
Abstract
The evolution of species is complex and subtle which always associates with the genetic variation and environment adaption during active/passive spread or migration. In crops, this process is usually driven and influenced by human activities such as domestication, cultivation and immigration. One method to discover this process is to analyze the genetic diversity of those crops in different regions. This research first assessed the similarity and differentiation between genetic diversity of genotype and phenotype in 768 world-wild cowpea germplasm which were collected by USDA and US breeding programs. Totally 1048 genotyping by sequencing (GBS) derived single nucleotide polymorphisms (SNPs) and 17 agronomic traits were used to analyze the genetic diversity, distance, cluster and phylogeny. The group differentiation was analyzed based on both the genotype distances from 1048 SNP markers and the phenotypic (Mahalanobis) distance D2 from 11 traits. A consistent result of diversity in genotype (polymorphism information content, PIC) and phenotype (Shannon and Simpson index) indicated that the East Africa and South Asia sub-continents were the original and secondary regions of cowpea domestication. Both dendrograms built by genetic distance present relationship among different regions, and the Mantel coefficient showed medium correlation level (r = 0.58) between genotype and phenotype. The information of both genotypic and phenotypic differentiations may help us to understand evolution and migration of cowpea more comprehensively and also will inform breeders how to use cowpea germplasm in breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cowpea (Vigna unguiculata L. Walp.), an annual herbaceous legume plant, is widely distributed in tropical and subtropical regions. All plant parts can be used as food with high nutritional value, providing protein, vitamins, and minerals. Cowpea pod is consumed as the vegetable, and its cereal used grain contains 23–25% protein and 50–67% starch (Agah et al. 2017). What’s more, cowpea is very efficient in nitrogen fixing, and plays an important role in agro-ecosystem. Cowpea has been considered to be cultivated from wild type at 3000 years ago in Africa (Scott 1977; Scowcroft and Gibson 1975). After transferred to Asia, cowpea evolved into two subspecies known as ssp. biflora and ssp. sesquipedalis that are important vegetables in East-south Asia. Now cowpea is the major food and fodder to the livelihoods of millions poor people in less-developed countries in tropical and subtropical regions (Behura et al. 2015).
Genetic diversity is a broad concept, which can be described by many aspects. Essentially, the genetic diversity is built upon the information from whole biosphere to summarize all development, variation, similarity and difference (Nei 1972). Generally, we prefer to shrink this concept in a certain species to reveal the total numbers of genetic characteristics in the inherited component. In addition, genetic diversity is the base of biodiversity, in another words, it is the material of evolution and mutation for species to adopt environment variations (Simmonds 1976). Meanwhile, the diversity can be improved by evolution and mutation as well. We prefer to describe the evolution as a process that converts variation within a population into the variations between populations. A population with more diversity means it is likely to have more chances to possess the variations of genes that adapt to current environment. The individuals with those advantage genes tend to survive and produce offspring containing context genes (Smith 1989). The natural selection, genetic drift, gene flow, mutation, and recombination create the divergence and change of populations in different circumstances. The important part of genetic diversity and population structure research is to study the similarity and difference within populations under multiple spaces and times. This study may help understanding the situation and history of development, evolution, distribution, spread and domestication in a certain species (Celik et al. 2016b; Wang et al. 2015; Caruso et al. 2008; Chen et al. 2015).
A comprehensive and abundant world-wild germplasm collection is the optimum material to describe the genetic diversity among or within different populations from multiple geographies. Meanwhile introducing the new accessions from germplasm collection into breeding program is also the finest way to increase genetic diversity in cultivated crops. Regards to the former experience, it is impractical to describe the whole collections comprehensively and accurately, even if the information in both phenotypic and genotypic levels is absolutely doable, due to labor and time constraints. To build a core germplasm collection which represented most accessions of whole collection in an obvious small scale is considered to be an alternative for exploiting the genetic resource efficiently (Odong et al. 2013). Many core collections in germplasm storage have been established for most important crop like rice (Oryza sativa L.) (Li et al. 2010), common bean (Phaseolus vulgaris L.) (Deulvot et al. 2010), barley (Hordeum vulgare) (Munoz-Amatriain et al. 2014), peanut (Arachis hypogaea L.) (Holbrook et al. 1993), sweet potato (Ipomoea batatas L.) (He et al. 1995), potato (Solanum tuberosum L.) (Su et al. 2017), cassava (Mannihot esculanta Crantz L.) (de Oliveira et al. 2014), maize (Zea mays L.) (Lu et al. 2009), and soybean (Glycine max L.) (Grant et al. 2010). Theoretically, the core collection ought to retain genetic diversity of the major germplasm by only approximate 10% accessions. The International Institute for Tropical Agriculture (IITA, http://www.iita.org/) collected 15,122 unique cowpea samples from 88 countries and constituted a core collection with 2062 accessions. Meanwhile, the cowpea core collection has also been established by USDA (United State Department of Agriculture) with 720 accessions selected from 7737 cowpea accessions (USDA-ARS Griffin, GA http://www.ars-grin.gov/ars.htm).
We collected 716 cowpea germplasm accessions including core collection and special germplasm from GRIN-USDA originally from 58 countries in six main regions (North American, Latin American, Asia, African, Europe and Oceania), plus 52 US cultivars in this study. The objective of this research was to analyze the genetic diversity and characterize the differentiation by both genotype and phenotype among and within different regions in the whole world-wide cowpea germplasm collection.
