
Abstract
Quantitative trait locus (QTL) mapping lays the foundation for marker-assisted selection (MAS) for lint yield and fiber quality in cotton (Gossypium hirsutum). Boll-related traits affect yield and fiber quality, but few studies have focused on boll-related traits. Here, we detected QTLs related to cotton boll, yield and fiber quality traits using intraspecific F2 and F2:3 populations from the cross AQ × 08-10604. A total of 91 QTLs for 17 traits related to boll, yield and fiber quality in the F2 and F2:3 populations were mapped, including 37 significant QTLs. Six pairs of common QTLs were detected, including two pairs for boll coat weight (BCW) in the same or similar positions on Chr. A10 and D1, two pairs for boll length (BL) in similar positions on Chr. A10 and A13, with a higher percentage of phenotype variation and two pairs for boll diameter (BD) in similar positions on Chr. A10 and D1. These results suggest that the traits BCW, BL and BD have high levels of stability. Five QTL clusters for the same or different traits were also identified on Chr. A10 (2), A13, D1 and D5. We also detected 64 epistatic QTLs for boll- and yield-related traits that play important roles in genetic variation. Correlation analysis revealed significant positive correlations between seed yield and boll number per plant and between boll weight and BCW, BL and BD. The results of this study enhance our understanding of the genetic basis of boll-related traits and will enable further MAS of upland cotton.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cotton is one of the most important economic crops worldwide. Gossypium hirsutum is a tetraploid species, accounting for 95 % of the world cotton yield. The challenge facing cotton breeders is how to meet the increasing demands of the textile industry. Due to the negative genetic correlation between fiber quality and lint yield, performing conventional breeding procedures to further improve fiber quality while simultaneously emphasizing yield has become increasingly difficult. The development of molecular markers has made it possible for crop breeders to employ rapid, precise alternative approaches to conventional selection schemes for improving both economic and agronomic traits of crops (Tanksley and Hewitt 1988).
Genetic linkage maps lay the foundation for exploiting quantitative trait loci (QTLs) conferring yield-, fiber quality- and boll-related traits in cotton. Since the first genetic map was constructed by Reinisch et al. (1994) using an interspecific cross of G. hirsutum × G. barbadense, an increasing number of interspecific genetic maps have been constructed throughout the world (Guo et al. 2007; Lacape et al. 2003; Rong et al. 2004; Yu et al. 2011). In addition, many QTLs for lint yield and fiber quality traits have been mapped (Chee et al. 2005; He et al. 2005, 2007; Jiang et al. 1998; Lacape et al. 2010; Mei et al. 2004; Ren et al. 2002; Rong et al. 2007). Numerous genetic studies have shown that both cotton yield traits and fiber quality traits are quantitative traits that are controlled by multiple minor genes, and their phenotypes are affected by hereditary and environmental factors. However, the current interspecific maps and mapped QTLs have very limited use in conventional cotton breeding.
Shappley et al. (1996) first successfully constructed an intraspecific genetic linkage map with molecular markers in upland cotton. Subsequently, several intraspecific genetic linkage maps were constructed for G. hirsutum (Lin et al. 2009; Ulloa and Meredith 2000; Ulloa et al. 2002, 2005; Zhang et al. 2005, 2009), which contain 73–604 loci. Using these maps, the precise locations of many QTLs have been determined (Chen et al. 2009; Guo et al. 2006; Li et al. 2014; Liu et al. 2012; Ning et al. 2014; Paterson et al. 2003; Qin et al. 2008, 2009; Shao et al. 2014; Shen et al. 2005, 2007; Sun et al. 2012; Ulloa and Meredith 2000; Ulloa et al. 2005; Wang et al. 2006, 2007; Wu et al. 2009; Zhang et al. 2005, 2009, 2010, 2012a, b). For fiber quality, for example, Qin et al. (2009) discovered eight QTLs for fiber strength (FS), micronaire (MIC) and fiber elongation (FE) simultaneously in two populations. Shen et al. (2005) identified 39 QTLs for fiber quality, including 11 QTLs for FL, 10 for FS, nine for MIC and nine for FE. Zhang et al. (2009) detected 13 QTLs for fiber quality and mapped the QTLs on their corresponding chromosomes, including four for fiber length (FL), two for FS, two for MIC, three for fiber length uniformity (FU) and two for FE, respectively. Zhang et al. (2012a) identified 63 QTLs affecting fiber quality, including 11 for FE, 16 for FL, nine for MIC, 10 for FS and 17 for FU, explaining 8.1–55.8 % of the total phenotypic variance. Using three populations, Shao et al. (2014) detected 77 QTLs, including 19 for FL, 14 for FU, 17 for MIC, 10 for FE and 17 for FS. Sun et al. (2012) identified 50 QTLs for fiber quality, including 10 for FS, 10 for FL, 10 for MIC, eight for FU and 12 for FE. For cotton yield traits, Guo et al. (2006) mapped three QTLs for lint percentage (LP) on the A03 linkage group and chromosome 6. Shen et al. (2007) identified five QTLs for boll weight (BW), six for LP, five for seed index (SI), five for boll number per plant (BN), one for seed cotton yield and four for lint yield (LY), respectively. Zhang et al. (2010) detected seven QTLs for five yield traits, including one for BN, two for BW, one for LP, one for SY and two for LY. Ning et al. (2014) discovered 13 QTLs for SY, nine for LP, 12 for BW and five for SI. These studies have demonstrated that many cotton yield traits are closely related to each other and the corresponding genes may be linked to produce multiple effects on final traits (Mauricio 2001). Since cotton fiber quality is controlled by multiple genes, which are vulnerable to environmental effects, progress to simultaneously improve cotton yield and fiber quality traits using conventional breeding methods has been slow. To date, several hundred QTLs related to fiber quality traits have been mapped, and more new QTLs are still being identified. However, the lower variation present in intraspecific populations versus interspecific populations has limited the development of genetic linkage maps, and the resulting lower-density genetic linkage maps still fail to meet the needs of QTL-assisted crop breeding.
