. 2020). Wright et al. (1998) employed restricted fragment length polymorphism (RFLP) markers to map quantitative trait loci (QTLs) using four different F2 populations generated from crosses between G. barbadense Pima S-7 and resistant Upland cotton lines. As such, B12 was first mapped to chromosome c14 (D02) and later confirmed by Rungis et al. (2002) using amplified fragment length polymorphism (AFLP) and simple sequence repeat (SSR) markers. Xiao et al. (2010) further fine-mapped B12 on D02 within a 3.4-cM region flanked by 4 SSR markers and 4 single nucleotide polymorphism (SNP) markers. In the QTL mapping study, Wright et al (1998) mapped six major QTLs for resistance to races 2 and 4, including two QTLs on c20 (i.e., D10, corresponding to B2 and B3) and four QTLs (on c05/A05, c14/D02, c20/D10 and LGD02) corresponding to b6. Interestingly, QTLs corresponding to B3 and b6 explained 53–56% of the phenotypic variation in reaction to races 7 and 18. Therefore, BB resistance genes other than B12 may confer partial resistance to Xcm race 18. Elassbli et al. (2021b) conducted a genome-wide association study (GWAS) involving over 330 Upland cotton germplasm accessions. The results showed that 55 SNPs were associated with resistance to BB race 18 on different chromosomes including A01, A05, D02, D08 and D10. Further segregation analysis was conducted in 95 F2 populations between resistant and susceptible parents to study segregation ratios for BB resistance (Elassbli et al. 2021c). Results showed that all F1 were resistant, and 65 F2 populations exhibited the expected 3:1 resistant/susceptible ratio, indicating a dominant resistance gene conferring resistance to Xcm race 18. Further, all the resistant germplasm tested possessed the B12 gene based on DNA markers analysis, indicating that the B12 gene is widely distributed in U.S. Upland cotton (Elassbli et al. 2021c). In another most recent study, Gowda et al. (2022) reported a major resistance locus (BB-13) on D02 based on GWAS of the same set of Upland cotton accessions used by Elassbli et al. (2021b), followed by linkage mapping in a bi-parental recombinant inbred line (RIL) population. However, no major resistance gene has been identified in the A-subgenome.
In general, G. barbadense is often considered to be more susceptible to Xcm compared to G. hirsutum (Brown and Ware 1958; Brinkerhoff 1970; Elassbli et al. 2021a), while a broad spectrum of disease susceptibility exists among Upland cotton germplasm. The quantitative response of resistance to Xcm indicated that multiple genetic factors come into play, including additive, dominant, and epistatic gene interactions, all contributing to the overall resistance (Bird 1960; El-Zik and Bird 1967, 1970; Innes 1969, 1974, 1983; Wallace and El-Zik 1989; Zhang et al. 2020). Modifier genes play a crucial role in influencing how major resistance genes are expressed in different genetic backgrounds. The combination of several resistance genes resulted in a high level of resistance to Xcm (Brinkerhoff 1970; Bird 1982; Zhang et al. 2020). Thus, the cultivation of resistant cotton cultivars is an effective strategy for mitigating disease epidemics and managing BB. However, it is important to note that as environmental conditions change, the stability of resistant cultivars may also vary. The specific combination of resistance genes and their quantity influence how effectively cotton resists the disease under different environmental conditions (Knight and Hutchinson 1950).
The A-genome Gossypium taxa, a subgroup of cotton species, exhibits near-immunity to Xcm. This level of resistance was deemed valuable enough to prompt efforts to introduce those resistance traits into cultivated tetraploid cotton (Knight 1953a). The immunity observed in A-genome cottons aligns with the likelihood that Xcm originated in the Old World (Knight 1948), particularly Africa (Follin 1981, 1983). The A-genome species has been used as a primary source of resistance (Zhang et al. 2020), providing a valuable pool of genetic traits that can be harnessed for the improvement of cultivated tetraploid cotton. However, the resistance of the cultivated diploid species to Xcm race 18 has not been explicitly evaluated or reported, and no genetic or QTL study has been reported on resistance to Xcm race 18 in the A-genome, although the race is currently the most prevalent. There is also a lack of development and reporting of introgressed Upland or G. barbadense lines from the diploid species that manifest race 18 resistance or immunity. This presents a compelling opportunity for genetic exploration and molecular investigations.
The hypothesized approach involves conducting controlled crosses between resistant and susceptible lines within diploid species, to elucidate the genetic basis of immunity. By employing advanced molecular markers, resistance genes can be transferred to Upland cotton through carefully designed interspecific hybridizations. Therefore, this study aimed to evaluate an association panel of 245 diploid accessions for resistance to Xcm race 18, identify QTLs for BB resistance, and transfer the BB resistance to tetraploid cotton.
Materials and methods
Plant materials, inoculations, and evaluations of BB resistance
The association mapping panel for the study of BB resistance in diploid cotton comprised 245 accessions of G. arboreum selected from the National Plant Germplasm System (NPGS) cotton collection (https://npgsweb.ars-grin.gov) (Wallace et al. 2009). The 245 accessions, detailed in Table S1, represented a diversity of landraces, conventional cultivars, and breeding lines released over a long period of breeding history (Li and Erpelding 2016; Li et al. 2018; Abdelraheem et al. 2024). To minimize within-accession variation, self-pollinated bolls from a single plant were chosen for each accession, and the increased seeds were utilized for both evaluating BB resistance and DNA extraction.
To assess BB resistance within this panel, two independent tests each with two replicates were conducted using a randomized complete block design (RCBD) in a greenhouse at the Fabian Garcia Research Center, New Mexico State University, Las Cruces, NM, USA. Each test included 10 seedlings per genotype per replicate. Planting was done in 10-cm plastic pots, with 10 seeds per pot. Susceptible and resistant controls, Acala 1517-18 GLS (PI688431, Zhang et al. 2019) and FiberMax (FM) 2334GLT (BASF), respectively, were included. The pots were filled with Miracle-Gro Moisture Control Potting Mix 2 CF (Scotts Co., Marysville, OH, USA).