Materials and methods
Plant materials
A total of 768 cowpea genotypes were used in this study including 716 USDA GRIN cowpea germplasm accessions and 52 USA cowpea cultivars. Among the 716 USDA germplasm accessions, 502 accessions were from the cowpea core-collection and the other 214 accessions were selected based on their special traits such as cowpea mosaic virus resistance, bacterial leaf bight resistance, Fusarium wilt resistance, iron chlorosis tolerance, low phosphorus and rock phosphorus tolerance, and seed antioxidant content. All seeds of the GRIN germplasm and US cultivars were obtained from USDA Plant Genetic Resources Conservation Unit at Griffin, GA and the 716 cowpea germplasm accessions were originally collected from 58 countries (Kamphuis et al. 2012; Singh et al. 2007, 2010) (Table 1, S1).
Field experiment and phenotyping
The 768 cowpea accessions were planted by a randomized complete block design (RCBD), with three replications in the Fayetteville Research and Extension Experimental Center, University of Arkansas, AR. The planting plot was set as 10 × 3 ft growing, 20 plants in a row-plot during both 2014 and 2015. The weed control, water and fertilizer application were followed as the regular management practice.
Totally 17 agronomical traits were investigated and recorded based on the criteria by USDA procedure description with modifications (https://npgsweb.ars-grin.gov/gringlobal/cropdetail.aspx?type=descriptor&id=188). The 17 traits consisted of (1) five quantitative traits: seed-weight, pod-length, pod-number, maturity, and plant-height; (2) six oligogenic traits: plant-habit, pod-position, pod-placement, seed-shape, flower-color, and vine; and (3) six typical qualitative traits: pod-color, seed-pattern, seed-color, seed-pattern color, leave-shape and seed-texture. The evaluating and coding for the 17 traits are described in the supplementary Table S2.
Genotyping
For DNA extraction, all cowpea were planted in 12*7 cell germination trays. Five unique seeds of each accession were seeded in a cell of tray, and then the leaf tissues from five seedling plants for each genotype were bulk sampled. After freeze-drying, the CTAB method was applied for DNA extraction (Qin et al. 2017). DNA sequencing was conducted using genotyping by sequencing (GBS) (Elshire et al. 2011) that was done by HiSeq 2000 in Beijing Genome Institute (BGI). Sequence assemble, mapping and SNP call were done by BGI using SOAP family software (http://soap.genomics.org.cn/). The SOAPaligner/soap2 (http://soap.genomics.org.cn/) was used to align the short-read to cowpea genome reference (cowpea_Genome_ 0.03.fa) and SOAPsnp v 1.05 was used for SNP calling (Li 2011). The cowpea_Genome_0.03.fa (6750 scaffolds or contigs) (http://harvest-blast.org/) was kindly provided by Dr. Timothy J. Close in University of California Riverside, CA, USA.
Statistical analysis
Genetic distance was calculated by allele-sharing method (Neuman et al. 2000). Phylogenetic reconstruction was based on the un-weighted pair-group method using arithmetic average (UPGMA) method implemented in PowerMarker version 2.7 (Liu and Muse 2005). The population structure based clustering group (Q) was inferred by model-based program STRUCTURE using the burn-in of 10,000, run length of 50,000, and a model allowing for admixture and correlated allele frequencies(Earl 2012). The tree to visualize the phylogenetic distribution of accessions and ancestry groups was constructed and bootstrapped 1000 times of re-sampling by using MEGA 6 (Tamura et al. 2007). Principal coordinate analysis (PCoA), that summarizes the major patterns of variation in multi-locus data set, was performed with Tassel software version 5.0 (Bradbury et al. 2007). PowerMarker was also used to calculate the average number of alleles, gene diversity, and polymorphism information content (PIC) values. Fst indicative of ancestral relationship between genetic groups was calculated using an AMOVA approach in Arlequin V2.000 (Excoffier et al. 2005).
Seventeen phenotypic traits were used to evaluate the genetic diversity by Shannon (H′) and Simpson index (1-D).
Mahalanobis distance as a measurement to calculate genetic differentiation among the groups (Ichihashi et al. 2005) by 11 traits, seed-weight, pod-length, pod-number, maturity, plant-height, plant-habit, pod-position, pod-placement, seed-shape, flower-color, and vine. Analysis of variance (ANOVA) and Canonical discriminant analysis were performed by the JMP Genomics to test the differences among the groups based on 11 traits (Sall et al. 2012). Eventually, the correlation of genetic structure differentiations resulting from the genotypic markers with phenotypic traits was assessed using the Mantel test (Mantel 1967) performed by PowerMarker.
Results
Genotypic diversity and differentiation
In this study, 768 accessions were collected from 6 continents including 11 geography populations: North American, Latin American, Asia (East Asia, Indian and Central West Asia), Africa (South Africa, East Africa, and West Africa), Europe, Oceania and American Cultivar. The SNP parameters of each group were estimated for the major allele frequency, gene diversity, heterozygosity, and PIC (Table 1). The genetic diversity and PICs varied from 0.35 and 0.32 for India to 0.21 and 0.17 for Europe, respectively, indicating genetic variation in each group. What is noteworthy is that some populations in same continents have significant difference in genetic diversity and PICs, such as the sub-populations of Asia and Africa.
The ANOVA for the molecular markers showed that the majority of genetic variance was within the populations with 90.4% of total variation, while 9.6% variation was observed among the populations. Pairwise genetic distances between each population based on allele sharing of the 768 accessions showed the medium differentiation. The mean genetic distance of all group pairs were ranging from 0.074 between North American and Latin American to 0.183 between Oceania and Central West Asia, East Africa and Europe (Table 2).
