
Abstract
Bacterial blight (BB), caused by Xanthomonas citri pv. malvacearum (Xcm), is a destructive disease to cotton production in many countries. In the U.S., Xcm race 18 is the most virulent and widespread race and can cause serious yield losses. Planting BB-resistant cotton cultivars is the most effective method of controlling this disease. In this study, 335 U.S. Upland cotton accessions were evaluated for resistance to race 18 using artificial inoculations by scratching cotyledons on an individual plant basis in a greenhouse. The analysis of variance detected significant genotypic variation in disease incidence, and 50 accessions were resistant including 38 lines with no symptoms on either cotyledons or true leaves. Many of the resistant lines were developed in the MAR (multi-adversity resistance) breeding program at Texas A&M University, whereas others were developed before race 18 was first reported in the U.S. in 1973, suggesting a broad base of resistance to race 18. A genome-wide association study (GWAS) based on 26,301 single nucleotide polymorphic (SNP) markers detected 11 quantitative trait loci (QTL) anchored by 79 SNPs, including three QTL on each of the three chromosomes A01, A05 and D02, and one QTL on each of D08 and D10. This study has identified a set of obsolete Upland germplasm with resistance to race 18 and specific chromosomal regions delineated by SNPs for resistance. The results will assist in breeding cotton for BB resistance and facilitate further genomic studies in fine mapping resistance genes to enhance the understanding of the genetic basis of BB resistance in cotton.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton (Gossypium spp.) is grown in over 84 countries for the textile industry. Most of the cotton crop is harvested from tetraploid Upland cotton (G. hirsutum, 2n = 4x = 52), which originated from merging of two diploid A and D genomes (Wendel et al. 1992; Wendel and Cronn 2003). There are many limiting factors in cotton production including biotic stresses such as bacterial blight (BB). BB is caused by the gram-negative bacterium Xanthomonas citri pv. malvacearum (Xcm). Xcm infects the aerial part of plants at all stages and generates water-soaking lesions on leaves, stems, and bolls of susceptible plants, followed by premature leaf senescence and reduced lint yield and fiber quality. Using 11 differential cotton cultivars, 22 physiological races of Xcm have been described worldwide (Hunter et al. 1968; Brinkerhoff 1970; Verma and Singh 1974; Hillocks 1992; Delannoy et al. 2005; Jalloul et al. 2015; Phillips et al. 2017). At present, race 18 is the most frequently encountered and most virulent race in the U.S. (El-Zik and Thaxton 1994; Zhang et al. 2020a).
The effective management of the disease in cotton in the 1970s until recently was achieved by planting of acid-delinted seeds and growing of resistant cultivars in the US. The replacement of BB resistant cultivars with susceptible transgenic Bollgard II (with two Bt genes) cultivars led to the spread of Xcm race 18 and significant economic losses to cotton farmers in the southern U.S. Cotton Belt including Arkansas and Mississippi (Phillips et al. 2017; Wheeler 2018; Zhang et al. 2020a). Therefore, routinely screening commercial cultivars for resistance to Xcm race 18 by artificial inoculation in field plots or greenhouses is important (Wheeler et al. 2007; Wheeler and Dever 2020; Allen 2020; Elassbli et al. 2021).
Currently, more than 22 major BB resistance B genes have been reported in cotton (Zhang et al. 2020a). Most of them are major genes including B1 to B12. The B12 gene was first identified in an African Upland cotton germplasm “S295”, and it confers resistance to all the Xcm races, including race 18 (Follin et al. 1988; Wallace and El-Zik 1989). Using restricted fragment length polymorphic (RFLP) markers and quantitative trait locus (QTL) mapping, B12 was mapped to chromosome c14 (D2) first by Wright et al. (1998) and later confirmed by Rungis et al. (2002) using amplified fragment length polymorphic (AFLP) and simple sequence repeat (SSR) markers. Xiao et al. (2010) further fine mapped B12 on D2 within a 3.4-cM region flanked by 4 SSR markers and 4 single nucleotide polymorphic (SNP) markers. In the quantitative trait locus (QTL) mapping study, Wright et al (1998) mapped six other QTL for resistance to races 2 and 4, including two QTL on c20 (i.e., D10, corresponding to B2 and B3) and four QTL (on c05/A05, c14/D02, c20/D10, and LGD02,) corresponding to b6. Interestingly, QTL corresponding to B3 and b6 also explained 53–56% of the phenotypic variation in reaction to a race 7 and 18 mixture. Therefore, BB resistance genes other than B12 may confer partial resistance to Xcm race 18. No linkage analysis of BB resistance genes has been reported in cotton since 2010 (Said et al. 2013, 2015a, b).