In addition, expression analysis of genes related to cotton fiber development at different stages and in different tissues has shown that this process is regulated in a highly complicated manner involving numerous genes (Taliercio and Boykin 2007; Xu et al. 2008; Al-Ghazi et al. 2009; Paterson et al. 2012), which also suggests that more QTLs should be identified in populations derived from crosses between cotton cultivars with wide genetic backgrounds (Shao et al. 2014). Meanwhile, epistasis, the interaction between alleles from two to more loci, may play an important role in evolutionary and quantitative variation in crops (Lou et al. 2009; Malmberg et al. 2005; Wang et al. 2010; Xing et al. 2002; Xu and Jia 2007; Xu et al. 2009; Yu et al. 1997; Zhang et al. 2001; Zhang et al. 2012a). QTL mapping is increasingly used to explore the role of epistasis in the genetic basis of complex quantitative traits (Li et al. 2009; Mohan et al. 2009). In cotton, QTL mapping analysis of epistatic effects has only been performed to examine a few plant architectural traits in intraspecific populations (Wang et al. 2006; Li et al. 2014). Wang et al. (2006) identified three epistatic QTLs for plant height (PH), three for fruit branch length (FBL) and three for fruit branch number (FBN). Li et al. (2014) detected a total of 54 pairs of epistatic QTLs (E-QTLs) for ten plant architecture traits, which exhibit additive-by-additive (AA), additive-by-dominant (AD), dominant-by-additive (DA) and dominant-by-dominant (DD) interactions, including five for PH, seven for FBL, eight for FBN and so on.
It is well known that the boll is one of the most important productive organs of cotton, and boll size and boll number are two important yield components; boll shape also affects fiber quality. For instance, Tang and Xiao (2014) recently showed that boll length makes the largest contribution to the largest proportion of phenotypic, additive and dominance variances for fiber length, while boll width makes the largest contribution to phenotypic and additive variances and the second largest contribution to dominance-by-environment interaction variance for micronaire. Ashraf and Ahmad (2000) suggested that boll length plays an important role in cotton breeding. However, few such studies have focused on boll-related traits due to a lack of proper QTL mapping varieties.
In the current study, two extremely distinctive upland cotton lines were crossed to construct F2 and F2:3 populations for QTL mapping. The paternal parent, 08-10604, a highly inbred line from a cross between G. hirsutum race yucatanense (directly introduced from Mexico) and G. hirsutum cv Sumian 8, was developed by our institute and it possesses extremely low boll weight (<2 g), many bolls per plant and low fiber quality (short fiber length, low fiber strength and coarse fibers), while the maternal parent, AQ, exhibits high boll weight (>6 g), fewer bolls per plant and superior fiber quality (high fiber strength and fine fibers). Here, we investigated 17 traits in cotton. Seven boll- and five yield-related traits of the two parents, F1 population, F2 individuals and F2:3 families were investigated at maturity, except for the first and last plant of each row. Five fiber quality traits were also investigated in the F2 and F2:3 populations. The boll-related traits included boll length (BL), boll diameter (BD), boll coat weight (BCW), locule number per boll (BLN), BW, seed number per boll (BSN), lint weight per boll (BLW). The yield-related traits included SI, LP, SY, LY and BN. The fiber quality traits included FL, FS, MIC, FE and FU. Boll length (BL) represents the longest part of a cotton boll from top to bottom, while BD is measured at the widest part of a cotton boll. The objectives of the present study were to (1) identify new QTLs with major effects on the 17 traits and QTLs with epistatic effects on the 12 boll- and yield-related traits, (2) analyze the common QTLs for boll-related traits and (3) summarize the QTL clusters for boll-related traits. This study provides a theoretical basis for genetic structure analysis and marker-assisted selection for high yield in cotton.
Materials and methods
Materials
Two upland cottons with significant differences in boll-related traits were used to develop F2 and F2:3 populations. The female parent was AQ (G. hirsutum), which has superior characters such as tall plants and large (but few) bolls. The male parent was 08-10604, a highly inbred line from a cross between G. hirsutum race yucatanense (directly introduced from Mexico) and G. hirsutum cv Sumian 8, which was then backcrossed with G. hirsutum acc. Tai8033. In 2011, 330 F2 individuals were planted and self-pollinated. The resulting F2:3 families were planted in 2012.
Methods
All materials were randomly planted in Jiangpu Breeding Station of Nanjing Agricultural University (Nanjing, China). The plots were 5 m long and 0.8 m apart, with a plant spacing of 40 cm. A total of 285 F2:3 family lines, along with P1, P2 and F1, were randomly planted, with two replicates in a single-row plot. The field management measures essentially followed normal agricultural practices.
Boll traits including BL and BD were measured at maturity using a digital caliper (Lugong, Shanghai Jiuliang Hardware Company, Shanghai, China). Fiber samples were collected from bolls at the internal middle parts of plants. F2 individuals and F2:3 family lines in the middle of each row were tagged for scoring, harvested and sent to the Cotton Quality Supervision, Inspection and Testing Center of the Ministry of Agriculture, China for tests of FL (cm), FS (cN/tex), MIC, FU and FE using an Uster HVI 900. The BCW and BW were measured and the BLN was counted in bolls from the internal, middle parts of the plants for 330 F2 individuals, and a total of 285 F2:3 family lines, along with P1, P2 and F1 in the middle of each row, were tagged for scoring and harvesting.
Genomic DNA was extracted from young leaves of the 330 F2 individuals, F1 and two parents using the improved CTAB method (Paterson et al. 1993). A total of 8,200 simple sequence repeat (SSR) primer pairs were chosen according to several cotton genetic maps (Lacape et al. 2003; Rong et al. 2004; Guo et al. 2007; Yu et al. 2007) and used to screen the parents for polymorphisms. These SSR primer sequences are available at http://www.cottonmarker.org. SSR-PCR amplifications were performed using a Programmable Thermal Controller (MJ Research), and PCR product electrophoresis and silver staining were conducted as described by Zhang et al. (2000, 2002).