In order to verify the subdivisions, a model-based clustering method for multi-loci genotype data was employed to infer the population structure and assign individuals to cluster (Q) using STUCTURE. The most probable structure number of (K) was calculated based on Evanno’s method (Evanno et al. 2005). The genetic background was estimated by Q value for each accession. As the result, totally three gene pools (Q1, Q2, and Q3) were demonstrated to be the best division for all 768 accessions/cultivars in our former reports (Qin et al. 2016, 2017; Shi et al. 2016; Xiong et al. 2016).
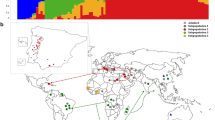
In all tested 768 accessions, the percentages of gene pool in each population were different. These differences were used to classify the geography population in this study. All Europe (100%) accessions are grouped into Q1 pool, after that the North American, Latin American and USA Cultivars also showed the relative high Q1 ratio (more than 40%), by contrast the ratio of Q2 pool in each population was far less than Q1, that only USA Cultivar and Africa have more than 40% Q2 accessions. The Q3 pool was minority (less than 25%) in all populations except the Oceania, which got 88% Q3 accessions; and Asia accessions, which were close to even distribution of three pools. The genetic distance was calculated to distinguish seven continent populations and to build a phylogenetic tree which divided those populations into three main branches where, Europe, North American, Latin American, and Asia were grouped together in branch 1 as high Q1 group; the Oceania alone comprised the second one as high Q3 group; Africa and Cultivars were placed in another branch as high Q2 group. The cowpea populations in the same branch and similar Q ratio revealed close genetic background.
As well as the corresponding Q ratio and phylogeny among continent populations, the similar result was also found among 11 regional populations. The dendrogram (Fig. 1) divided the six populations including North America, Latin America, Oceania, East Africa, India, and South Africa into one main branch, which shown high Q3 ratio (more than 21%). While the rest five populations including West Africa, Cultivar, East Asia, Central West Asia, and Europe were grouped into the other branch, which was marked as low Q3 group. The consistency of structuring with phylogeny in both continent and regional populations improved the reliability of our study.

a Dendrogram of differentiation and genetic structure ratios (blue for Q1, red for Q2, yellow for Q3, and green for admixture) among 6 continents plus one group consisted of America cowpea cultivars, and b dendrogram of differentiation and genetic structure ratios among 11 regions. Only more than 0.50 bootstrap values are shown
The results of principal coordinate analysis in Tassel 5 are marked by different geographical indications. As can be seen from Fig. 2, most accessions from the same origin tend to cluster, and there were obvious cross-overlapping among populations. Although some sub-populations are from the same continent, their distributions in the binary scatter-plots are distinctly separated from each other, like the accessions from Central-West Asia, India and East Asia. The US Cultivars and other American (North American and Latin American) populations are also significantly different in the scatter plot. Moreover for the two African sub-populations, both West and East Africa in the figure, there is almost no overlap between the two regions. However there are also the opposite phenomenon, as in a continent South Africa and East Africa have a very similar distribution, adding that those accessions from North America and Latin America have a similar phenomenon. It is noteworthy that, there is a clear separation between American cultivar and local American species, while the accessions from western Africa indicated closer relation with it by large-scale overlap in scatter-plots.

Principal coordinate analysis based on 1048 SNPs in 768 accessions/cultivars
Phenotype diversity and differentiation
Totally 17 phenotypic traits were collected and recorded with standards to analyze and estimate genetic diversity among geography populations. The Shannon–Weaver diversity index (H′) and Simpson index (1-D) were calculated for the qualitative and quantitative characters to compare diversity between the different characters and among various districts. The high consistency (r = 0.97, P < 0.01) was found between Shannon (H′) and Simpson index (1-D) in the 17 traits based on Mantel text (Table 3). Higher diversity was observed in Asia populations, for eight traits, including pod-number, plant-high, pod-length, seed-weight, plant-habit, seed-color, seed-pattern color and seed-shape, with the highest diversity score in this population set. And the population of Africa had the most diversity in maturity, plant habit, and seed shape. Except the population from Europe whose diversity was limited by population size, cultivar populations seemed to be the narrowest one in trait diversity, on the account of the preference to yield (pod number, seed weight), harvest (plant habit, maturity) and market (seed color, texture and shape) during the breeding. Additionally, high level variable coefficient (CV) of 11 continuous traits was observed in Asia and Africa populations (Table 3), which also implied the high genetic diversity.
All 768 accessions/cultivars were marked with colors according to different geographic regions, which were conducted by using Canonical discriminant analysis of 11 continuous traits (Fig. 3). The first four significant canonical discriminant functions (CAN) explained 92.0% total phenotype differentiation, and 44.9% of first CAN and 23.1% of the second CAN function, respectively. According to the discriminant analysis, almost all accessions can be grouped into their own geography populations that tended to distinguish with others but overlaps. The accessions from Africa and Asia are most widely distributed, indicating rich phenotype diversity. Quite a number of Africa accessions were more similar to wild cowpea that had massive vegetative growth with poor harvest. The Asia populations contained more diversity of pod and seed traits, which may be caused by local market desires. Dense and mixed grouping occurred in most sub-populations from same continent especially in Latin and North American. However East Asia and East Africa clearly separated with other same continent populations. The dendrogram of differentiation classified the populations to three main branches based on Mahalanois distance (Fig. 4). The populations in the same branch were also with the high overlap degree in discriminant analysis in this study, which implied similar genetic background, vice versa. Basically, the ‘Cultivar’ group was independent and stayed alone and separated from others, however it still modestly related to Asia and Africa populations (Table 2). The accessions from South Asia subcontinent were high relevant to Central East Africa accessions, as well as the short genetic distance between Latin and North American was observed.