The traditional linkage mapping, based on bi-parental crosses to create genetic variation in a segregating population, is limited by genetic differences between two parental lines. An alternative approach to overcome this drawback is the genome-wide association study (GWAS). GWAS depends on linkage disequilibrium (LD) and uses a sample of lines from a broad breeding population, unrelated by any specific crossing design (Zhu et al. 2008). GWAS can efficiently associate genotypes with phenotypes in natural populations and jointly detect natural allelic variations and candidate genes in one study. GWAS is a powerful tool to detect the locations of genetic factors responsible for complex traits because of its high resolution with an acceptable cost and without the need for pedigree information (Flint-Garcia et al. 2005). GWAS has been used in genetic studies of many important crops including cotton. In cotton, GWAS has been employed to identify QTL related to important agronomic traits, including fiber quality and yield traits (e.g., Islam et al. 2016; Fang et al. 2017; Wang et al. 2017; Du et al. 2018; Ma et al. 2018; Naoumkina et al. 2019; Nie et al. 2020), seed oil content (Ma et al. 2019), early maturity traits (e.g., Su et al. 2018), resistance to verticillium wilt and fusarium wilt (e.g., Li et al. 2017; Zhang et al. 2020b; Abdelraheem et al. 2020a), and tolerance to drought and salt (e.g., Sun et al. 2019; Abdelraheem et al. 2020b; Li et al. 2020).
The objectives of this study were to evaluate an association mapping (AM) panel of 335 elite U.S. Upland cotton accessions for resistance to Xcm race 18, and to identify QTL for BB resistance through GWAS using a total of 26,301 polymorphic SNPs from the CottonSNP63K array (Hulse-Kemp et al. 2015).
Materials and methods
Plant materials
The association mapping (AM) panel used in this study consisted of 335 U.S. Upland cotton accessions and was previously genotyped using SSR markers (Tyagi et al. 2014). The AM panel contained cotton cultivars and breeding lines developed or grown in 14 U.S. cotton states between the early 1900s and 2005. The seed source was from the National Cotton Germplasm Collection, USDA-ARS, College Station, Texas, USA. The genotypes in the AM panel were arranged in a randomized complete block design with two replications. Ten seed for each genotype was sown in a 10-cm pot as a replication on July 4, 2016 in the greenhouse at New Mexico State University, Las Cruces, NM. The five commercial Upland cultivars, namely FM 2334GLT, FM 9058F, FM 2484B2F, PHY 375 WRF, and FM 1830GLT, were used as resistant checks, while four Pima genotypes (G. barbadense), namely PHY 805 RF, PHY 811 RF, PHY 841 RF, and NMSI E2032, were used as susceptible checks (Elassbli et al. 2021).
Bacterial culture, inoculation, and evaluation of bacterial blight resistance
The Xcm culture was identified as race 18 (Wheeler et al. 2007; Wheeler 2018). Bacteria were maintained and cultured on the ATCC medium: 1829 carrot potato dextrose agar at 30 ˚C for 3–5 days until a uniform culture covered the petri dishes. The seedlings were inoculated 3 week post-planting using the toothpick scratching method (Bird 1982). Briefly, a toothpick was dipped in a bacterial slime, and then the lower surface of a cotyledon was scratched. After inoculation, seedlings were placed in a sealed container containing water at the bottom to maintain 99% relative humidity at 23–25 °C for 48 h. The inoculated plants were then returned to the greenhouse and not watered for 1 day to increase the intensity of symptoms. The temperature of the greenhouse was maintained at an average 30–35 °C. Cotyledons were screened at 13 and 19 day post-inoculation (dpi), and true leaves were screened at 43 dpi on Aug 2nd, Aug 8th, and Sep 17th, respectively. Plant responses to Xcm infections were evaluated on an individual plant basis, as follows: 0 was recorded for resistance (R) with no water-soaking symptoms and 1 for susceptibility (S) with water-soaking symptoms. Disease incidence (DI, %) was then calculated on a plot basis, using the following formulae:
Statistical analysis
The DI for each line on a replication basis was used to perform an analysis of variance (ANOVA) using SAS software version 9.4 PROC GLM (2012 SAS Institute Inc., Cary, NC, USA). Genotypes and replications were considered random in the analysis.
GWAS analysis
The AM panel accessions were previously genotyped using the CottonSNP63K array (Hulse-Kemp et al. 2015). The same CottonSNP63K array was also used in other studies (Hinze et al. 2017; Abdelraheem et al. 2020a). The population structure and linkage disequilibrium were previously estimated based on 26,301 SNPs by Abdelraheem et al. (2020a), using the Bayesian clustering algorithm in the program STRUCTURE (Pritchard et al. 2000). The GWAS was performed using a mixed linear model (MLM) with TASSEL software (Bradbury et al. 2007), to identify associations between SNP markers and BB resistance. A false discovery rate (FDR) adjusted P-value threshold was calculated for BB resistance by dividing the P value at 0.05 with the total number of SNPs. The SNPs above the threshold were considered significant. A QTL was declared when four significant SNPs were detected within a 10 Mb region (Said et al. 2013). To identify predicted genes within a QTL region, the TM-1 genome sequence (ZJU-improved_v2-1_a1) was used (www.cottongen.com).). To estimate the interval for each QTL, a drop-off method to a nearest left and a nearest right significant marker from an apparent peak SNP marker was used.