JoinMap 3.0 (Van Ooijen 2001) was used to construct a complete linkage map. A logarithm of odds (LOD) threshold of 4.0 and a 50 cM maximal distance were used to determine all linkage groups. The major QTLs and their effects were determined with WinQTLCart2.5 software (Wang et al. 2005) using the composite interval mapping (CIM) method (Zeng 1994). QTLs with LOD values between 2.0 and 3.0 were defined as suggestive QTLs (Lander and Kruglyak 1995), and QTLs with LOD values no less than the threshold value (calculated by a permutation test with 1,000 repeats) were defined as significant QTLs (Churchill and Doerge 1994). QTLs for the same trait across different generations were defined as “common” QTLs when their confidence intervals overlapped. Epistatic QTLs (E-QTLs) were also detected using the mapping of additive, dominance and digenic epistasis genes in biparental populations (BIP) functionality of the inclusive composite interval mapping (ICIM) software IciMapping ver. 3.2 (Wang et al. 2012), The ICIM-EPI mapping method in BIP functionality has high detection efficiency and is used specifically for estimating digenic epistasis genes in biparental populations, even if the effect of a single QTL is minor. The probability in stepwise regression was set at 0.0001 and the scanning step was 5 cM. A LOD threshold score of 5.0 was used to declare significant E-QTLs.
The name of each QTL includes a “q” followed by an abbreviation of the trait name, the population type, the chromosome or linkage group and a serial number to distinguish different QTLs of the same trait on the same chromosome. Linkage groups were assigned to chromosomes based on anchored markers in the dense linkage map (Han et al. 2004; Guo et al. 2007). Linkage groups that could not be assigned to any chromosome were designated as “LGXX” (where LG indicates linkage group and XX is the serial number).
Results
Marker analysis and map construction
We chose 8,200 SSR markers to screen for polymorphisms between the two parents, AQ and 08-10604. Approximately 3.22 % (264/8,200) of the SSR primers showed polymorphisms. These polymorphic primers were then used to screen F2 individuals, and 284 loci were obtained, including 155 codominant loci (1:2:1) and 129 dominant loci (1:3 or 3:1). There were 45 loci (17.31 %) that showed segregation distortion, and 37 loci were mapped to chromosomes or linkage groups.
A total of 230 loci were successfully assigned to 28 linkage groups on the 16 chromosomes of the cotton genome using JoinMap3.0 software at a LOD ≥4. The total length of the map was 1,531.94 cM, with an average inter-marker distance of 6.66 cM. The average distance of adjacent markers was 6.24 cM in the A-subgenome, covering 692.48 cM, and 6.78 cM in the D-subgenome, covering 718.76 cM (Table 1).
Trait performance
A T test showed that the boll-, yield- and fiber quality-related traits were significantly different between the two parents, except for the lint percentage trait (Table 2). Among the 17 traits examined, the values of 14 traits were higher in AQ than in 08-10604, except for BN, FE and MIC. There were significant or highly significant differences in traits between AQ and 08-10604, except for LY, suggesting that the parents used in this study were appropriate for searching for genes responsible for boll-related and fiber traits. The variation coefficients of traits such as BLN, BL and BD were lower, indicating that these traits were relatively stable.
Correlations (Tables 3, 4) between boll-related traits and the other traits in the F2 and F2:3 populations were analyzed using SPSS20.0 software. In the F2:3 population (Table 4), significant positive correlations were observed between BD, BL, BCW, BLN and BW (0.613**, 0.447**, 0.725**, 0.431**), while significant negative correlations existed between BD, BL, BCW, BLN and BN (−0.232**, −0.202**, −0.230**, −0.203**) and between BW and BN (−0.261**). Furthermore, there were significant positive correlations between FL, FS and BL (0.162**, 0.241**), while significant negative correlations were observed between BD, BL, BCW and MIC (−0.220**, −0.208**, −0.224**). Boll-related traits not only contribute significantly to cotton yield, but they also contribute to fiber quality traits. In the F2 population, BLN had the highest contribution (12.63 %), followed by BL, BW and BCW. However, in F2:3, BL had the highest contribution (15.93 %), followed by LP, BSN and BCW (Table 5). In the F2 and F2:3 populations, BW had notable positive correlations with the traits BCW, BL, SI, BLN and BD, while BW and BLW also had considerable positive correlations. Since it is difficult to accurately deduce the relationships between characters through correlation analysis, BW-related traits should be further analyzed using regression equations (Table 6). According to the analysis of regression equations and path coefficients, BCW had the highest positive contribution to BW. In the F2 population, BW increased by 0.426 g, which was accompanied by a 1 g increase in BCW. BW increased by 0.230 g as a result of a 1 mm increase in BD. BW increased by 0.136 g, while the BLN increased by 1. However, BW decreased by 0.061 g while SI increased by 1 g. In the F2:3 population, BW increased by 0.369 g, which was accompanied by an increase in BCW of 1 g. BW increased by 0.208 g as a result of a 1 mm increase in BD. BW increased by 0.180 g while the BLN increased by 1. Unlike the F2 population, in the F2:3 population, BW increased by 0.197 g while SI increased by 1 g.
QTLs for boll-, fiber quality- and yield-related traits
We performed QTL analysis using CIM via WinQTLCart2.5. Many QTLs related to almost all traits were detected in the F2 and F2:3 populations. A total of 91 M-QTLs were detected in the F2 and F2:3 populations for 17 traits using CIM, including 37 significant QTLs with LOD values greater than or equal to the threshold value calculated by a permutation test with 1,000 repeats. We detected 18 and 19 significant QTLs in the F2 and F2:3, respectively (Fig. 1; Table 5). Moreover, six pairs of common QTLs and five QTL clusters affecting boll-related traits were found in both populations (Table 7). A total of 64 pairs of E-QTLs exhibiting additive-by-additive (AA), additive-by-dominant (AD), dominant-by-additive (DA) and dominant-by-dominant (DD) interactions were detected for all measured traits in both populations (Table 8), namely, 39 for boll- and 25 for yield-related traits, but no interaction for fiber quality traits was detected. The phenotype variation (PV) explained by all E-QTLs ranged from 7.852 to 34.251 %.