Inoculation was performed using a newly developed backpack sprayer method (Zhang et al. 2024). Briefly, a Xcm race 18 culture was maintained and cultured on the ATCC medium (carrot potato dextrose agar) at 30 °C for 3–5 days until a uniform culture covered the petri dishes. The culture was then transferred to a liquid ATCC medium until the bacterial concentration of 106 ml−1 was reached. The organoslicone non-ionic wetting agent, Silwet L-77 (Loveland Industries, Greeley, CO, USA) was added to the inoculum at 0.25% (v/v) and mixed before spraying using a backpack sprayer (Model #190,479, Husqvarna 4-Gallon Plastic Backpack Sprayer, Charlotte, NC, USA). Seedlings at 3 weeks post planting were topically sprayed with the inoculum until water dripping on the edge of leaves became apparent. Following inoculation, seedlings were kept at 99% relative humidity at 28–35 °C for 4 days in a sealed container with water at the bottom. Seedlings were then watered daily to maintain soil moisture without insect control. The response of the cotton seedlings to Xcm infections was evaluated on an individual plant basis at 21-day post-inoculation (dpi), using a rating scale from 0 to 5 for foliar disease severity ratings (DSR) on a plant basis, where 0 for a healthy plant with no water-soaked lesions, 1 for a plant with a few water-soaked lesions, 2 for a plant with a low number of water-soaked lesions, 3 for a plant with a moderate number of water-soaked lesions, 4 for a plant with a high number of water-soaked lesions, and 5 for a plant with a very high number of water-soaked lesions (Zhang et al. 2023). The disease incidence (DI, percentage of infected plants with symptoms) and average DSR (the sum of the DSR divided by the number of plants) were calculated. Depending on mean DSR, an accession was subjectively rated as immune with no susceptible plants (DSR = 0.0), highly resistant (HR at 0 < DSR ≤ 1), moderately resistant (MR at 1 < DSR ≤ 2), partially resistant (PR at 2 < DSR ≤ 2.5), partially susceptible (PS at 2.5 < DSR ≤ 3), and highly susceptible (HS, with DSR > 3.5).
The plot data were then subjected to a separate and a combined analysis of variance (ANOVA) for the two tests using SAS 9.3 (SAS Institute Inc., Cary, NC, USA). The PROC GLM statistical approach was employed to assess the statistical significance of various sources of variation. Broad-sense heritability (h2) was calculated using variance components, as the following: h2 = genotypic variance/[(genotypic variance + (genotype × test variance/n) + (error variance/nr)], where r = number of replicates and n = number of tests.
Genotyping of G. arboreum accessions
DNA extraction was performed on the entire set of 245 accessions, and this procedure followed the protocols specified in the DNeasy Plant Mini kit (Qiagen, Valencia, CA) manufacturer’s instructions. To quantify the extracted DNA, a PicoGreen dsDNA Assay kit (Molecular Probes, Inc., Eugene, OR) was utilized. This involved loading 1 μL of DNA to a 1% agarose gel for electrophoresis using 1 × TBE running buffer.
The genotyping of the panel was conducted through a genotyping-by-sequencing (GBS) approach at the Institute for Genomic Diversity, Cornell University, Ithaca, New York, utilizing the method originally proposed by Elshire et al. (2011). Following this, polymorphic SNPs were identified using the TASSEL-GBS pipeline (Glaubitz et al. 2014) within the TASSEL software, following the approach detailed by Li and Erpelding (2016).
In summary, the sequencing reads underwent alignment to the reference genome of G. arboreum (cultivar Shixiyal, ‘SXY1’ genome CRI-uplated_V1) (Du et al. 2018)) using the Burrows–Wheeler Aligner (Li and Durbin 2009). Tags that uniquely aligned to positions in the genome were retained for subsequent SNP identification. SNPs were further filtered with criteria of minimum minor allele frequency of 0.05, a minimum taxa coverage of 0.8, a minimum site coverage of 0.1 and a maximum heterozygosity ratio of 0.2. As a result, 7009 SNPs were employed for the GWAS (Li et al. 2014; Li and Erpelding 2016).
Genome-wide association analysis and identification of candidate genes
For GWAS, TASSEL 5 software (www.maizegenetics.net/bioinformatics/tasselindex.htm) was utilized (Bradbury et al. 2007). A mixed linear model (MLM) association test was conducted by simultaneously accounting for multiple levels of population structure (Q-matrix) and K-matrix (MLM (Q + K)). To reduce the number of false positives, the principal component analysis MLM (PCA + K) mixed linear model correcting for both Q-matrix and K-matrix was used. A QTL was declared when two or more significant SNPs (at P < 0.001) were found within a 1 to 5 Mb region. A false discovery rate (FDR) adjusted P-value for DSR was obtained for a multiple comparison correction (Benjamini and Hochberg 1995). The Bonferroni threshold for SNP significance was set at P < 1.43 × 10−4 (p = 1/n, where n is the number of SNPs, − log 10(1/7009) ≈ 3.85) (Li et al. 2013; Yang et al. 2015; Liu et al. 2006). The R software package “Cmplot” was employed to generate Manhattan plots (Turner 2014).
To identify candidate genes for BB resistance, annotated genes that were within a QTL region from the GWAS were searched through the cotton functional genomic database (CottonFGD, https://cottonfgd.org/) (Zhu et al. 2017). The function of the identified genes was determined through gene annotation. Then, the annotated genes within the QTL region were filtered based on relevant gene ontology terms, including response to fungus or biotic stimulus. Genes that had expression levels greater than 1.0 FPKM (fragments per kilobase of million mapped reads) were considered as candidate genes for BB resistance.