Canonical discriminant analysis (CDA) using 12 phenotypic traits among structural groups in 768 accessions/cultivars

a Dendrogram of differentiations among 6 continents plus one group consisted of America cowpea cultivars, b dendrogram of differentiation among 11 regions. Only more than 0.50 bootstrap values are shown
Relationships between genotype and phenotype differentiation
The dendrogram based on the Mahalanobis distance (D2) using the 11 phenotypic traits (Fig. 4) didn’t fully match with the dendrogram based on the genotype differentiation using 1028 markers (Fig. 2). However, all dendrograms and scatter diagrams revealed clear and consistent relations among the seven regions/groups: Cultivar, India, East Asia, West Africa, East Africa, Latin American and North American. Mantel analysis has been widely used to compare the genetic correspondence between genotypic and phenotypic differentiation (Gizaw et al. 2007). In this study, genetic distance derived from the genotypes was correlated with the distance derived from phenotype in medium level (r = 0.53, P < 0.02), indicating the matching of dendrograms and similarity of scatter diagrams between genotype and phenotype.
Discussion
The genotype and phenotype for genetic analysis
Compared to the rice, corn, soybean, barley, wheat, and other major cereal or economic crops, high-throughput sequencing and large-scale SNP development in cowpea started very late and there was no research reports involving SNP markers until 2009 (Cai et al. 2008; Shirasawa et al. 2007; Ching et al. 2002; Rostoks et al. 2005; Yanagisawa et al. 2003; Muchero et al. 2009b). GBS is considered to be one of the most advanced and efficient technology for SNP developing, which can obtain massive genetic markers without deep sequencing or understanding of genetic information in aim crop (Uncu et al. 2016; Schroder et al. 2016; Celik et al. 2016a; Ariani et al. 2016; Arbelaez et al. 2015). The 5808 SNPs derived from 768 accessions using GBS in this study had a similar nucleotide ratio with former studies (Huynh et al. 2013; Muchero et al. 2009a). The large number of missing and misreading in those SNPs wouldn’t be ignored by consideration of the non-reference sequencing in GBS. Therefore, only 1028 markers were left for future research by removing the rare (less than 5%) loci, high ratio (more than 20%) missing and high (more than 5%) heterozygous nucleotide (Huynh et al. 2013).
The analysis of phenotypic traits is an important method to estimate and evaluate the relationship and genetic diversity among accessions or groups (Jaaska and Jaaska 1988). Even if the phenotype record and detection are easily affected by different readers or inspectors, while as a classical taxonomy method, it cannot only reflect the genetic difference between accessions/groups, but also can reveal the adaptability of environment in multiple regions (Li et al. 2017; Zeka et al. 2015). For calculating the Simpson and Shannon index, all 17 traits used the assignment data for diversity analysis (Darling et al. 2016; Peng et al. 2014). The ANOVA, CV dendrograms and discriminant analysis were calculated based on original data of 11 continuous traits. The three-level assignment method was used to evaluate seven traits as oligogene controlled traits to simplify and unify the detection, however, the virtual instance may be more complicated than this assumption (Cobb et al. 2013).
Genotype diversity and relationship
Genotype diversity has been partially described in our former report (Xiong et al. 2016), while here we not only analyzed the populations under the continents but also involved the difference among each continent for more comprehensive understanding and recognizing in cowpea diversity. Among all continents, the most richness of diversity was observed in Africa population, in particular, the highest diversity is found in East part of Africa while slightly reduced in western and southern populations. The wider genetic diversity found in East Africa conformed previous reports that the eastern Africa was the center of domestication of cowpea and western Africa was a distinct gene pool which was derived by immigrant wild cowpea from eastern part (Baudoin and Maréchal 1985; Singh and Rachie 1985; Smartt 1986; Fatokun et al. 1993; Hartl et al. 1997). The next most diversity was observed in Asia population, especially in India (even more than East Africa) which was considered to be the secondary domestication place around the word according to the dendrogram and former reports (Bower 2007; Mahalakshmi et al. 2007; Timko et al. 2007). More humidity and short daytime may change the genetic structure of the alien crop. Along with the spreading to the eastern and western, the diversities were found to decline in both areas, for human or nature selections may narrow down the variation of imported cowpea (Xu et al. 2012). The medium diversity and similar genetic background were revealed in Latin and North American, where had the shortest genetic distance with Indian. This fact strongly recommended a direct import or transport routes from India to American continent. The “Cultivar” was distinguished with other populations except the one from West Africa, which had a long-term cooperation with USA in cowpea breeding since 1940s (Behura et al. 2015; Fang et al. 2007b). This breeding program based import may explain the distinction and differentiation between the Cultivars and accessions in the same continent.
Phenotype diversity and relationship
Both Simpson and Shannon indexes revealed the extraordinary diversity in Asia and Africa populations using 17 traits. Especially Africa populations, not only include bunch of breeding type resource, but also contained semi-wild type accessions. The maturity of Africa are wildly distributed from 23 to 214, meanwhile a lot of typical wild-type features such as evergreen, perennial, perpetual blooming and crack pod can be found in Africa populations(Behura et al. 2015). Their grain polymorphism is also the most widely with richness in pattern color and had the similar distribution in all kinds of grain type. According to the traditional evolution theory and past reports, the Africa was confirmed to be the original domestication place in this study (Kapoor-Vijay and White 1992). The high diversity was also observed in Asia accessions, which were closer to cultivated cowpea generally. The most agricultural diversities were focused in India population, which was closely related to the population from east Africa. The slight decline was found in east Asia population, which contained abundant ssp. Biflora and ssp. Sesquipedlias that are significantly distinguished with Africa type accessions, which may be caused by the considerable climate change (Krishna 2014). The humid weather and short daytime may extend the maturity and pod drying time, which made the pod have more time to lengthen and keep fresh (Kanchikerimath and Singh 2001; Osman et al. 1998). In addition, the preference of long and fresh pod in local diet changed the cowpea tending to a vegetable crop in Asia. The diversities of populations in North and Latin America were relatively low, which were difficult to differentiate from each other. Meanwhile, the American accessions are closer to some Asian resources but properly distinct with African populations and obviously different from USA commercial varieties (Cultivar group). The cultivars have very narrow agriculture diversity including plant-habit, vine, maturity, plant-high and seed-color for those advanced yield and harvest traits were selected for breeding during the domestications (Fang et al. 2007a; Santos and Boiteux 2013). The Cultivars in our study were closer to the accessions from South and West Africa, which implied a direct import from Africa for USA breeding projects (Singh et al. 1997).