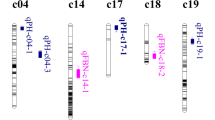
The locations of 91 M-QTLs on the linkage map
Boll-related traits
A total of 58 M-QTLs were detected in the F2 and F2:3 populations for seven boll-related traits using CIM, including 30 significant QTLs with LOD values greater than or equal to the threshold value calculated by a permutation test with 1,000 repeats. We detected 17 and 13 significant QTLs in the F2 and F2:3, respectively (Fig. 1; Table 5). Only common QTLs and QTL clusters for boll-related traits were found in both populations (Table 7).
Boll coat weight
Thirteen M-QTLs (nine significant QTLs) were detected in the F2 and F2:3 populations. In the F2 population, among seven M-QTLs affecting BCW, five significant QTLs were detected, including qBCW-F 2 -A10-1, qBCW-F 2 -A13-1, qBCW-F 2 -A13-2, qBCW-F 2 -D1-1 and qBCW-F 2 -D1-2, which explained 6.40–9.51 % of PV, with LOD scores of 4.46–7.34. In the F2:3 population, among six M-QTLs, four significant QTLs were detected, namely, qBCW-F 2:3 -A10-1, qBCW-F 2:3 -A10-2, qBCW-F 2:3 -D1-1 and qBCW-F 2:3 -D1-2, which explained 7.07–13.72 % of PV, with LOD scores of 5.05–6.27. The favorable alleles of all 13 M-QTLs originated from AQ. Among the 13 QTLs, two pairs of QTLs for BCW, qBCW-F 2:3 -A10-2 and qBCW-F 2 -A10-1 and qBCW-F 2:3 -D1-1 and qBCW-F 2 -D1-1, were found on Chr. A10 and Chr. D1, respectively, in both populations at the same (50.01 cM) or similar positions (52.61, 53.61 cM) in the same marker interval. These two pairs of common QTLs contributed positive additive effects from AQ. In addition, seven pairs of E-QTLs for BCW were also detected, which are distributed on nine chromosomes or linkage groups and displayed −0.082 to 0.090 AA effects, −0.224 to 0.428 AD effects, −0.218 to 0.210 DA effects and −0.767 to 0.210 DD effects, explaining 14.745–33.718 % of PV.
Boll length
Twelve M-QTLs (nine significant QTLs) were detected in the F2 and F2:3 populations. In the F2 population, among seven M-QTLs affecting boll length, six significant QTLs were detected, namely, qBL-F 2 -A10-1, qBL-F 2 -A10-2, qBL-F 2 -A6-1, qBL-F 2 -D5-1, qBL-F 2 -D5-2 and qBL-F 2 -D5-3, which explained 3.68–9.91 % of PV, with LOD scores of 3.89–11.74. In the F2:3 population, among five M-QTLs, three significant QTLs were detected, namely, qBL-F 2:3 -A10-1, qBL-F 2:3 -A10-2 and qBL-F 2:3 -D1-1, which explained 3.12–15.93 % of PV, with LOD scores of 4.41–8.44. Among of 12 M-QTLs from AQ, two common pairs of QTLs, including qBL-F 2:3 -A10-1 and qBL-F 2 -A10-2 (in the same marker interval, NAU1297-290-NAU1297-400) and qBL-F 2:3 -A13-1 and qBL-F 2 -A13-1 (in the same marker interval, NAU6699-400-NAU5110-490), were detected in both populations. Meanwhile, six pairs of E-QTLs were detected. These E-QTLs, which are distributed on nine chromosomes or linkage groups, displayed −0.670 to 1.556 AA effects, −0.874 to 3.104 AD effects, −2.230 to 2.256 DA effects and −1.608 to 4.876 DD effects, accounting for 7.852–27.413 % of PV.
Boll diameter
Ten M-QTLs (six significant QTLs) were detected in the F2 and F2:3 populations. In the F2 population, among five M-QTLs affecting BD, three significant QTLs were detected, namely, qBD-F 2 -A3-2, qBD-F 2 -D1-1 and qBD-F 2 -D1-2, which explained 4.29–6.94 % of PV, with LOD scores of 4.88–8.43. In the F2:3 population, among five M-QTLs, three significant QTLs were detected, namely, qBD-F 2:3 -A5-1, qBD-F 2:3 -D1-1 and qBD-F 2:3 -D1-2, which explained 4.33–7.15 % of PV, with LOD scores of 3.76–5.06. Among the 10 M-QTLs from AQ, two common pairs of QTLs, qBD-F 2:3 -A10-2 and qBD-F 2 -A10-1 (in very close positions, 105.51 and 101.51 cM, respectively) and qBD-F 2:3 -D1-1 and qBD-F 2 -D1-1 (in the same marker interval, NAU6539-400-MNL2921-180), were detected in both populations. In addition, five pairs of E-QTLs, which are distributed on eight chromosomes or linkage groups, displayed −0.630 to 0.547 AA effects, −0.257 to 2.343 AD effects, −1.400 to 1.284 DA effects and −4.165 to 3.649 DD effects, accounting for 9.244–28.680 % of PV (Table 8).
Locule number per boll
Six M-QTLs (two significant QTLs) were detected in the F2 and F2:3 populations. Among three M-QTLs affecting BLN, two significant QTLs were detected in the F2:3 population, namely, qBLN-F 2:3 -D5-1 and qBLN-F 2:3 -D5-2, which explained 5.08 and 6.48 % of PV, respectively, with LOD scores of 4.26 and 4.47, originating from AQ. In the F2 population, no significant QTL for BLN was detected, although three M-QTLs, qBLN-F 2 -D10-1, qBLN-F 2 -D1-1 and qBLN-F 2 -D1-2, were detected, which explained 3.81–12.63 % of PV, with LOD scores of 2.58–3.26. Four M-QTLs contributed positive effects from AQ while the other two M-QTLs (qBLN-F 2 -D10-1 and qBLN-F 2 -D10-2) contributed positive additive effects from 08-10604. Moreover, five pairs of E-QTLs for BLN were detected. These E-QTLs, which are distributed on six chromosomes or linkage groups, displayed −0.104 to 0.067 AA effects, −0.108 to 0.685 AD effects, −0.108 to 0.685 DA effects and −1.629 to 1.246 DD effects, explaining 15.359–31.953 % of PV.