Evaluation of a tri-species population involving G. arboreum for BB resistance
A tri-species introgression population developed from Upland cotton and two diploid species, G. arboreum and G. aridum was screened for BB resistance. The development of this population was previously described (Sacks and Robinson 2007). Briefly, a synthetic hexaploid from a cross of G. hirsutum/G. aridum was crossed with five Asiatic cotton accessions- A1-024 (PI408782), A2-019 (PI129723), A2-087 (PI417895), A2-113 (PI529740), and A2-190 (PI615699) to produce fertile tri-species tetraploid (G. hirsutum/G. aridum/G. arboreum, AADD). The synthetic tri-species hybrids were backcrossed twice with MD51ne (Meredith 1993). The resulted BC2F1 were advanced to BC2F7 through a single boll descent method in each generation, in which one boll from each plant was collected and bulked to advance to the next generation. The bulked seeds of BC2F7, together with the susceptible Upland check Acala 1517-18 GLS and the resistant Upland check FM 2334GLT, were used for evaluation of the BB resistance in a greenhouse at Las Cruces, New Mexico, using the procedure described above.
Results and discussion
Response of diploid astatic to BB Xcm race 18
The artificial inoculation was highly successful as all the plants from the resistant Upland cotton check FM 2334GLT displayed resistant responses with no water-soaked lesions at an average DSR and DI of 0 and 0%, respectively, while Acala 1517-18 GLS was highly susceptible with an average DSR and DI of 3.9 and 100%, respectively. The DSR among the diploid accessions for BB resistance ranged from 0 to 4 with an average of 0.47, while DI ranged from 0 to 100% with an average of 16.86% (Supplementary Table 1). Averaging across the two tests, many accessions had significantly and consistently lower DSR and a higher level of resistance including immunity and high resistance, similar to the resistant check (FM 2334GLT); and most of the susceptible accessions were more susceptible than the susceptible check (Acala1517-18 GLS) in both tests. We used the method described by Elassbli et al. (2021a, b) to classify the 245 diploid accessions into different categories based on DSR evaluated across the two tests at 21 dpi, 152 accessions (62%) were immune (Immune, with DSR = 0), with no symptomatic plants observed at 21 dpi (Supplementary Table 1). In addition, 34 accessions (14%) were highly resistant. Fourteen accessions (6%) were considered as MR. Eight accessions (3%) were PR, while 10 accessions were PS, and seven accessions (3%) were HS. This result indicates that a majority of the diploid accessions (> 80%) exhibited varying degrees of resistance to Xcm race 18, with a significant portion being classified as immune or HR. Therefore, a favorable genetic diversity for BB resistance within the diploid cotton germplasm was observed. This is consistent with Knight (1953a) that the A-genome Gossypium taxa show near-immunity to Xcm (with race designation unknown), which is deemed sufficiently valuable to impel introgression into the cultivated tetraploid cotton. The Xcm immunity of A-genome cotton supported the probable Old-World origin of the pathogen (Knight 1948; Knight and Hutchinson 1950; Follin 1981, 1983). The notion that A-genome diploids may have evolved resistance over an extended period of exposure to the pathogen before polyploid formation occurred adds an evolutionary perspective to the observed resistance (Wright et al. 1998). In addition, the spraying inoculation method gave consistent results between the two tests. Spraying bacterial suspension onto the lower surfaces of leaves was the most frequent method of inoculation used. With this method, the inoculum was forced through the open stomata into the substomatal cavity. Therefore, it was most effective when most of the stomata were open (Bird and Blank 1951). The existence of quantitative genetic variation in BB resistance among the 245 Asiatic diploid accessions suggests the potential for identifying and harnessing genetic factors contributing to BB resistance in the diploid panel.
Responses to BB infections in each plant were assessed based on DSR symptoms from 0 for no symptoms to 5 for very high levels of water-soaked lesions at 21 dpi. The combined analysis of variance is shown in Table 1 and indicates that DSR was significant (at P < 0.001) in test, genotype, and genotype × test interaction. Therefore, a further analysis was conducted on an individual test basis. In both Tests 1 and 2, DSR showed a significant genotypic variation (at P < 0.001). Despite being conducted in the same greenhouse at different periods, utilizing distinct inoculum preparations and inoculations, along with other uncontrollable factors in the greenhouse, genotype × test interactions were observed. However, it is noteworthy that the coefficient of variation (CV) remained relatively low and consistent, ranging from 11.88 to 13.15 in Test 1 and Test 2, respectively (Table 1). Broad-sense heritability estimates for DSR in the two tests were similar, ranging from 0.65 in Test 1 to 0.66 in Test 2, and the estimate was 0.57 in a combined ANOVA from the two tests (Table 1). Wright et al. (1998) reported moderate to high heritability estimates ranging from 0.58 to 0.90 in four mapping populations tested for resistance to race 18 based on qualitative screening. Even for high-heritability traits such as those, analysis of quantitative phenotype can improve the reliability of genetic mapping. Therefore, in this study, we treated the response to BB as a quantitative instead of a qualitative trait to narrow down the genomic regions, because of a qualitative scoring led to map inflation ranges for QTL detection and map to large gaps in linkage groups or at the telomere of the chromosomes and/or the linkage groups (Wright et al. 1998; Said et al. 2013; Abdelraheem et al. 2017). Moving forward, further investigation into specific genetic factors governing BB resistance is needed.
QTLs mapped for BB resistance in the diploid panel
Mean DSRs for each accession from Tests 1 and 2 and the average from the two tests were used for GWAS based on 7009 polymorphic SNP markers using the MLM model, which accounts for both population structure and kinship. Seventy-two significant SNPs were detected for BB resistance based on DSR (Fig. 1; Table 2), leading to the identification of 9 major QTLs distributed on 7 chromosomes, each explaining 17–41% of the phenotypic variation (PVE) in the diploid panel (Table 2). Based on mean DSR across the two tests, 3 QTLs including one each on chromosomes A01, A05 and A12 were detected. In Test 1, 3 QTLs were detected, including one each on A05, A10 and A13; and in Test 2, 3 QTLs with one each on A02, A06 and A12 were also detected. One common QTL on A01 (qBB-A2-A05-1) was detected between Test 1 and the overall means across the two tests, while two QTLs (qBB-A2-A12-1 and qBB-A2-A12-2) were detected on chromosome A12 based on results in Test 2 and the overall means across the two tests.