Differentiation between genotype and phenotype
Assessment of differentiation and relationship between genotype and phenotype has never failed to fascinate geneticist. In this study, the similar genetic diversities were found between genotype using 1028 SNPs and phenotype based on 17 agriculture traits. The phenotypic dendrogram based on the Mahalanobis distance moderately matched with the genotypic dendrogram based on the genetic distance. Despite of the very detailed relevance between each population, the main processes of cowpea spreading were clear and explicit. According to the high consistent diversity and relation analysis, it is concluded that (1) the east Africa and South Asia sub-continents were identified to be the original and second domestication region of cultivated cowpea; (2) the cowpea continued spreading to adjacent regions from two domestication centers in both Africa and Asia continents, respectively; (3) the cowpea was transferred to America from South Asia sub-continent by agriculture communication of two colonies of Great Briton; and (4) the US breeding programs may have imported the crucial resource from West Africa and East Asia directly. However, the obvious confliction between phenotype and genotype were found in Oceania and Europe, which had a close relation in phenotype but adverse in genotype. This incompatibility may be caused by the leak of accessions in both populations.
Genetic differentiation assessed by the genotypic markers among geography populations was correlated with the phenotypical differentiation assessed by the agriculture traits in medium level (Mantel test, r = 0.52, P < 0.001) compared with other crops (Li et al. 2010). The decreased correlation can be explained by the sensitivity of some traits to environmental influence, which results in low heritability. The low heritability makes it hard to discriminate lines in the population. A non-neutral evolutionary history for some traits can also give a low correlation between phenotypic distance and genetic distance.
Potential benefit of genotypic and phenotypic diversity
Study of genetic diversity has practical significance for the fluctuation of genetic diversity is the result from the crop evolution. Research of genetic diversity can reveal the species history and the impact of human activities during the evolution. Those facts have proven that human activity increased the species evolution rate towards a single direction, especially in the process of breeding, which is actually the process of dramatic decline of genetic diversity. During long-term and pursuit of high-yield cultivation, some elite resources were used excessively, which caused a narrower genetic background of massive cultivars in specific regions, which is harmful to future agriculture (Zhou et al. 2012; Bauchet et al. 2017). Cowpea spreads all over the world, so the process of domestication should arouse our attention and consideration. At first, when cowpea was transported to Asia, its growth period prolonged greatly due to climate change, which was considered to be a negative breeding trait. However, it makes the cowpea turn to an important vegetable crop in Asia, which proved an example to broaden genetic diversity during the cultivation. We habitually consider the genetic diversity as a finite resource, but the diversity itself is being created. On one hand, it is the result of the adaption or evolution to the environment, on the other hand it is a result of modification by human. The history of cowpea domestication is more than 3000 years (Faris 1965), while as a crop, the cowpea is used for most purposes in agriculture, including cereal, vegetable, fodder, crop rotation, nitrogen fixation and artifact. According to context phenomenon, we may have this conclusion: “human domestication will never reduce the genetic diversity, but the single aiming will”. In recent years, development and use of functional crops get more and more attention in the world. Those accessions with specific function and specific nutrients not only made a prominent contribution in their own aspect, but also broaden the width of the spectrum of breeding.