Results
Analysis of variance
The artificial inoculation was highly successful in that, all the individual plants from the resistant Upland cotton controls- FM 2334GLT, FM 9058F, FM 2484B2F, PHY 375 WRF, and FM 1830GLT, displayed resistant responses with no water-soaked symptoms, while all the plants from four Pima genotypes- PHY 805 RF, PHY 811 RF, PHY 841 RF, and NMSI E2032, were susceptible. An analysis of variance detected a significant (P < 0.0001) genotypic variation in average DI among the 335 Upland cotton accessions in all the three screening periods (13, 19 and 43 dpi; Table 1). Therefore, significant genetic variation in BB resistance existed which provided the statistical basis for further GWAS.
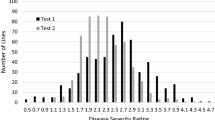
The mean genotypic DI among the three evaluated dpi was significantly correlated with coefficients of correlation ranging from 0.899 to 0.934 (P < 0.0001). The result indicated that screenings at 13 and 19 dpi on inoculated cotyledons were highly consistent. The cotyledon screenings were also highly congruent with the screening of true leaves, suggesting that Xcm infections spread from cotyledons to true leaves as seedlings grew. Therefore, it is valid to inoculate cotyledons by scratching and then evaluate cotton responses to Xcm infections on inoculated cotyledons or non-inoculated true leaves, or both.
Resistant germplasm lines against Xcm race 18
The mean DI for each accession is shown in Supplementary Table 1. Using an accepted method of classifying cotton for BB resistance into different categories (Elassbli et al. 2021) based on DI evaluated across the three dpi, 42 accessions were highly resistant (HR, with DI < 5%), including 40 lines with no symptomatic plants observed in all three dpi. In addition, 7 lines fell into the moderately resistant category (MR, with DI = 5–30%), including four lines with zero DI on cotyledons and two lines showing no symptoms on true leaves. One line was partially resistant (PR, with DI = 30–50%). A total of 131 accessions were highly susceptible (HS, with DI > 95%), including 56 lines with 100% DI at all three screenings; 136 lines were moderately susceptible (MS, with DI = 80–95%); and 19 lines were partially susceptible (PS, with DI = 50–80%). Therefore, most of the accessions evaluated in this study were susceptible to Xcm race 18. However, variation in responses to BB was commonly observed as small but various proportions of resistant plants were detected in many overall susceptible lines; and the reverse was true for some resistant lines. Therefore, segregation for BB resistance within obsolete Upland cotton germplasm is common, due likely to residual genetic variation when the lines were developed and outcrossing when seeds were increased before and after seeds were maintained by the National Germplasm Collection.
The results for the 50 resistant lines (HR, MR and PR) are listed in Table 2. Although these resistant obsolete accessions were developed by different breeding programs, 26 (53% of the resistant accessions) lines or cultivars were bred by the multi-adversity resistance (MAR) program at Texas A&M University which had a long history of breeding Upland cotton for resistance to Xcm from the 1950s to late 1990s. Some of the resistant MAR lines released from this program were also used by other breeding programs, such as the University of Arkansas and FiberMax. Surprisingly, for most of the other resistant lines that were developed by different breeding programs, resistance to Xcm 18 was not a target trait for selection, because the disease was not an issue at the time, and no lines with known resistance from Texas A&M University were used.
To ensure the accuracy of the results, all the 50 resistant lines, together with 17 susceptible accessions, were selected and retested using the same procedure under the same conditions. The results for both replicated tests were consistent (Supplementary Table 2), and the coefficient of correlation was highly significant between the two tests (0.906, P < 0.0001).
QTL identification of resistance to Xcm race 18
Through GWAS for the AM panel using 26,301 polymorphic SNPs from the CottonSNP63K array, a total of 11 QTL anchored by 79 SNPs on five chromosomes (A01, A05, D02, D08, and D10) were identified to confer resistance to Xcm race 18 (Table 3; Fig. 1). A01, A05 and D02 each carried three QTL. The three QTL on A01 were identified at the regions of 15.5–24.6 Mb with 215 predicted genes (GH_A01G0924–GH_A01G1139), 39.9–53.4 Mb with 95 predicted genes (GH_A01G1283–GH_A01G1378), and 77.1–94.6 Mb with 131 predicted genes (GH_A01G1550–GH_A01G1681), each explaining an average of 25.7, 19.7, and 18.5% of the phenotypic variation (PV) in BB resistance, respectively. On A05, the three QTL were identified at the intervals of 34.2–40.5 Mb with 177 predicted genes (GH_A05G2903–GH_A05G3080), 51.2–57.1 Mb with 37 predicted genes (GH_A05G3236–GH_A05G3273), and 86.6–90.6 Mb with 22 predicted genes (GH_A05G3602–GH_A05G3624), each explaining an average of 22.8, 19.0, and 29.0% of the PV, respectively. The three QTL on D02 were located at the intervals of 24.0–28.9 Mb with 91 predicted genes (GH_D02G1045–GH_D02G1136), 37.6–44.3 Mb with 130 predicted genes (GH_D02G12356–GH_D02G1384), and 51.2–63.0 Mb with 320 predicted genes (GH_D02G1541–GH_D20G1861), each explaining 33.0, 32.0, and 33.9% of the PV, respectively. The three QTL on D02 explained the highest proportion of the PV among all the QTL detected. A QTL was identified on D08 within a 2 Mb region at the interval of 2.96–4.91 Mb with 155 predicted genes (GH_D08G0333–GH_D08G0488), which explained an average of 18.3% of the PV. Interestingly, on D10, the QTL was identified within a narrow chromosomal region (63.32–63.36 Mb), which explained 22.5% of the PV for BB resistance. Within this region, there were only two predicted genes- GH_D10G2525 and GH_D10G2526, encoding for an uncharacterized but possible cytochrome P450 protein and a hypothetic protein, respectively.