A Manhattan plot showing chromosomal regions of SNPs with significant association with bacterial blight resistance based on disease severity ratings in the 245 diploid cotton accessions in two tests including mean across the two tests (bottom), Test 1 (middle), and Test 2 (top)
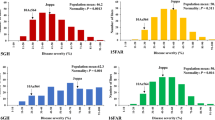
Haplotype analysis was performed on a chromosome basis for several QTLs to explore the importance of the QTLs for resistance to BB. On chromosome A01, 142 blocks were detected. Among those regions, LD block 42 has four SNPs (S1_459999186, S1_459999197, S1_460021209, and S1_460021239) within 22 kb, forming three linkage haplotype blocks. Results showed that > 80% of the accessions (197) carrying the major haplotype (ACGT) had a DSR of 0.34 which significantly reduced BB infections compared to the accessions (49) carrying the haplotypes (TTTG or ACTG) with DSR values two and four-folds higher, respectively (Fig. 2). Within the major haplotype, out of 197 accessions, 127 considered immune with 0% of DI, while 12 accessions considered MR and HS to BB resistance (Table S1).

The mean phenotype values of the 245 diploid accessions with major and minor alleles/haplotypes in significant loci on chromosomes A01, A02, A05, A06, A12 and A13 for the bacterial blight resistance in diploid cotton based on disease severity rating
On chromosome A02, five SNPs (S1_303501817, S1_303505134, S1_303507674, S1_303507711, and S1_303507839) were clustered within 6 kb with three haplotypes. Notably, 147 accessions with the haplotype (GCCAT) had a DSR of 0.61, with 114 accessions classified as immune and HR, and 33 accessions considered MR and HS. Seventy accessions carrying the haplotype (CTTGG) had significantly lower DSR, with 64 accessions classified as immune and HR, and five accessions were MR and HS (Fig. 2).
In addition, on chromosome A12, four SNPs (S1_627126363, S1_627126459, S1_627134046, and S1_627275529) were clustered within 149 kb, forming a major (TAAC) and a minor (GTTA) haplotype. The 134 accessions carrying the major haplotype exhibited a DSR of 0.56, with 85 accessions classified as immune, 33 as HR, and 16 as MR and HS (Fig. 2). The result indicates the complexity of the genetic basis for resistance to Xcm race 18 and suggests the existence of major and minor alleles/haplotypes in both resistant and susceptible accessions with multiple genetic variants contributing to the observed phenotypic differences. Further, it highlights the importance of considering the interactions between alleles on different genetic backgrounds. In addition, BB resistance in the diploid may be controlled by multiple genes, each with different alleles. These genes could interact in complex ways to influence the phenotype, and other genetic or environmental factors could also play a role in BB resistance. Wright et al. (1998) indicated that the complexity of the b6 resistance, involving 4 genes (Qb6a, Qb6b, Qb6c, Qb6d) with very different dosage effects and derived from different parents was an unexpected discovery that demonstrates a horizontal component of bacterial blight resistance. In addition, selection pressure acting on the diploid species has maintained diversity in the genetic variants associated with the trait.
In this study, 9 major QTLs were identified in the A genome for the first time, all considered novel due to the lack of gene mapping studies for validation in the diploid species. Notably, QTLs on chromosomes A01 and A05 were consistent with findings by Elassbli et al. (2021b) and Wright et al. (1998). Wright et al. (1998) mapped six major QTLs for resistance to races 2 and 4, including two QTLs on c20 (i.e., D10, corresponding to B2 and B3) and four QTLs (on c05/A05, c14/D02, c20/D10, LGD02) corresponding to b6. Interestingly, QTL corresponding to B3 and B6 explained 53–56% of the phenotypic variation in reaction to races 7 and 18. Therefore, BB resistance genes other than B12 may confer partial resistance to Xcm race 18. In addition, it is worthwhile to study the homoeologous relationship between the QTLs identified on A02 and A10 in this study and these on D02 and D10 reported by Wright et al. (1998) and Elassbli et al. (2021b)
Despite varying degrees of resistance observed in D-genome diploid cotton taxa against Xcm, complete immunity is not exhibited, challenging their role as primary sources of resistance genes (Knight 1948). However, Wright et al. (1998) noted that among seven resistance genes from tetraploid cotton, six genes (86%) were mapped to D-subgenome chromosomes. Wright et al. (1998) further explained that this may be due to the differences in evolutionary rates between subgenomes, gene conversion or other intergenomic exchanges that escaped detection by genetic mapping, or other factors. Most BB resistance genes were traditionally identified through Mendelian genetic studies using biparental crosses, and these B genes exhibit resistance to different Xcm races (Zhang et al. 2020). In addition to the well-documented B12 in S295, conferring resistance to all US Xcm races (Wallace and El-Zik 1989), our study suggests that resistance to race 18 may be conferred by other major B genes. However, the specific B genes possessed by the identified resistant accessions remain unknown, including whether they have a homoeologous gene to B12 in addition to b6 and B4. Thus, conducting segregation analysis and gene mapping emerges as a potent strategy to map and validate those genes, providing a comprehensive understanding of the genetic architecture within the diploid species for BB resistance (Elassbli et al. 2021c).