Lint weight per boll
Five M-QTLs (two significant QTLs) were detected in the F2 and F2:3 populations. In the F2 population, of the two M-QTLs, one significant QTL was detected, qBLW-F 2 -A3-1, which explained 4.13 % of PV, with a LOD score of 3.38. In the F2:3 population, among the three M-QTLs, one significant QTL was detected, qBLW-F 2:3 -D5-1, which explained 11.65 % of PV, with a LOD score of 3.75. Five M-QTLs contributed positive effects from AQ while the other M-QTL (qBLW-F 2 -A3-1) contributed positive additive effects from 08-10604. Two pairs of E-QTLs for BLW, including interactions between two loci on Chr. A10 and Chr. A9 and between two loci on Chr. A10 and Chr. D1, were also detected in the F2 population. These E-QTL pairs demonstrated 0.133 and −0.076 AA effects, −0.197 and 0.390 AD effects, 0.121 and −0.403 DA effects and −0.268 and 0.603 DD effects, explaining 26.070 and 29.861 % of PV, respectively.
Boll weight
Nine M-QTLs (three significant QTLs) were detected in the F2 and F2:3 populations. Among seven M-QTLs, three significant QTLs were detected in the F2 population, qBW-F 2 -A10-1, qBW-F 2 -D5-1 and qBW-F 2 -D5-2, which explained 3.37–9.86 % of PV, with LOD scores of 4.09–5.44. In the F2:3 population, no significant QTL was detected for this trait, although two M-QTLs, qBW-F 2:3 -A10-1 and qBW-F 2:3 -D13-1, were detected, which explained 7.19 and 7.17 % of PV, with LOD scores of 2.70 and 3.05, respectively. Eight M-QTLs had alleles with positive effects from AQ while the other M-QTL (qBW-F 2 -A9-1) contributed positive additive effect from 08-10604. Six pairs of E-QTLs for BW were also detected. These E-QTLs, which are distributed on six chromosomes or linkage groups, displayed −0.249 to 0.288 AA effects, −0.886 to 0.321 AD effects, −0.571 to 0.877 DA effects and −0.814 to 0.694 DD effects, explaining 13.849–33.539 % of PV.
Seed number per boll
Three M-QTLs were detected in the F2 and F2:3 populations. There were no significant QTLs for this trait. One M-QTL, qBSN-F 2 -A3-1, was detected in the F2 population, which explained 6.50 % of PV, with a LOD score of 3.19, originating from 08-10604. In the F2:3 population, two M-QTLs were detected, including qBSN-F 2:3 -A10-1, with the favorable alleles originating from AQ, and qBSN-F 2:3 -A9-1, with the favorable alleles originating from 08-10604, which explained 7.89 and 14.30 % of PV, with LOD scores of 2.56 and 3.64, respectively. Eight pairs of E-QTLs for BSN were also found. These E-QTLs, which are distributed on ten chromosomes or linkage groups, displayed −2.321 to 2.074 AA effects, −1.989 to 6.707 AD effects, −1.296 to 4.359 DA effects and −10.173 to 3.793 DD effects, explaining 11.101–34.351 % of PV.
Meanwhile, QTL clusters of boll-related traits were also observed in this study. A total of five QTL clusters of boll-related traits were found on Chr. A10, A13, D1 and D5 (Table 7). For example, Chr. A10 contains two QTL clusters, i.e., A10-cluster-1 at 35.81–77.71 cM and A10-cluster-1 at 86.51–105.51 cM, carrying 12 and four QTLs, respectively. Chr. A13 contains one QTL cluster carrying four QTLs, i.e., A13-cluster at 48.51–58.21 cM. Chromosome D1 contains one QTL cluster carrying 11 QTLs, i.e., D1-cluster at 51.61–97.01 cM. Finally, Chr. D5 contains one QTL cluster carrying ten QTLs, namely, D5-cluster at 96.91–150.01 cM. These results suggest that genes controlling boll-related traits may be linked or may exhibit pleiotropy.
Fiber quality-related traits
CIM revealed a total of 22 M-QTLs in the F2 and F2:3 populations for five traits, including four significant QTLs with LOD values greater than or equal to the threshold value calculated by a permutation test with 1,000 repeats; these QTLs were only detected in the F2:3 (Fig. 1; Table 5).
Fiber elongation
Five M-QTLs for FE were detected in the F2 and F2:3 populations. There were no significant QTLs for this trait. Three M-QTLs were detected in the F2 population, including qFE-F 2 -A10-1, qFE-F 2 -A12-1 and qFE-F 2 -D5-1, which explained 3.08–5.27 % of PV, with LOD scores of 2.55–3.37; the positive additive effects originated from AQ, except for qFE-F 2 -D5-1. In the F2:3 population, two M-QTLs were detected, qFE-F 2:3 -A13-1 and qFE-F 2:3 -LG02-1, which explained 4.90 and 8.49 % of PV, respectively, with LOD scores of 2.81–3.48; the favorable alleles of two M-QTLs originated from 08-10604.
Fiber length
Four M-QTLs (one significant QTL) were detected in the F2 and F2:3 populations. Among three M-QTLs, one significant QTL, qFL-F 2:3 -A6-1, was detected in the F2:3 population, which explained 10.89 % of PV, with a LOD score of 5.26. In the F2 population, one M-QTL, qFL-F 2 -A13-1, was detected, which explained 3.96 % of PV, with a LOD score of 2.86. The favorable alleles of three M-QTLs originated from 08-10604, except for qFL-F 2:3 -LG03-1, where it originated from AQ.
Fiber strength
Five M-QTLs (one significant QTL) were detected in the F2 and F2:3 populations. Among three M-QTLs, one significant QTL, qFS-F 2:3 -D1-1, was detected in the F2:3population, which explained 3.78 % of PV, with a LOD score of 4.09. In the F2 population, two M-QTLs, qFS-F 2 -A12-1 and qFS-F 2 -D5-1, were detected, which explained 4.31–7.36 % of PV, with a LOD score of 2.58–4.96. Three M-QTLs had alleles with positive effects from AQ while the other two M-QTLs (qFS-F 2:3 -D1-2 and qFS-F 2 -A12-1) contributed positive additive effects from 08-10604.