References
Agah S, Kim H, Mertens-Talcott SU, Awika JM (2017) Complementary cereals and legumes for health: synergistic interaction of sorghum flavones and cowpea flavonols against LPS-induced inflammation in colonic myofibroblasts. Mol Nutr Food Res. https://doi.org/10.1002/mnfr.201600625
Arbelaez JD, Moreno LT, Singh N, Tung CW, Maron LG, Ospina Y, Martinez CP, Grenier C, Lorieux M, McCouch S (2015) Development and GBS-genotyping of introgression lines (ILs) using two wild species of rice, O. meridionalis and O. rufipogon, in a common recurrent parent, O. sativa cv. Curinga. Mol Breed. 10.1007/s11032-015-0276-7
Ariani A, Teran JCBMY, Gepts P (2016) Genome-wide identification of SNPs and copy number variation in common bean (Phaseolus vulgaris L.) using genotyping-by-sequencing (GBS). Mol Breed. 10.1007/s11032-016-0512-9
Bauchet G, Grenier S, Samson N, Bonnet J, Grivet L, Causse M (2017) Use of modern tomato breeding germplasm for deciphering the genetic control of agronomical traits by Genome Wide Association study. Theor Appl Genet. https://doi.org/10.1007/s00122-017-2857-9
Baudoin J, Maréchal R (1985) Cowpea taxonomy, origin and germplasm. Cowpea research, production and utilization. Wiley, Chichester, pp 3–9
Behura R, Kumar S, Saha B, Panda MK, Dey M, Sadhukhan A, Mishra S, Alam S, Sahoo DP, Sugla T, Sahoo L (2015) Cowpea [Vigna unguiculata (L.) Walp]. Methods Mol Biol 1223:255–264. https://doi.org/10.1007/978-1-4939-1695-5_20
Bower A (2007) African American foodways: explorations of history and culture. University of Illinois Press, Champaign
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Cai CM, Van K, Kim MY, Jun TH, Shin JH, Cho SY, Lee YS, Lee SH (2008) SNP discovery, linkage analysis and microsynteny in tentative consensus sequences derived from roots cDNA in a supernodulating soybean mutant. Euphytica 164(1):189–197. https://doi.org/10.1007/s10681-008-9702-9
Caruso M, Federici CT, Roose ML (2008) EST-SSR markers for asparagus genetic diversity evaluation and cultivar identification. Mol Breed 21(2):195–204. https://doi.org/10.1007/s11032-007-9120-z
Celik I, Bodur S, Frary A, Doganlar S (2016a) Genome-wide SNP discovery and genetic linkage map construction in sunflower (Helianthus annuus L.) using a genotyping by sequencing (GBS) approach. Mol Breed. https://doi.org/10.1007/s11032-016-0558-8
Celik I, Camci H, Kose A, Kosar FC, Doganlar S, Frary A (2016b) Molecular genetic diversity and association mapping of morphine content and agronomic traits in Turkish opium poppy (Papaver somniferum) germplasm. Mol Breed. https://doi.org/10.1007/s11032-016-0469-8
Chen HL, Liu LP, Wang LX, Wang SH, Wang ML, Cheng XZ (2015) Development of SSR markers and assessment of genetic diversity of adzuki bean in the Chinese germplasm collection. Mol Breed. https://doi.org/10.1007/S11032-015-0383-5
Ching A, Caldwell KS, Jung M, Dolan M, Smith OS, Tingey S, Morgante M, Rafalski AJ (2002) SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. https://doi.org/10.1186/1471-2156-3-19
Cobb JN, Declerck G, Greenberg A, Clark R, McCouch S (2013) Next-generation phenotyping: requirements and strategies for enhancing our understanding of genotype-phenotype relationships and its relevance to crop improvement. Theoret Appl Genet 126(4):867–887. https://doi.org/10.1007/s00122-013-2066-0
Darling KF, Schweizer M, Knudsen KL, Evans KM, Bird C, Roberts A, Filipsson HL, Kim JH, Gudmundsson G, Wade CM, Sayer MDJ, Austin WEN (2016) The genetic diversity, phylogeography and morphology of Elphidiidae (Foraminifera) in the Northeast Atlantic. Mar Micropaleontol 129:1–23. https://doi.org/10.1016/j.marmicro.2016.09.001
de Oliveira EJ, Ferreira CF, da Silva Santos V, de Jesus ON, Oliveira GA, da Silva MS (2014) Potential of SNP markers for the characterization of Brazilian cassava germplasm. Theor Appl Genet 127(6):1423–1440. https://doi.org/10.1007/s00122-014-2309-8
Deulvot C, Charrel H, Marty A, Jacquin F, Donnadieu C, Lejeune-Hénaut I, Burstin J, Aubert G (2010) Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genom 11(1):468
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. https://doi.org/10.1371/journal.pone.0019379
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Fang J, Chao C-CT, Roberts PA, Ehlers JD (2007a) Genetic diversity of cowpea [Vigna unguiculata (L.) Walp.] in four West African and USA breeding programs as determined by AFLP analysis. Genet Resour Crop Ev 54(6):1197–1209
Fang JG, Chao CCT, Roberts PA, Ehlers JD (2007b) Genetic diversity of cowpea [Vigna unguiculata (L.) Walp.] in four West African and USA breeding programs as determined by AFLP analysis. Genet Resour Crop Ev 54(6):1197–1209. https://doi.org/10.1007/s10722-006-9101-9
Faris DG (1965) The origin and evolution of the cultivated forms of Vigna Sinensis. Can J Genet Cytol 7(3):433–452. https://doi.org/10.1139/g65-058
Fatokun C, Danesh D, Young N, Stewart E (1993) Molecular taxonomic relationships in the genus Vigna based on RFLP analysis. Theor Appl Genet 86(1):97–104
Gizaw S, Van Arendonk JAM, Komen H, Windig JJ, Hanotte O (2007) Population structure, genetic variation and morphological diversity in indigenous sheep of Ethiopia. Anim Genet 38(6):621–628. https://doi.org/10.1111/j.1365-2052.2007.01659.x