Identification of candidate genes for BB resistance QTL
In this study, a total of 150 genes were identified within eight QTL regions, of which 32 were found to be related to disease resistance and considered as candidate genes (Supplementary Table 2). The QTL qBB-A2-A01 at 68–70 Mb co-localized with a gene encoding U-box domain-containing protein 4 which had a role in disease defense. This QTL region was close to the reniform nematode (RN) resistant gene encoding wound-induced protein 1-like (Cotton_A_00283 at 70 Mb) (Li et al. 2018). Therefore, it appeared that the BB resistance QTL and the reniform nematode (RN) resistance QTL locus were closely linked (ca. 0.39 Mb apart). One major QTL qBB-A2-A05 at 25.8–25.9 Mb region contained a candidate gene (Ga05G2600) eoncoding chalcone synthase 2 (CHS2) and is highly expressed in leaves and stems. CHS2 is a key enzyme in the biosynthesis of diverse flavonoids involved in disease resistance, nodulation, and pigmentation in different crops that respond to challenges by pathogens. Flavonoids are secondary metabolites involved in plant development and defense. Recently, Zu et al. (2023) conducted a transcriptome analysis of F6 RILs and the parent lines following Fusarium oxysporum f. sp. vasinfectum (FOV) infection. The findings highlighted the significant role of CHI genes in cotton in response to FOV infection. This gene has also been reported for BB resistance in common bean (Phaseolus vulgaris L.) (Cox et al. 2021). Two QTLs (qBB-A2-A06 and qBB-A2-A12) were associated with one gene each within the leucine-rich repeat (LRR) protein kinase family. Within this family, one prominent group consists of genes encoding LRR domain-containing receptor-like kinases (LRR-RLKs), which are known as resistance (R) genes and play a crucial role in plant immune responses (Jones and Dangl 2006). These R genes, upon detecting invading pathogens, can initiate a cascade of defense-related responses, ultimately leading to enhanced plant resistance. In cotton, previous studies have identified LRR domain containing genes that confer resistance in interactions with Fusarium wilt, Verticillium wilt, and plant-parasite nematodes (Li et al. 2015a, b; Abdelraheem et al. 2020). The findings presented in this study align with the hypothesis that R genes could be pivotal regulators in cotton’s resistance to BB. However, the mechanisms remain largely unknown and there are no R genes cloned in cotton against any pathogens. Cotton presents challenges to cloning R genes due to a long growth cycle, a large genome size with an allopolyploid nature, and a relatively long and technically challenging crop transformation process (DeFeyter et al. 1993; Cox et al. 2019). Thus, further functional molecular studies are necessary to validate and confirm this hypothesis, providing a deeper understanding of the specific mechanisms through which R genes contribute to resistance in cotton against BB.
Interestingly, on chromosome A13, at the major QTL qBB-A2-A13, 10 genes were detected within 2 Mb, eight of which (Ga13G1095, Ga13G1096, Ga13G1097, Ga13G1098, Ga13G1099, Ga13G1100, Ga13G1101, and Ga13G1104) responded to auxin, which plays an important role in plant–microbe interactions, including interactions between plant hosts and pathogenic microorganisms that cause disease. It is now well-established that indole-3-acetic acid (IAA), the most well-studied form of auxin, promotes disease in many plant-pathogen interactions (Kunkel and Johnson 2021). Recently, studies have shown that IAA can act both as a plant hormone that modulates host signaling and physiology to increase host susceptibility and as a microbial signal that directly impacts the pathogen to promote virulence. To make things even more complex, this signaling network can be further modulated by several other hormones, including other auxin, abscisic acid (ABA), and gibberellins (GAs). Not surprisingly, plant pathogens have evolved mechanisms for taking advantage of this regulatory crosstalk as a strategy for promoting pathogenesis, for example, by producing hormones or other virulence factors that modulate hormone signaling (Spoel and Dong 2008; Kazan and Manners 2009; Robert-Seillaniantz et al. 2011; Kazan and Lyons 2014; Ma and Ma 2016).
The BB resistance in a tri-species population involving G. arboreum and G. aridum
Of the five diploid Asiatic accessions used in producing the tri-specific hybrid, two accessions (A2-019 and A2-190) were included in the GWAS panel for evaluation for Xcm race 18 resistance, while the other three accessions were not included due to seed availability. Results showed that both accessions tested were HR and MR, respectively, to Xcm race 18.
A total of 731 seedlings of the tri-species introgression population were evaluated for BB resistance, and 221 plants (32.2%) displayed typical water-soaked lesions. As a comparison, the resistant control FM 2334GLT and the susceptible control Acala 1517-18 GLS incurred 1 and 100%, DI, respectively. The differences among the three groups were significant (Fig. 3), indicating that there existed BB resistance in the introgression population. The BB resistance was not detected in the Upland cotton parents used to develop the tri-specific population. G. aridum was identified to possess a resistance gene Renari transferred to Upland cotton for reniform nematode (Rotylenchulus reniformis) resistance (Romano et al. 2009). Therefore, the resistance detected in the introgression population is likely transferred from the diploid Asiatic cotton, although resistance from G. aridum could not be ruled out because its response to Xcm race 18 has not been reported. Recombinant inbred lines from the bulk introgression population have been developed and are being used for segregation analysis and QTL/gene mapping for BB resistance.

Evaluation of resistance to bacterial blight race 18 in an introgressed trispecific population derived from Gossypium hirsutum/G. aridum/G. arboreum, as compared to resistant Upland check FM 2334GLT and susceptible Upland cotton Acala 1517-18 GLS, based on disease incidence (DI)
Conclusions
This study utilized a panel of 245 accessions of G. arboreum to study the genetic basis of BB resistance in a diploid species. Many accessions were immune to BB race 18; thus, genetic resources possessing different BB resistance genes should be utilized for developing resistant cultivars to BB. The identification of QTLs on chromosomes A01, A02, A05, A06, A10, A12 and A13 suggest multiple genetic loci conferring resistance to Xcm race 18. The homologous or homoeologous relationships between the QTLs on A01, A02, A05 and A10 identified in the diploid A genome from this study and these reported in tetraploid cotton should be investigated. Because susceptible G. arboreum lines exist, genetic and molecular studies can be performed to identify B gene(s) that confer immunity to BB in resistant by susceptible crosses within the diploid species. Through careful selection, breeding, and genotyping, researchers can ultimately identify and isolate specific resistance genes within the diploid Gossypium species. Markers can then be used to trace their transfer to Upland cotton through interspecific hybridizations. Therefore, segregation analysis and gene mapping could be a powerful tool for mapping and validating resistance genes to give a comprehensive understanding of the genetic architecture for BB resistance within the diploid species. The development of portable markers closely linked to those resistance genes would further enhance their utility in marker-assisted selection for BB resistance.