Fiber uniformity
Three M-QTLs were detected in the F2 and F2:3 populations. There were no significant QTLs for this trait. One QTL, qFU-F 2 -A12-1, was detected in the F2 population, which explained 4.53 % of PV, with a LOD score of 2.62, originating from AQ. In the F2:3 population, two M-QTLs, qFU-F 2:3 -D1-1 and qFU-F 2:3 -D1-2, were detected, which explained 6.52 and 3.43 % of PV, with LOD scores of 2.76 and 2.94, respectively, originating from AQ.
Micronaire
Five M-QTLs (two significant QTLs) were detected in the F2 and F2:3 populations. Among four M-QTLs, two significant QTLs were detected in the F2:3 population, including qMIC-F 2:3 -A9-1, which originated from 08-10604, and qMIC-F 2:3 -D1-1, which originated from AQ, which explained 7.14 % and 11.3 % of PV, respectively, with LOD scores of 3.59–5.22. In the F2 population, one M-QTL, qMIC-F 2 -D7-1, was detected, which explained 9.85 % of PV, with a LOD score of 4.05, originating from AQ.
Yield-related traits
Eleven QTLs related to three yield-related traits were detected in the F2 and F2:3 populations, except for BN and SY, using CIM, including three significant QTLs with LOD values greater than or equal to the threshold value calculated by a permutation test with 1,000 repeats, which were detected only in the F2:3 (Fig. 1; Table 5). However, 25 pairs of E-QTLs exhibiting AA, AD, DA and DD interactions were detected for yield-related traits in both populations (Table 8).
Lint percentage
Six M-QTLs (two significant QTLs) were detected in the F2:3 population. Two significant QTLs, qLP-F 2:3 -A6-1 and qLP-F 2:3 -D5-2, were detected, which explained 6.02 and 15.19 % of PV, with LOD scores of 4.91 and 5.98, respectively. All QTLs from AQ were responsible for the increase in LP. One pair of E-QTLs was detected. These E-QTLs are distributed on two chromosomes or linkage groups and displayed −0.030 AA effects, −0.043 AD effects, 0.014 DA effects and 0.020 DD effects, accounting for 20.773 % of PV.
Seed cotton yield
Two pairs of E-QTLs, including interactions between two loci on Chr. A3 and Chr. D8 and between two loci on Chr. D1 and Chr. D8, were detected in the F2:3. These E-QTLs demonstrated 11.502 and −12.416 AA effects, 2.727 and −19.917 AD effects, 3.003 and 14.343 DA effects and 10.370 and 8.181 DD effects, explaining 13.692 and 21.831 % of PV, respectively.
Lint yield
One M-QTL, qLY-F 2:3 -D10-1, was detected in the F2:3 population, which explained 6.72 % of PV, with a LOD score of 2.60. The favorable alleles originated from 08-10604. In addition, six pairs of E-QTLs were found, which are distributed on ten chromosomes or linkage groups and displayed −6.949 to 2.180 AA effects, −12.855 to 5.798 AD effects, −11.122 to 9.879 DA effects and −16.669 to 14.745 DD effects, explaining 10.586–33.594 % of PV.
Seed index
Four M-QTLs (one significant QTL) were detected in the F2 and F2:3 populations. Among three M-QTLs, one significant QTL, qSI-F 2:3 -D1-1, was detected in the F2:3 population, which explained 3.79 % of PV, with a LOD score of 5.32; the favorable alleles of the three M-QTLs originated from AQ. In the F2 population, one M-QTL, qSI-F 2 -D9-1, was detected, which explained 3.41 % of PV, with a LOD score of 2.86, originating from 08-10604. Meanwhile, seven pairs of E-QTLs were detected, which are distributed on nine chromosomes or linkage groups and displayed −0.549 to 0.366 AA effects, −1.312 to 0.912 AD effects, −0.626 to 0.912 DA effects and −2.661 to 1.573 DD effects, explaining 13.962–28.186 % of PV.
Boll number per plant
Nine pairs of E-QTLs were detected, which are distributed on 10 chromosomes or linkage groups and displayed −1.159 to 2.819 AA effects, −16.753 to 16.104 AD effects, −11.910 to 3.923 DA effects and −18.791 to 14.007 DD effects, accounting for 12.511–33.610 % of PV.
Epistasis-QTLs for interactions between boll and yield-related traits
In addition, we further observed two groups of interacting marker intervals, each of which simultaneously controlled two traits. The interacting marker intervals NAU2300-600-HAU1997-1000 on Chr. A13 and NAU5121-180-Gh354-150 on Chr. D5 influenced both SI and BD. The interacting marker intervals NAU4907-1000-HAU1785-400 on Chr. D5 and cgr5834-180-BNL786-130 on Chr. D1 influenced both BCW and BLN. Five groups of interacting marker intervals were detected, each of which had interactions on the same chromosome. Chr. A9 contains one pair of interacting marker intervals for BCW, BNL1672-100-NAU6130-200 and cgr6692-150-NAU490-500. Chr. A13 contains one pair of interacting marker intervals for BW, HAU0539-210-CIR221-150 and NAU6122-320-BNL1421-200. Chr. D1 contains one pair of interacting marker intervals for LY, MNL2921-180-Gh216-100 and TME03-200-JESPR-221-190. Chr. D5 contains two pairs of interacting marker intervals, including one for SI, Gh354-150-JESPR-204-220 and NAU779-600-TMC05-190 and one for BN, NAU4907-1000-HAU1785-400 and BNL3347-150-NAU5121-180. In addition, some marker intervals had interactions with other multiple marker intervals to control different traits. For example, the marker interval NAU5035-250-NAU1167-500 had interactions with multiple marker intervals including cgr6692-150-NAU490-500, BNL1667-150-HAU3297-300, cgr6680-200-NAU5439-230 and HAU2738-300- NAU3201-220 to control four traits, BCW, BLN, BD and SY, respectively.