Grant D, Nelson RT, Cannon SB, Shoemaker RC (2010) SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res 38(1):D843–D846. https://doi.org/10.1093/nar/gkp798
Hartl DL, Clark AG, Clark AG (1997) Principles of population genetics, vol 116. Sinauer Associates, Sunderland
He G, Prakash CS, Jarret RL (1995) Analysis of genetic diversity in a sweetpotato (Ipomoea batatas) germplasm collection using DNA amplification fingerprinting. Genome 38(5):938–945
Huynh B-L, Close TJ, Roberts PA, Hu Z, Wanamaker S, Lucas MR, Chiulele R, Cissé N, David A, Hearne S, Fatokun C, Diop NN, Ehlers JD (2013) Gene pools and the genetic architecture of domesticated cowpea. Plant Genome. https://doi.org/10.3835/plantgenome2013.03.0005
Ichihashi H, Honda K, Wakami N (2005) Robust PCA with intra-sample outlier process based on fuzzy Mahalanobis distances and noise clustering. In: Fuzz-IEEE 2005: Proceedings of the IEEE International Conference on Fuzzy Systems, pp 640–645
Jaaska V, Jaaska V (1988) Isoenzyme variation in the generaPhaseolus and Vigna (Fabaceae) in relation to their systematics: aspartate aminotransferase and superoxide dismutase. Plant Syst Evol 159(3–4):145–159
Kamphuis LG, Gao L, Singh KB (2012) Identification and characterization of resistance to cowpea aphid (Aphis craccivora Koch) in Medicago truncatula. BMC Plant Biol 12:101. https://doi.org/10.1186/1471-2229-12-101
Kanchikerimath M, Singh D (2001) Soil organic matter and biological properties after 26 years of maize-wheat-cowpea cropping as affected by manure and fertilization in a Cambisol in semiarid region of India. Agr Ecosyst Environ 86(2):155–162. https://doi.org/10.1016/S0167-8809(00)00280-2
Kapoor-Vijay P, White J (1992) Global biodiversity strategy. Conservation biology: a training manual for biological diversity and genetic resources. Commonwealth Science Council, pp 13–22
Krishna K (2014) Cowpea farming zones of Africa and Asia. Agroecosystems: soils, climate, crops, nutrient dynamics, and productivity. CRC Press, Boca Raton, pp 139–146
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21):2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li X, Yan W, Agrama H, Hu B, Jia L, Jia M, Jackson A, Moldenhauer K, McClung A, Wu D (2010) Genotypic and phenotypic characterization of genetic differentiation and diversity in the USDA rice mini-core collection. Genetica 138(11–12):1221–1230. https://doi.org/10.1007/s10709-010-9521-5
Li CQ, Song L, Zhu YJ, Zhai YJ, Wang QL (2017) Genetic diversity assessment of upland cotton variety resources in china based on phenotype traits and molecular markers. Crop Sci 57(1):290–301. https://doi.org/10.2135/cropsci2016.03.0200
Liu KJ, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2129. https://doi.org/10.1093/bioinformatics/bti282
Lu Y, Yan J, Guimaraes CT, Taba S, Hao Z, Gao S, Chen S, Li J, Zhang S, Vivek BS, Magorokosho C, Mugo S, Makumbi D, Parentoni SN, Shah T, Rong T, Crouch JH, Xu Y (2009) Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet 120(1):93–115. https://doi.org/10.1007/s00122-009-1162-7
Mahalakshmi V, Ng Q, Lawson M, Ortiz R (2007) Cowpea [Vigna unguiculata (L.) Walp.] core collection defined by geographical, agronomical and botanical descriptors. Plant Genet Resour 5(03):113–119. https://doi.org/10.1017/S1479262107837166
Mantel N (1967) Correction. Biometrics 23(4):854–1000
Muchero W, Diop NN, Bhat PR, Fenton RD, Wanamaker S, Pottorff M, Hearne S, Cisse N, Fatokun C, Ehlers JD (2009a) A consensus genetic map of cowpea [Vigna unguiculata (L) Walp.] and synteny based on EST-derived SNPs. Proc Natl Acad Sci 106(43):18159–18164
Muchero W, Matthews WC, Diop NN, Bhat PR, Wanamaker S, Ehlers JD, Close TJ, Roberts PA (2009b) Qtl mapping of root-knot nematode resistance in cowpea (Vigna Unguiculata) using est-derived Snp markers. J Nematol 41(4):361–362
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Neuman RJ, Saccone NL, Holmans P, Rice JP, Sun LW (2000) Clustering methods applied to allele sharing data. Genet Epidemiol 19:S57–S63
Odong TL, Jansen J, van Eeuwijk FA, van Hintum TJ (2013) Quality of core collections for effective utilisation of genetic resources review, discussion and interpretation. Theor Appl Genet 126(2):289–305. https://doi.org/10.1007/s00122-012-1971-y
Osman M, Emmingham WH, Sharrow SH (1998) Growth and yield of sorghum or cowpea in an agrisilviculture system in semiarid India. Agroforest Syst 42(1):91–105. https://doi.org/10.1023/A:1006147926915
Peng L, Ru M, Wang BQ, Wang Y, Li B, Yu J, Liang ZS (2014) Genetic diversity assessment of a germplasm collection of Salvia miltiorrhiza Bunge. based on morphology, ISSR and SRAP markers. Biochem Syst Ecol 55:84–92. https://doi.org/10.1016/j.bse.2014.01.020
Qin J, Shi AN, Xiong HZ, Mou BQ, Motes D, Lu WG, Miller JC, Scheuring DC, Nzaramba MN, Weng YJ, Yang W (2016) Population structure analysis and association mapping of seed antioxidant content in USDA cowpea (Vigna unguiculata L. Walp.) core collection using SNPs. Can J Plant Sci 96(6):1026–1036. https://doi.org/10.1139/cjps-2016-0090
Qin J, Shi AN, Mou BQ, Bhattarai G, Yang W, Weng YJ, Motes D (2017) Association mapping of aphid resistance in USDA cowpea (Vigna unguiculata L. Walp.) core collection using SNPs. Euphytica. https://doi.org/10.1007/s10681-016-1830-z