References
Abdelraheem A, Liu F, Song M, Zhang JF (2017) A meta-analysis of quantitative trait loci for abiotic and biotic stress resistance in tetraploid cotton. Mol Genet Genom 292:1221–1235
Abdelraheem A, Elassbli H, Zhu Y, Kuraparthy V, Hinze L, Stelly D, Zhang JF (2020) A genome-wide association study uncovers consistent quantitative trait loci for resistance to Verticillium wilt and Fusarium wilt race 4 in the US Upland cotton. Theor Appl Genet 133:563–577
Abdelraheem A, Zhu Y, Zeng L, Stetina S, Zhang JF (2024) A genome-wide association study for resistance to Fusarium wilt (Fusarium oxysporum f. sp. vasinfectum) race 4 in diploid cotton (Gossypium arboreum) and resistance transfer to tetraploid Gossypium hirsutum. Mol Genet Genom 299:30
Allen SJ, West KD (1991) Predominance of race 18 of Xanthomonas campestris pv. malvacearum on cotton in Australia. Plant Dis 75:43–44
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bird LS (1960) Developing cotton immune to bacterial blight. In: Proceedings of the cotton improvement conference 12, National Cotton Council, Memphis, TN, 16–23
Bird LS (1982) The MAR (multi-adversity resistance) system for genetic improvement of cotton. Plant Dis 66:172–176
Bird LS (1986) Half a century of dynamics and control of cotton diseases: bacterial blight. Proc Beltwide Cotton Res Conf Cotton Dis Counc 46:24–33
Bird LS, Blank LM (1951) Breeding strains of cotton resistant to bacterial blight. Bull Texas Agric Exp Stn 736:25
Bird LS, Smith HE (1961) Bacterial blight of cotton. MP-534. The Agricultural and Mechanical College of Texas, Texas Agric. Exp. Stn.–Texas Agric. Extension Serv., College Station, TX
Bradbury PJ, Zhang Z, Kroon DE, Casstevens RM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brinkerhoff LA (1970) Variation in Xanthomonas malvacearum and its relation to control. Ann Rev Phytopathol 8:85–110
Brinkerhoff LA, Verhalen LM, Mamaghani R, Johnson WM (1979) Inheritance of an induced mutation for bacterial blight resistance in cotton. Crop Sci 19:901–903
Brown HB, Ware JO (1958) Cotton. McGraw-Hill, New York, pp 176–179
Cox KL Jr, Babilonia K, Wheeler T, He P, Shan L (2019) Return of old foes-recurrence of bacterial blight and Fusarium with of cotton. Curr Opin Plant Biol 50:95–103
Cox LD, Munholland S, Mats L, Zhu H, Crosby WL, Lukens L, Pauls KP, Bozzo GG (2021) The induction of the isoflavone biosynthesis pathway is associated with resistance to common bacterial blight in Phaseolus vulgaris L. Metabolites 11:433
DeFeyter R, Yang YO, Gabriel DW (1993) Gene-for-genes interactions between cotton R genes and Xanthomonas campestris pv. malvacearum Avr genes. Mol Plant Microbe Interact 6:225–237
Delannoy E, Lyon BR, Marmey P, Jalloul A, Daniel JF, Montillet JL, Essenberg M, Nicole M (2005) Resistance of cotton towards Xanthomonas campestris pv. malvacearum. Annu Rev Phytopathol 43:63–82
Du X, Huang G, He S et al (2018) Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat Genet 50:96–802
Elassbli H, Abdelraheem A, Zhu Y, Teng T, Sanogo S, Wheeler TA, Wedegaertner T, Zhang JF (2021a) Evaluation and analysis of commercial cultivars and elite breeding lines for resistance to the bacterial blight pathogen race 18 in cotton. Euphytica 21:21
Elassbli H, Abdelraheem A, Zhu Y, Teng Z, Wheeler TA, Kuraparthy V, Hinze L, Stelly DM, Wedegaertner T, Zhang JF (2021b) Evaluation and genome-wide association study of resistance to bacterial blight race 18 in U.S. Upland cotton germplasm. Mol Genet Genomics 296:719–729
Elassbli H, Zhu Y, Abdelraheem A, Wheeler TA, Wedegaertner T, Zhang JF (2021c) Genetic analysis of resistance to bacterial blight Race 18 and B12-linked marker analysis in U.S. Upland cotton. Crop Sci 61:3458–3468
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
El-Zik KM, Bird LS (1967) Inheritance of resistance to races of Xanthomonas malvacearum (E. F. Sm.) Dowson in cotton. In: Proceedings of the Beltwide cotton production research conference, pp 216–217
EL-Zik KM, Bird LS (1970) Effectiveness of specific genes and gene combinations in conferring resistance to races of Xanthomonas malvacearum in Upland cotton. Phytopathol 60:441–447
El-Zik KM, Thaxton PM (1995) Breeding for resistance to bacterial blight of cotton in relation to races of the pathogen. In: Forrester N, Constable G (eds) Challenging the future: proceedings of the world cotton research conference-1. CSIRO, Brisbane Australia, pp 236–241
Follin JC (1981) Evidence of a race of Xanthomonas campestris pv malvacearum (E.F. Smith) Dow. Which is virulent against the gene combination B2B3 in Gossypium hirsutum L. Coton Fibres Trop 36:34–35
Follin JC (1983) Races of Xanthomonas campestris pv malvacearum (Smith) Dye in Western and Central Africa. Coton Fibres Trop 38:277–280
Follin JC, Girardot B, Mangano V, Benitez R (1988) New results on inheritance of immunity to bacterial blight (Xanthomonas campestris pv. malvacearum (Smith) Dye, race 18 and 20) in the cotton plant (Gossypium hirsutum L.). Coton Et Fre Tropic 43:167–175
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q et al (2014) TASSEL-GBS: a high-capacity genotyping by sequencing analysis pipeline. PLoS ONE 9:e90346
Gowda SA, Shrestha N, Harris TM, Phillips AZ, Fang H, Sood S, Zhang K, Bourland F, Bart R, Kuraparthy V (2022) Identification and genomic characterization of major effect bacterial blight resistance locus (BB-13) in Upland cotton (Gossypium hirsutum L.). Theor Appl Genet 135:4421–4436