Manhattan plot for QTL identified for resistance to bacterial blight race 18 in 335 US Upland cotton accessions based on MLM
To reduce the number of predicted genes, a drop-off method from the peak of LOD score within each QTL region was used. As a result, the total number of predicted genes for the 11 QTL was reduced from 1315 to 361 (Table 4). Except two regions- the qBB-A01-3 QTL with no apparent peak containing 131 genes and the qBB-D02-3 QTL with 70 genes, the number of predicted genes was reduced to 40 or below for each QTL with an average of 18 genes per QTL. Six of the QTL regions contained genes between 2 and 15. However, candidate genes were not identified in this study due to the lack of further experiments.
Discussion
Obsolete germplasm is an essential source for increasing genetic variability of Upland cotton. In this study, an AM panel consisted of 339 obsolete U.S. Upland accessions released between the early 1900s and 2005 were used for GWAS of resistance to BB caused by Xcm race 18 using 26,301 polymorphic SNP markers. As a result, 11 QTL were identified to confer resistance to BB. This study represented the first investigation on screening a large set of obsolete U.S. Upland cotton germplasm for Xcm race 18 resistance and GWAS on BB resistance in cotton.
In this greenhouse evaluation using a scratching method on the lower surface of cotyledons, 50 accessions were found to be resistant, while many other lines also had low observed percentages of resistant seedlings. Although a few plants that escaped from infections may not be completely ruled out, the results were highly consistent between two screening dpi on cotyledons and between the inoculated cotyledons and non-inoculated true leaves. Furthermore, a retest using the 50 resistant and 17 susceptible accessions confirmed the results. Some of these accessions were previously reported to be resistant to BB. For example, H1330, Arkot 8102 and Arkot 8606 were resistant to races 1, 2, 7, and 18 (Bourland 1996; Bourland et al. 1997; Bourland and Benson 2002). Using a mixture of races 1, 2, 7, and 10 or a mixture 1, 2 and 18, Bayles and Verhalen (2007) reported that Cascot L-7, GP 1005, GP 3755, Lankart 511, Paymaster HS 200, and several MAR cultivars (Tamcot CAB-CS, Tamcot CAMD-E, Tamcot CD3H, Tamcot SP-21 and Tamcot SP-37H) were resistant. Many lines developed from the MAR program were highly resistant to Xcm race 18. These lines included CABCSV506S-1–94, CABD3CABCH-1–89, CABD3HP3S-1–90, and CAHUGLBBCS-1–88. HGPICG14QH-1–94 was a line developed from the MAR-7B germplasm pool and was resistant to all U.S. Xcm races (Thaxton and El-Zik 2004). The MAR program maintained high resistance levels in its germplasm to the U.S. Xcm races (Thaxton and El-Zik 1998). Many of resistant Tamcot cultivars observed in this study were reported to be resistant to all Xcm races (1–18) in the U.S., including Tamcot CAB-CS, Tamcot CAMD-E, Tamcot CD3H, Tamcot Pyramid, Tamcot SP-21, Tamcot SP-23, Tamcot SP-37, and Tamcot SP-37H (Bayles and Verhalen 2007; Bird 1976, 1979; Zhang et al. 2020a).
Quantitative traits should be evaluated in multiple environments due to environmental factors and genotype × environment interactions. However, this requirement did not apply to this current study, because cotton responses to Xcm infections were evaluated qualitatively (with or without the typical water-soaking symptom) not quantitatively, and approximately 20 plants for each germplasm accession were artificially screened. The accuracy in screening the association mapping panel for BB resistance was assessed three times on cotyledons and true leaves during different growth stages, followed by consistent results from reevaluation of selected resistant and susceptible accessions. Because of segregation or contamination for BB resistance in many accessions, we did not observe 0 or 100% DI, as expected for true breeding lines. As such, DI was used to treat BB resistance as a quantitative trait, not a Mendelian trait. This phenomenon of heterogeneous status within germplasm accessions has been observed in many other crops such as soybean (Mihelich et al. 2020). Therefore, phenotyping in this study was the most reliable for any quantitative trait or qualitative trait (20 plants genotype−1). The QTL detected in this study should be stable across environments.