Rostoks N, Mudie S, Cardle L, Russell J, Ramsay L, Booth A, Svensson JT, Wanamaker SI, Walia H, Rodriguez EM, Hedley PE, Liu H, Morris J, Close TJ, Marshall DF, Waugh R (2005) Genome-wide SNP discovery and linkage analysis in barley based on genes responsive to abiotic stress. Mol Genet Genom 274(5):515–527. https://doi.org/10.1007/s00438-005-0046-z
Sall J, Lehman A, Stephens ML, Creighton L (2012) JMP start statistics: a guide to statistics and data analysis using JMP. SAS Institute, Cary
Santos CA, Boiteux LS (2013) Breeding biofortified cowpea lines for semi-arid tropical areas by combining higher seed protein and mineral levels. Genet Mol Res 12(4):6782–6789. https://doi.org/10.4238/2013.December.16.4
Schroder S, Mamidi S, Lee R, McKain MR, McClean PE, Osorno JM (2016) Optimization of genotyping by sequencing (GBS) data in common bean (Phaseolus vulgaris L.). Mol Breed. https://doi.org/10.1007/S11032-015-0431-1
Scott SJ (1977) Nitrogen-fixation in cowpea (Vigna-Unguiculata-(L) Walp) as influenced by available nitrate levels. HortScience 12(4):388–389
Scowcroft WR, Gibson AH (1975) Nitrogen-fixation by rhizobium associated with tobacco and cowpea cell-cultures. Nature 253(5490):351–352
Shi AN, Buckley B, Mou BQ, Motes D, Morris JB, Ma JB, Xiong HZ, Qin J, Yang W, Chitwood J, Weng YJ, Lu WG (2016) Association analysis of cowpea bacterial blight resistance in USDA cowpea germplasm. Euphytica 208(1):143–155. https://doi.org/10.1007/s10681-015-1610-1
Shirasawa K, Maeda H, Monna L, Kishitani S, Nishio T (2007) The number of genes having different alleles between rice cultivars estimated by SNP analysis. Theor Appl Genet 115(8):1067–1074. https://doi.org/10.1007/s00122-007-0632-z
Simmonds NW (1976) Evolution of crop plants. Longman Group Ltd., Harlow
Singh SR, Rachie KO (1985) Cowpea research, production and utilization. Wiley, New York
Singh B, Chambliss O, Sharma B (1997) Recent advances in cowpea breeding. Adv Cowpea Res 3049
Singh P, Prasuhn D, Yeh RM, Destito G, Rae CS, Osborn K, Finn MG, Manchester M (2007) Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J Control Release 120(1–2):41–50. https://doi.org/10.1016/j.jconrel.2007.04.003
Singh SK, Kakani VG, Surabhi GK, Reddy KR (2010) Cowpea (Vigna unguiculata [L.] Walp.) genotypes response to multiple abiotic stresses. J Photochem Photobiol B 100(3):135–146. https://doi.org/10.1016/j.jphotobiol.2010.05.013
Smartt J (1986) Cowpea research, production and utilization. Edited by S. R. Singh and K. O. Rachie. Chichester, New York, etc.: John Wiley and Sons (1985), pp. 460, £22.50. Exp Agric 22(04):431. https://doi.org/10.1017/s001447970001468x
Smith JM (1989) Evolutionary genetics. Oxford University Press, Oxford
Su W, Wang L, Lei J, Chai S, Liu Y, Yang Y, Yang X, Jiao C (2017) Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing. PLoS ONE 12(2):e0172066. https://doi.org/10.1371/journal.pone.0172066
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24(8):1596–1599. https://doi.org/10.1093/molbev/msm092
Timko MP, Ehlers JD, Roberts PA (2007) Cowpea. In: Pulses, sugar and tuber crops. Springer, New York, pp 49–67
Uncu AO, Frary A, Karlovsky P, Doganlar S (2016) High-throughput single nucleotide polymorphism (SNP) identification and mapping in the sesame (Sesamum indicum L.) genome with genotyping by sequencing (GBS) analysis. Mol Breed. https://doi.org/10.1007/S11032-016-0604-6
Wang QB, Zhang L, Zheng PJ (2015) Genetic diversity and evolutionary relationship analyses within and among Raphanus species using EST-SSR markers. Mol Breed. https://doi.org/10.1007/s11032-015-0261-1
Xiong HZ, Shi AN, Mou BQ, Qin J, Motes D, Lu WG, Ma JB, Weng YJ, Yang W, Wu DX (2016) Genetic diversity and population structure of Cowpea (Vigna unguiculata L. Walp). PLoS ONE. https://doi.org/10.1371/journal.pone.0160941
Xu P, Wu X, Wang B, Luo J, Liu Y, Ehlers J, Close T, Roberts P, Lu Z, Wang S (2012) Genome wide linkage disequilibrium in Chinese asparagus bean (Vigna unguiculata ssp. sesquipedialis) germplasm: implications for domestication history and genome wide association studies. Heredity 109(1):34–40
Yanagisawa T, Kiribuchi-Otobe C, Hirano H, Suzuki Y, Fujita M (2003) Detection of single nucleotide polymorphism (SNP) controlling the waxy character in wheat by using a derived cleaved amplified polymorphic sequence (dCAPS) marker. Theor Appl Genet 107(1):84–88. https://doi.org/10.1007/s00122-003-1235-y
Zeka D, Sedlak P, Sedlakova V, Sharma K, Vejl P (2015) Phenotype and molecular diversity evaluation of some wild 2n Solanum species (super series Rotata). Chil J Agr Res 75(2):147–151. https://doi.org/10.4067/S0718-58392015000200002
Zhou H, Muehlbauer G, Steffenson B (2012) Population structure and linkage disequilibrium in elite barley breeding germplasm from the United States. J Zhejiang Univ Sci B 13(6):438–451. https://doi.org/10.1631/jzus.B1200003
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiong, H., Qin, J., Shi, A. et al. Genetic differentiation and diversity upon genotype and phenotype in cowpea (Vigna unguiculata L. Walp.). Euphytica 214, 4 (2018). https://doi.org/10.1007/s10681-017-2088-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-2088-9