Green JM, Brinkerhoff LA (1956) Inheritance of three genes for bacterial blight resistance in Upland cotton. Agron J 48:481–485
Hillocks RJ (1992) Bacterial blight. In: Hillocks RJ (ed) Cotton disease. CAB International, Wallingford, pp 39–85
Hunter RE, Brinkerhoff LA, Bird LS (1968) The development of a set of upland cotton lines for differentiating races of Xanthomonas malvacearum. Phytopath 58:830–832
Hussain T (1984) Prevalence and distribution of Xanthomonas campestris pv. malvacearum races in Pakistan and their reaction to different cotton lines. Trop Pest Manag 30:159–162
Innes NL (1965a) Inheritance of resistance to bacterial blight of cotton. I. Allen (Gossypium hirsutum) derivatives. J Agric Sci 64:257–271
Innes NL (1965b) Resistance to bacterial blight of Cotton: the genes B9 and B10. Exp Agric 1:189–191
Innes NL (1965c) Inheritance of resistance to bacterial blight of cotton II Intra-Herbaceum Crosses. J Agric Sci 64:433–437
Innes NL (1965d) Sakel strains of cotton highly resistant to bacterial blight. Euphytica 14:135–138
Innes NL (1966) Inheritance of resistance to bacterial blight of cotton. III. Herbaceum resistance transferred to tetraploid cotton. J Agric Sci 66:433–439
Innes NL (1969) Inheritance of resistance to bacterial blight of cotton IV Tanzania Selections. J Agric Sci 72:41–51
Innes NL (1974) Resistance to bacterial blight of cotton varieties homozygous for combinations of B resistance genes. Ann Appl Biol 78:89–98
Innes NL (1983) Bacterial blight of cotton. Biol Rev 58:157–176
Innes NL, Brown SJ (1969) A quantitative study of the inheritance of resistance to bacterial blight in Upland cotton. J Agric Sci 73:15–23
Jalloul A, Sayegh M, Champion A, Nicole M (2015) Bact blight of cotton. Phytopathol Mediterr 54:3–20
Jones JDG, Dangl JL (2006) The plant immune system. Nature 444:323–329
Kazan K, Lyons R (2014) Intervention of phytohormone pathways by pathogen effectors. Plant Cell 26:2285–2309
Kazan K, Manners JM (2009) Linking development to defense: auxin in plant–pathogen interactions. Trends Plant Sci 14:373–382
Knight RL (1944) The genetics of blackarm resistance. IV. G. punctatum (Sch. and Thon.) crosses. J Genet 46:1–27
Knight RL (1948) The genetics of blackarm resistance VII Gossypium arboreum L. J Genet 49:109–116
Knight RL (1950) The genetics of blackarm resistance VIII Gossypium barbadense. J Genet 50:67–76
Knight RL (1953a) The genetics of blackarm resistance. IX. The gene B6m from G. arboreum. J Genet 51:270–275
Knight RL (1953b) The genetics of blackarm resistance. X. The gene B7 from Stoneville 20. J Genet 51:515–519
Knight RL (1954) The genetics of blackarm resistance XI Gossypium anomalum. J Genet 52:466–472
Knight RL (1963) The genetics of blackarm resistance. XII. Transference of resistance from Gossypium herbaceum to G. barbadense. J Genet 58:328–346
Knight RL, Clouston TW (1939) The genetics of blackarm resistance. I. Factors B1 and B2. J Genet 38:133–159
Knight RL, Hutchinson JB (1950) The evolution of blackarm resistance in cotton. J Genet 50:36–58
Kunkel BN, Johnson JMB (2021) Auxin plays multiple roles during plant-pathogen interactions. Cold Spring Harb Perspect Biol 13:a040022
Lagiere R (1960) La bacte´riose du cotonnier (Xanthomonas malvacearum) (E.F. Smith) Dowson dans le monde et en Re´publique Centrafricaine (Oubangui-Chari). ICRT, Paris, p 252
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–wheeler transform. Bioinformatics 25:1754–1760
Li R, Erpelding JE (2016) Genetic diversity analysis of Gossypium arboreum germplasm accessions using genotyping-by-sequencing. Genetica 144:535–545
Li H, Peng ZY, Yang XH et al (2013) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45:43–50
Li R, Rashotte AM, Singh NK, Lawrence KS, Weaver DB, Locy RD (2015a) Transcriptome analysis of cotton (Gossypium hirsutum L.) genotypes that are susceptible, resistant, and hypersensitive to reniform nematode (Rotylenchulus reniformis). PLoS ONE 10:e0143261
Li R, Rashotte AM, Singh NK, Weaver DB, Lawrence KS, Locy RD (2015b) Integrated signaling networks in plant responses to sedentary endoparasitic nematodes: a perspective. Plant Cell Rep 34:5–22
Li R, Erpelding JE, Stetina SR (2018) Genome-wide association study of Gossypium arboreum resistance to reniform nematode. BMC Genet 19:52
Liu D, Guo X, Lin Z, Nie Y, Zhang X (2006) Genetic diversity of Asian cotton (Gossypium arboreum L.) in China evaluated by microsatellite analysis. Genet Resour Crop Evol 53:1145–1152
Ma KW, Ma W (2016) Phytohormone pathways as targets of pathogens to facilitate infection. Plant Mol Biol 91:713–772
Meredith WR (1993) Registration of `MD51ne’ cotton. Crop Sci 33:141
Phillips AZ, Berry JC, Wilson MC, Vijayaraghavan A, Burke J, Imani Bunn J, Allen TW, Wheeler T, Bart RS (2017) Genomics-enabled analysis of the emergent disease cotton bacterial blight. PLoS Genet 13:1–23
Robert-Seilaniantz A, MacLean D, Jikumaru Y, Hill L, Yamaguchi S, Kamiya Y, Jones JD (2011) The microRNA miR393 re-directs secondary metabolite biosynthesis away from camalexin and towards glucosinolates. Plant J 67:218–231