Discussion
The F2 population has the most complete genetic composition of all populations and can provide the most abundant genetic information; theoretically, this population can be applied to QTL and genetic effect analysis. However, the F2 represents a temporarily separating population, and its trait performance cannot be repeated among generations, which greatly limits the application of the F2 population to QTL mapping. Although the genetic compositions of the F2 and F2:3 populations differ, a highly positive correlation exists between these populations (Xu and Zhu 1994). The use of the average value of each line in the F2:3 population to estimate the phenotypes of F2 individuals can reduce the environmental error and improve the accuracy of QTL location. However, this technique underestimates the dominance and over-dominance effects of QTLs. Considering the advantages of the F2 and F2:3 populations, it is essential to use both populations for QTL analysis. Hence, both F2 and F2:3 populations were employed for QTL mapping in this study, and common QTLs were simultaneously detected in the same or similar positions, suggesting that these QTLs can be used for further verification and analysis and thus, for marker-assisted breeding. However, many previous studies have focused on QTL mapping of cotton yield and fiber quality traits, but few studies have examined traits related to cotton bolls, especially for boll weight-related traits such as BD, BL, BLN and BCW (except for BW), despite the fact that these traits play important roles in cotton breeding (Ashraf and Ahmad 2000; Tang and Xiao 2014). In this study, we detect 58 M-QTLs in the F2 and F2:3 populations for boll-related traits, including 13 BCW, 12 BL, 10 BD, six BLN, five BLW, nine BW and three BSN QTLs. Moreover, we also found six pairs of common QTLs conferring boll-related traits (including two pairs each for BCW, BL and BD, respectively), suggesting that these common QTLs have high reliability and can be utilized for MAS to improve boll weight.
In addition, we detected several boll-related QTL-rich regions with QTLs conferring yield-related or fiber traits on a few chromosomes. For instance, a region on Chr. A10 contains 17 M-QTLs (eight significant QTLs) controlling BCW, BL, BLN, BLW, BW, BSN, BD and FE. A region on Chr. A13 contains 10 M-QTLs (two significant QTLs), including those controlling BCW, BL, BW, FE, FL and LP. A region on Chr. D1 contains 17 M-QTLs (12 significant QTLs) controlling BCW, BL, BLN, BD, FS, FU, MIC and SI. A region on Chr. D5 contains 16 M-QTLs (nine significant QTLs) controlling BCW, BL, BLN, BLW, BW, BD, FE, FS and LP. The clustering of QTLs within linkages indicates that genes for different traits on the same chromosome are linked or that the phenotypes are due to pleiotropic effects of a single QTL, especially QTLs for boll weight traits, which is consistent with the results of correlation analysis. The synergistic alleles of QTLs of boll-related traits mainly came from AQ. For example, we found that the additive effect of the traits BL and BD originated from the same parent, AQ. Therefore, AQ can play an important role in improving boll weight. In this study, five QTL clusters for boll-related traits were found on Chr. A10, A13, D1 and D5. Unlike those for boll-related traits, the M-QTLs for fiber quality traits detected in this study were not clustered, although QTL clusters for fiber quality or plant architecture traits were previously been reported in cotton (Chen et al. 2009; Mei et al. 2004; Wang et al. 2006; Zhang et al. 2005, 2009, 2012a). Said et al. (2013) also detected QTL clusters comprising regions containing four or more QTLs for various traits (including fiber quality and others). Said et al. (2013) ascribed the different results from various studies to the use of different genetic populations, markers and marker densities, and testing environments.
Many previous studies have focused on QTL mapping for fiber quality and yield traits using intraspecific maps (Chen et al. 2009; Guo et al. 2006; Qin et al. 2008, 2009; Shao et al. 2014; Shen et al. 2005, 2007; Sun et al. 2012; Ulloa et al. 2005; Wang et al. 2007; Wu et al. 2009; Zhang et al. 2010, 2012a, 2005, 2009). However, it is difficult to compare the QTLs detected in these studies because few common markers exist in the diverse intraspecific populations employed, and the maps produced in these studies cover different chromosome regions of the cotton genome. Nonetheless, both the present study and the previous studies have revealed many common characteristics for QTLs conferring fiber quality and yield traits, and these QTLs for fiber traits were mapped to the same chromosomes in different populations. For example, some QTLs for fiber quality and yield traits detected in the current study were also mapped to the same chromosomes in previous studies, including three QTLs for FS (Shao et al. 2014; Sun et al. 2012; Wu et al. 2009), two QTLs for FL (Liang et al. 2013; Shao et al. 2014; Shen et al. 2005; Zhang et al. 2005, 2009, 2012a), four QTLs for MIC (Liang et al. 2013; Qin et al. 2008, 2009; Shao et al. 2014; Shen et al. 2005, 2007; Zhang et al. 2012a, 2013), two QTLs for FU (Shao et al. 2014; Zhang et al. 2012a), three QTLs for FE (Liang et al. 2013; Qin et al. 2008; Shao et al. 2014; Shen et al. 2005; Sun et al. 2012; Wang et al. 2007; Zhang et al. 2012a, 2013), six QTLs for BW (Ning et al. 2014; Shen et al. 2007; Xia et al. 2014; Zhang et al. 2010), four QTLs for LP (Liu et al. 2012; Wang et al. 2007; Wu et al. 2009; Zhang et al. 2009, 2013), one QTL for LY (Xia et al. 2014) and three QTLs for SI (Liu et al. 2012; Shen et al. 2007; Wang et al. 2007; Wu et al. 2009; Xia et al. 2014). These QTLs may be common QTLs for fiber quality and yield traits in upland cotton, which may be verified through the use of many more common markers in the future.
Furthermore, in the present study, some QTLs were not mapped to the same chromosomes as those of previous studies. For example, six QTLs for yield traits were detected on different chromosomes, including three QTLs (qBW-F2-A13-1, qBW-F2-A13-2 and qBW-F2-A9-1) for BW on Chr. A9 and A13, two QTLs (qLP-F2:3-A13-1 and qLP-F2:3-A13-2) for LP on Chr. A13 and one QTL (qSI-F2:3-D1-1#) for SI on Chr. D1. Moreover, one QTL (qFE-F2-A12-1) for fiber quality (FE) was also detected on Chr. A12. Our results indicate that these seven positive additive QTLs were from the elite parent AQ, and they are distributed on different chromosomes, implying that these QTLs are unique to upland cotton and may be useful for cotton improvement.