The African Upland cotton germplasm S295 was initially thought to be the major resistance source to Xcm race 18. Surprisingly, however, our present study demonstrated that sources of resistance to race 18 were much broader within the U.S. Upland germplasm pool. Of the 50 resistant accessions, many were developed before the detection of race 18 was first reported in 1973 (Phillips et al. 2017). In fact, some of them were developed prior to the MAR breeding or any BB resistance breeding program. For example, Acala 8 was a direct selection in 1917 from an introduced germplasm from Acala, Mexico, and Acala 1064 was developed at New Mexico State University in 1935 (Zhang et al. 2019). Clevewilt 6 was selected in 1937 and found to be moderately resistant to root-knot nematodes in Louisiana (Jones et al. 1958). Coker Wild #4 and Coker 201 were developed in South Carolina in 1928 and the early 1970s, respectively. PD-1 was a public release in 1985 from USDA-ARS, Pee Dee, SC (Culp et al. 1985); Paymaster HS 200 (Texas) and SG 747 (Mississippi) were developed by Delta and Pine Land Co. It is understandable that many of the resistant lines were from Texas, including Lankart 57 and Lankart 511 released in 1935 and 1984, respectively; and GP 1005, GP 3755, and GP 5479 developed in the 1970s and early 1980s. Of course, many resistant lines and cultivars were developed from Texas A&M University, such as Tamcot SP-21 and Tamcot SP-23 released in 1976. Screening more germplasm should provide more resistant lines or sources for breeding cotton resistant to Xcm 18.
The resistance reactions observed in resistant accessions are due to the presence of B genes on cotton chromosomes. Most of the BB resistance genes were identified using the traditional Mendelian genetic studies on biparental crosses (Zhang et al. 2020a). These B genes are resistant to different Xcm races. B12 was identified as a resistance gene to all the races including race 18 in African Upland S295 (Follin et al. 1988; Wallace and El-Zik, 1989; Wright et al. 1998; Xiao et al. 2010). Another surprising finding of our study is that some of the resistant accessions were previously found to carry resistant B genes other than B12. For example, Lankart 57 and Lankart 511 were documented to carry the B2B4b6 genes (Kappelman 1982). C5HUG2BES-2–87 was a glanded, glabrous plant type with the B2B3B7 genes (El-Zik and Thaxton 1997). Tamcot Pyramid also possessed these three genes and it also expressed some levels of resistance to race 18 (Brinkerhoff et al. 1984; Bayles and Johnson 1985; Verma 1986; Wright et al. 1998). Based on Wright et al. (1998), two QTL corresponding to B2 and B3 are located on the same chromosome- D10/c20, but not closely linked, and b6 was associated with several QTL on A05/c05, D02/c14, D10/c20 and LGD02; and the QTL corresponding to these genes contributed to phenotypic variation for resistance to Xcm race 18. Our current study identified three QTL on each of the three chromosomes (A01/c01, A05, and D02) and one QTL each on D08/c24 and D10 for resistance to Xcm race 18. Therefore, in addition to B12 in S295 which confers resistance to all the U.S. Xcm races (Wallace et al. 1989), some levels of resistance to race 18 may be provided by other major B genes. One of the QTL in D02 identified by this study may correspond to B12 and other QTL detected may correspond to other major B genes. However, it is currently unknown if the resistant germplasm lines identified in our study possess the B12 gene. A segregation analysis and B12-linked marker assay should be performed to address this question.
In this study, the number of predicted genes was identified within each of the 11 QTL regions, including 91 to 320 genes within each of 8 QTL regions and 2–37 genes with each of three QTL regions. Although a drop-down method allowed further narrowing of the QTL regions, the number of genes within each QTL region did not permit selection of candidate genes for further experiments in this study. Segregation for BB resistance within many accessions and the imbalance between resistant and susceptible genotypes further compromised the resolution and accuracy in delineating the QTL regions to a small number of genes. Segregation of BB resistance within many current commercial cultivars and elite breeding lines was also frequently observed (Wheeler and Dever 2020; Elassbli et al. 2021). Therefore, candidate genes in this study were not predicted and studied, although some were associated with responses to diseases or abiotic stresses. In addition, because there were still many predicted genes within the QTL regions for BB resistance, the present study did not perform GO analysis and RNA-seq to identify candidate genes with differential expression and sequence variation for the QTL. However, it should be studied using near-isogenic lines after a large genetic population with no or minimal germplasm contamination for BB resistance is used to further narrow chromosomal regions for resistance genes or QTL. Nevertheless, this study showed that only five (A01, A05, D02, D08, and D10) of 26 chromosome pairs and 11 chromosomal regions were involved in BB resistance, providing important information for targeted marker development and candidate gene analysis. For high resolution mapping of resistance genes or QTL for BB resistance, large segregating populations from bi-parental or multi-parental crosses between homozygous resistant and susceptible genotypes should be developed.