Romano GB, Sacks EJ, Stetina SR, Robinson AF, Fang DD, Gutierrez OA, Scheffler JA (2009) Identification and genomic location of a reniform nematode (Rotylenchulus reniformis) resistance locus (Renari) introgressed from Gossypium aridum into upland cotton (G. hirsutum). Theor Appl Genet 120:139–150
Rungis D, Llewellyn D, Dennis ES, Lyon BR (2002) Investigation of the chromosomal location of the bacterial blight resistance gene present in an Australian cotton (Gossypium hirsutum L.) cultivar. Aust J Agric Res 53:551–560
Sacks EJ, Robinson AF (2007) Development of tri-species backcross populations using a 2(ADD) hexaploid bridging line to introgress genes from A-genome diploids into Upland cotton. In: Proceedings of the world cotton research conference-4. Lubbock, TX 10–14 Sept. 2007. Intl. Cotton Advisory Committee, Washington, DC, Article No. 2000
Said JI, Lin Z, Zhang X, Song M, Zhang JF (2013) A comprehensive meat QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance on tetraploid cotton. BMC Genomics 14:776
Smith EF (1920) Bacterial diseases of plants. Saunders Co., Philadelphia
Spoel SH, Dong X (2008) Making sense of hormone crosstalk during plant immune responses. Cell Host Microbe 3:348–351
Thaxton PM, Brooks TD, El-Zik KM (2001) Race identification and severity of bacterial blight from natural infestations across the cotton belt. In: Proceedings of the Beltwide cotton conference, Anaheim, CA 9–13 Jan 2001. National Cotton Council America, Memphis, pp 137–138
Turner SD (2014) qqman: an R package for visualizing GWAS results using Q–Q and manhattan plots. Biorxiv, 005165
Verma JP (1986) Bacterial blight of cotton. Boca Raton, FL
Verma JP, Singh RP (1975) Recent studies on the bacterial diseases of fibre and oil seed crops in India. Current Trends in Plant Pathology 342:134–145
Wallace TP, El-Zik KM (1989) Inheritance of resistance in three cotton cultivars to the HVI isolate of bacterial blight. Crop Sci 29:1114–1119
Wallace TP, Bowman D, Campbell BT, Chee P, Gutierrez OA, Kohel RJ, McCarty J, Myers G, Percy R, Robinson F, Smith W, Stelly DM, Stewart JM, Thaxton P, Ulloa M, Weaver DB (2009) Status of the USA cotton germplasm collection and crop vulnerability. Genet Resour Crop Evol 56:507–532
Wheeler T (2018) Bacterial blight on cotton. In: Proceedings of the Beltwide cotton conference, San Antonio, TX, January 3–5, 2018. Natl. Cotton Counc. Am., Memphis
Wheeler TA, Sagaram US, Schuster GL, Gannaway JR (2007) Identification of factors that influence screening for bacterial blight resistance. J Cotton Sci 11:91–97
Wheeler TA, Harris T, Bart R, Woodward J, Isakeit T, Allen T, Kemerait RC (2022) Response of Xanthomonas citri pv. malvacearum isolates to cotton differing in susceptibility to the bacterium and their predicted type III effectors. Plant Health Prog 23:40–44
Wickens GM (1953) Bacterial blight of cotton. A survey of present knowledge, with particular reference to possibilities of control of the disease in African rain-grown countries. Empire Cotton Grow Rev 30:81–103
Wright RJ, Thaxton PM, El-Zik KM, Paterson AH (1998) D subgenome bias of Xcm resistance genes in tetraploid Gossypium (cotton) suggests that polyploidy formation has created novel avenues for evolution. Genetics 149:1987–1996
Xiao JH, Fang DD, Bhatti M, Hendrix B, Cantrell RG (2010) A SNP haplotype associated with a gene resistant to Xanthomonas axonopodis pv. malvacearum in Upland cotton (Gossypium hirsutum L.). Mol Breed 25:593–602
Yang Z, Nguyen B, Wright R (2015) Physical mapping B12 resistance in the cotton. Plant Cell Biotechnol Mol Biol 16:1–11
Zhang JF, Wedegaertner T, Idowu OJ, Sanogo S, Flynn R, Hughs SE, Jones DC (2019) Registration of glandless ‘Acala 1517–18 GLS’ cotton. J Plant Reg 13:12–18
Zhang JF, Bourland FM, Wheeler TA, Wallace TP (2020) Bacterial blight resistance in cotton: genetic basis and molecular mapping. Euphytica 216:111
Zhang JF, Zhu Y, Bourland F, Wheeler T (2024) Development of a quick and reliable spraying method to evaluate the US Upland cotton germplasm for resistance to bacterial blight race 18 and diagnostic DNA marker analysis. Phytopathology (in revision)
Zu Q, Deng X, Qu Y, Chen X, Cai Y, Wang C, Li Y, Chen Q, Zheng K, Liu X et al (2023) Genetic channelization mechanism of four chalcone isomerase homologous genes for synergistic resistance to Fusarium wilt in Gossypium barbadense L. Int J Mol Sci 24:14775
Zhu T, Liang C, Meng Z, Sun G, Meng Z, Guo S, Zhang R (2017) CottonFGD: an integrated functional genomics database for cotton. BMC Plant Biol 17:1–9
Acknowledgements
This research was supported in part by the U.S. Department of Agriculture, Agricultural Research Service. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
AA conducted the study, analyzed the results, and drafted the manuscript. YZ inoculated the plants and collected data for the study. LZ conceived the study and provided the tri-species population. SS and CD provided the seeds for the diversity panel. TW shared expertise in the pathogen and screening techniques. JZ conceived and directed the study. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abdelraheem, A., Zhu, Y., Zeng, L. et al. Identification of new genetic sources of resistance to bacterial blight race 18 in diploid Asiatic cotton and resistance transfer to tetraploid Upland cotton (Gossypium hirsutum). Euphytica 220, 85 (2024). https://doi.org/10.1007/s10681-024-03342-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-024-03342-1