References
Abdelraheem A, Thyssen GN, Fang DD, Jenkins JN, McCarty JC Jr, Wedegaertner ZJF (2020b) GWAS reveals consistent QTL for drought and salt tolerance in a MAGIC population of 550 lines derived from intermating of eleven Upland cotton (Gossypium hirsutum) parents. Mol Genet Genom. https://doi.org/10.1007/s00438-020-01733-2 (Online Oct 13, 2020)
Abdelraheem A, Elassbli H, Zhu Y, Kuraparthy V, Hinze L, Stelly D, Zhang JF (2020a) A genome-wide association study uncovers consistent quantitative trait loci for resistance to Verticillium wilt and Fusarium wilt race 4 in the US Upland cotton. Theor Appl Genet 133:563–577
Allen TW (2020) Cultivar response to inoculation with Xanthomonas citri subsp. malvacearum in Mississippi in 2019. In: Proc Beltwide Cotton Conf. p. 80–83. Austin, TX, January 8–10, 2020
Bayles MB, Johnson WM (1985) Growth of homologous and heterologous phytopathogenic bacteria in cotton lines susceptible, resistant, or immune to blight. Phytopathology 75:515–519
Bayles MB, Verhalen LM (2007) Bacterial blight reactions of sixty-one upland cotton cultivars. J Cotton Sci 11:40–51
Bird LS (1976) Registration of Tamcot SP21, Tamcot SP23 and Tamcot SP37 cottons. Crop Sci 16:884
Bird LS (1979) Registration of Tamcot CAMD-E Cotton (Reg. No. 74). Crop Sci 29:411
Bird LS (1982) The MAR (multi-adversity resistance) system for genetic improvement of cotton. Plant Dis 66:172–176
Bourland FM (1996) Registration of ‘H1330’ cotton. Crop Sci 36:813
Bourland FM, Benson NR (2002) Registration of Arkot 8606, an early-maturing cotton germplasm line. Crop Sci 42:1382
Bourland FM, McGowen RE, Johnson JT (1997) Registration of Arkot 8102, Arkot 8506, and Arkot 8514 germplasm lines of cotton. Crop Sci 37:1936
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler E (2007) TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brinkerhoff L (1970) Variation in Xanthomonas malvacearum and its relation to control. Annu Rev Phytopathol 8:85–110
Brinkerhoff LA, Verhalen LA, Johnson WM, Essenberg M, Richardson PE (1984) Development of immunity to bacterial blight of cotton and its implications for other diseases. Plant Dis 68:168–173
Culp TW, Moore RE, Pitner JB (1985) Registration of PD-1 cotton. Crop Sci 25:198
Delannoy E, Lyon BR, Marmey P, Jalloul A, Daniel JF, Montillet JL, Essenberg M, Nicole M (2005) Resistance of cotton towards Xanthomonas campestris pv. malvacearum. Ann Rev Phytopath 43:63–82
Du X, Huang G, He S, Yang Z, Sun G, Ma X, Li N, Zhang X, Sun J, Liu M, Jia Y, Pan Z, Gong W, Liu Z, Zhu H, Ma L, Liu F, Yang D, Wang F, Fan W, Gong Q, Peng Z, Wang L, Wang X, Xu S, Shang H, Lu C, Zheng H, Huang S, Lin T, Zhu Y, Li F (2018) Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat Genet 50:796–802
Elassbli H, Abdelraheem A, Zhu Y, Teng T, Sanogo S, Wheeler TA, Wedegaertner T, Zhang JF (2021) Evaluation and analysis of commercial cultivars and elite breeding lines for resistance to the bacterial blight pathogen race 18 in cotton. Euphytica 21:21
El-Zik K, Thaxton P (1994) Breeding for resistance to bacterial blight of cotton in relation to races of the pathogen. In: challenging the future: World Cotton Research Conference-1, Brisbane, Australia, pp 236- 241
El-Zik KM, Thaxton PM (1997) Registration of seven multi-adversity resistant (MAR-5) germplasm lines of upland cotton. Crop sci 37:1394–1395
Fang L, Wang Q, Hu Y, Jia Y, Chen J, Liu B, Zhang Z, Guan X, Chen S, Zhou B, Mei G, Sun J, Pan Z, He S, Xiao S, Shi W, Gong W, Liu J, Ma J, Cai C, Zhu X, Guo W, Du X, Zhang T (2017) Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nature Genet 49:1089–1098
Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064
Follin JC, Girardot B, Mangano V, Benitez R (1988) New results on inheritance of immunity to bacterial blight (Xanthomonas campestris pv. malvacearum (Smith) Dye, race 18 and 20) in the cotton plant (Gossypium hirsutum L.). Coton et Fibres Tropicales 43:167–175
Hillocks RJ (1992) Cotton diseases. CAB International, Wallingford, p 415
Hinze LL, Hulse-Kemp AM, Wilson IW, Zhu QH, Llewellyn DJ, Taylor JM, Spriggs A, Fang DD, Ulloa M, Burke JJ, Giband M, Lacape JM, Van Deynze A, Udall JA, Scheffler JA, Hague S, Wendel JF, Pepper AE, Frelichowski J, Lawley CT, Jones DC, Percy RG, Stelly DM (2017) Diversity analysis of cotton (Gossypium hirsutum L.) germplasm using the CottonSNP63K Array. BMC Plant Biol 17:37
Hulse-Kemp AM, Lemm J, Plieske J, Ashrafi H, Buyyarapu R, Fang DD, Frelichowski J, Stelly D (2015) Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. Genes Genome Genet 5:1187–1209
Hunter R, Brinkerhoff L, Bird LS (1968) The development of a set of Upland cotton lines for differentiating races of Xanthomonas malvacearum. Phytopathology 58:830–832
Islam MS, Thyssen GN, Jenkins JN, Zeng L, Delhom CD, McCarty JC, Fang DD (2016) A MAGIC population-based genome-wide association study reveals functional association of GhRBB1_A07 gene with superior fiber quality in cotton. BMC Genom 17:903
Jalloul A, Sayegh M, Champion A, Nicole M (2015) Bacterial blight of cotton. Phytopathol Mediterr 54:3–20
Jones JE, Wright SL, Bewsom LD (1958) Sources of tolerance to and inheritance of resistance to root-knot nematodes in cotton. In: 11th annual Cotton Improvement Conference, Dec 15–16, 1958. Houston, TX, USA. National Cotton Council of America, Memphis, TN. pp 34–39
Kappelman AJ (1982) Registration of three okra-leaf, frego-bract disease resistant cotton germplasms (Reg. No. GP 183, GP 184, and GP 185). Crop Sci 22:693–694
Li T, Ma X, Li N, Zhou L, Liu Z, Han H, Gui Y, Bao Y, Chen J, Dai X (2017) Genome-wide association study discovered candidate genes of Verticillium wilt resistance in upland cotton (Gossypium hirsutum L.). Plant Biotechnol J 15:1520–1532
Li B, Chen L, Sun W, Wu D, Wang M, Yu Y, Chen G, Yang W, Lin Z, Zhang X, Duan L, Yang X (2020) Phenomics-based GWAS analysis reveals the genetic architecture for drought resistance in cotton. Plant Biotechnol J 18:2533–2544
Ma Z, He S, Wang X, Sun J, Zhang Y, Zhang G, Wu L, Li Z, Liu Z, Sun G, Yan Y, Jia Y, Yang J, Pan Z, Gu Q, Li X, Sun Z, Dai P, Liu Z, Gong W, Wu J, Wang M, Liu H, Feng K, Ke H, Wang J, Lan H, Wang G, Peng J, Wang N, Wang L, Pang B, Peng Z, Li R, Tian S, Du X (2018) Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat Genet 50:803–813
Ma J, Liu J, Pei W, Ma Q, Wang N, Zhang X, Cui Y, Li D, Liu G, Wu M, Zang X, Song J, Zhang J, Yu S, Yu J (2019) Genome-wide association study of the oil content in upland cotton (Gossypium hirsutum L.) and identification of GhPRXR1, a candidate gene for a stable QTLqOC-Dt5-1. Plant Sci 286:89–97
Mihelich NT, Mulkey SE, Stec AO, Stupar RM (2020) Characterization of genetic heterogeneity within accessions in the USDA soybean germplasm collection. Plant Genome 3:e20000
Naoumkina M, Thyssen GN, Fang DD, Jenkins JN, McCarty JC, Florane CB (2019) Genetic and transcriptomic dissection of the fiber length trait from a cotton (Gossypium hirsutum L.) MAGIC population. BMC Genom 20:112
Nie X, Wen T, Shao P, Tang B, Nuriman-Guli A, Yu Y, Du X, You C, Lin Z (2020) High-density genetic variation maps reveal the correlation between asymmetric interspecific introgressions and improvement of agronomic traits in Upland and Pima cotton varieties developed in Xinjiang, China. Plant J 103:677–689
Phillips AZ, Berry JC, Wilson MC, Vijayaraghavan A, Burke J, Bunn JI, Allen TW, Wheeler T, Bart RS (2017) Genomics-enabled analysis of the emergent disease cotton bacterial blight. PLoS Genet 13:e1007003
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rungis D, Llewellyn D, Dennis E, Lyon B (2002) Investigation of the chromosomal location of the bacterial blight resistance gene present in an Australian cotton (Gossypium hirsutum L.) cultivar. Crop Pasture Sci 53:551–560
Said JI, Lin ZX, Zhang XL, Song MZ, Zhang JF (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom 14:776
Said JI, Knapka JA, Song MZ, Zhang JF (2015a) Cotton QTLdb: a cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum × G. barbadense populations. Mol Genet Genom 290:1615–1625
Said JI, Song MZ, Wang H, Lin Z, Zhang X, Fang DD, Zhang JF (2015b) A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol Genet Genom 290:1003–1025
Su J, Ma Q, Li M, Hao F, Wang C (2018) Multi-locus genome-wide association studies of fiber-quality related traits in Chinese early-maturity upland cotton. Front Plant Sci 9:1169
Sun H, Meng M, Yan Z, Lin Z, Nie X, Yang X (2019) Genome-wide association mapping of stress-tolerance traits in cotton. Crop J 7:77–88
Thaxton PM, El-Zik K (1998) Host plant resistance to pathogens in MAR cotton germplasm. In: Proceedings World Cotton Research Conference-2, Athens, Greece. pp 113–119
Thaxton PM, El-Zik KM (2004) Registration of eight multi-adversity resistant (MAR-7B) germplasm lines of upland cotton. Crop Sci 44:1494–1495
Tyagi P, Gore MA, Bowman DT, Campbell BT, Udall JA, Kuraparthy V (2014) Genetic diversity and population structure in the US upland cotton (Gossypium hirsutum L.). Theor Appl Genet 127:283–295
Verma JP (1986) Bacterial blight of cotton. CRC Press, Boca Raton
Verma J, Singh R (1974) Recent studies on the bacterial diseases of fibre and oil seed crops in India. Curr Trends Plant Pathol 342:134–145
Wallace T, El-Zik K (1989) Inheritance of resistance in three cotton cultivars to the HV1 isolate of bacterial blight. Crop Sci 29:1114–1119
Wang M, Tu L, Lin M, Lin Z, Wang P, Yang Q, Ye Z, Shen C, Li J, Zhang L, Zhou X, Nie X, Li Z, Guo K, Ma Y, Huang C, Jin S, Zhu L, Yang X, Min L, Yuan D, Zhang Q, Lindsey K, Zhang X (2017) Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat Genet 49:579–587
Wendel JF, Cronn RC (2003) Polyploidy and the evolutionary history of cotton. Adv Agron 78:139–186
Wendel JF, Brubaker C, Percival A (1992) Genetic diversity in Gossypium hirsutum and the origin of Upland cotton. Am J Bot 79:1291–1310
Wheeler T (2018) Bacterial blight on cotton. In: Proc Beltwide Cotton Conf., San Antonio, TX, January 3–5, 2018. Natl. Cotton Counc. Am., Memphis, TN
Wheeler TA, Dever JK (2020) Effects of verticillium wilt and bacterial blight on commercial varieties. pp 73–79. In: Proc Beltwide Cotton Conferences, Austin, TX, January 8–10, 2020
Wheeler TA, Sagaram US, Schuster GL, Gannaway JR (2007) Identification of factors that influence screening for bacterial blight resistance. J Cotton Sci 11:91–97
Wright RJ, Thaxton PM, El-Zik KM, Paterson AH (1998) D subgenome bias of Xcm resistance genes in tetraploid Gossypium (cotton) suggests that polyploidy formation has created novel avenues for evolution. Genetics 149:1987–1996
Xiao J, Fang D, Bhatti M, Hendrix B, Cantrell R (2010) A SNP haplotype associated with a gene resistant to Xanthomonas axonopodis pv. malvacearum in upland cotton (Gossypium hirsutum L.). Mol Breed 25:593–602
Zhang JF, Abdelraheem A, Flynn R (2019) Genetic gains of Acala 1517 cotton since 1926. Crop Sci 59:1052–1061
Zhang JF, Bourland FM, Wheeler TA, Wallace TP (2020a) Bacterial blight resistance in cotton: genetic basis and molecular mapping. Euphytica 216:111
Zhang J, Abdelraheem A, Thyssen GN, Fang DD, Jenkins JN, McCarty JC, Wedegaertner T (2020b) Evaluation and genome-wide association study of Verticillium wilt resistance in a MAGIC population derived from intermating of eleven Upland cotton (Gossypium hirsutum) parents. Euphytica 216:9
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgements
The authors would like to thank Cotton Incorporated and New Mexico State University Agricultural Experiment Station for funding support.
Funding
Cotton Incorporated (Grant 12-257) and New Mexico State University Agricultural Experiment Station.
Author information
Authors and Affiliations
Contributions
HE, AA, YZ and ZT performed the screening for bacterial blight resistance. AA and HE performed the GWAS analysis. HE drafted the manuscript. TW developed the screening techniques and assisted in germplasm screening. VK developed the association mapping panel. LH and DS genotyped part of the panel. TW, LH and DS edited the MS. JZ conceived and directed the study, and helped draft and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Disclaimer
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Elassbli, H., Abdelraheem, A., Zhu, Y. et al. Evaluation and genome-wide association study of resistance to bacterial blight race 18 in U.S. Upland cotton germplasm. Mol Genet Genomics 296, 719–729 (2021). https://doi.org/10.1007/s00438-021-01779-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-021-01779-w