Finally, epistasis, or interlocus interaction, is a type of gene interaction whereby one gene interferes with the phenotypic expression of another non-allelic gene. A considerable body of evidence from classical studies strongly suggests the prevalence of an epistatic effect on quantitative traits in genetic populations (Zhang et al. 2001). Based on heterosis research in rice, Yu et al. (1997) found that epistasis plays an important role in the inheritance of quantitative traits and heterosis. Xing et al. (2002) further reported that epistasis, in the form of additive-by-additive interactions, plays a highly important role in controlling the expression of yield and yield-component traits. Some studies have demonstrated that E-QTLs play an important role in the genetic control of plant architectural traits in cotton (Wang et al. 2006; Song and Zhang 2009) and other crops such as maize (Xu et al. 2009) and wheat (Wang et al. 2010). In this study, we identified 64 pairs of E-QTLs for 12 boll weight-related traits in both populations examined, including seven for BCW, six for BL, five for BLN, two for BLW, nine for BN, six for BW, eight for BSN, five for BSN, one for LP, six for LY, seven for SI and two for SY. Notably, two pairs of interacting marker intervals simultaneously control two traits. Moreover, some marker intervals have interactions with other multiple marker intervals to control different traits. However, no E-QTLs detected in the current study were mapped to the same chromosomes as those of previous studies. In addition, no E-QTLs for fiber quality were detected in the current study, which is inconsistent with a previous report employing different populations (Wang et al. 2013).
In conclusion, in the F2 and F2:3 populations, common QTLs were detected in the same and similar positions simultaneously, suggesting that these are major QTLs that can be used for further verification and analysis and thus, for marker-assisted breeding. QTL clusters were inferred and identified using the positions and distribution of QTLs along the Gossypium genome. The presence of QTL clusters indicates that genes pertaining to certain traits are more heavily concentrated in certain regions of the genome than in others. The markers associated with E-QTLs identified in the current study will be important for future breeding programs aimed at developing cotton cultivars.
References
Al-Ghazi Y, Bourot S, Arioli T, Dennis ES, Llewellyn DJ (2009) Transcript profiling during fiber development identifies pathways in secondary metabolism and cell wall structure that may contribute to cotton fiber quality. Plant Cell Physiol 50:1364–1381
Ashraf M, Ahmad S (2000) Influence of sodium chloride onion accumulation, yield components and fiber characteristics in salt-tolerant and salt-sensitive lines of cotton (Gossypium hirsutum L.). Field Crops Res 66(2):115–127
Chee P, Draye X, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum and Gossypium barbadense (cotton) by a backcross-self approach: I. Fiber elongation. Theor Appl Genet 111(4):757–763
Chen H, Qian N, Guo WZ, Song QP, Li BC, Deng FJ, Dong CG, Zhang TZ (2009) Using three overlapped RILs to dissect genetically clustered QTL for fiber strength on Chro. D8 in upland cotton. Theor Appl Genet 119:605–612
Churchill G, Doerge R (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Guo WZ, Ma GJ, Zhu YC, Yi CX, Zhang TZ (2006) Molecular tagging and mapping of quantitative trait loci for lint percentage and morphological marker genes in upland cotton. J Integr Plant Biol 48:320–326
Guo WZ, Cai CP, Wang CB, Han ZG, Song XL, Wang K, Niu XW, Wang C, Lu KY, Shi S, Zhang TZ (2007) A microsatellite-based, gene-rich linkage map reveals genome structure, function, and evolution in Gossypium. Genetics 176:527–541
Han ZG, Guo WZ, Song XL, Zhang TZ (2004) Genetic mapping of EST-derived microsatellites from the diploid Gossypium arboreum in allotetraploid cotton. Mol Genet Genomics 272:308–327
He DH, Lin ZX, Zhang XL, Nie YC, Guo XP, Feng CD, Stewart MD (2005) Mapping QTLs of traits contributing to yield and analysis of genetic effects in tetraploid cotton. Euphytica 144:141–149
He DH, Lin ZX, Zhang XL, Nie YC, Guo XP, Zhang YX, Li W (2007) QTL mapping for economic traits based on a dense genetic map of cotton with PCR-based markers using the interspecific cross of Gossypium hirsutum × Gossypium barbadense. Euphytica 153:181–197
Jiang CX, Wright RJ, El-Zik KM, Paterson AH (1998) Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc Natl Acad Sci USA 95(8):4419–4424
Lacape JM, Nguyen TB, Thibivilliers S, Bojinov B, Courtois B, Cantrell R, Burr B, Hau B (2003) A combined RFLP SSR AFLP map of tetraploid cotton based on a Gossypium hirsutum × Gossypium barbadense backcross population. Genome 46:612–626
Lacape JM, Llewellyn D, Jacobs J, Arioli T, Becker D, Calhoun S, Al-Ghazi Y, Liu S, Palaï O, Georges S, Giband M, de Assunção H, Barroso PAV, Claverie M, Gawryziak G, Jean J, Vialle M, Viot C (2010) Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol 10:132
Lander E, Kruglyak K (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Li JZ, Fu CY, Zhang HL, Liu LF, Mu P, Li ZC (2009) QTL mapping and QTL 9 environment interactions of appearance quality in upland rice introgression lines under upland and lowland environments. J Agric Biotechnol 17:651–658
Li CQ, Song L, Zhao HH, Wang QL, Fu YZ (2014) Identification of quantitative trait loci with main and epistatic effects for plant architecture traits in upland cotton (Gossypium hirsutum L.). Plant Breed 133:390–400
Liang QZ, Hu C, Hua H, Li ZH, Hua JP (2013) Construction of a linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Chin Sci Bull 58:3233–3243
Lin ZX, Zhang YX, Zhang XL, Guo XP (2009) A high-density integrative linkage map for Gossypium hirsutum. Euphytica 166(1):35–45
Liu RZ, Wang BH, Guo WZ, Qin YS, Wang LG, Zhang YM, Zhang TZ (2012) Quantitative trait loci mapping for yield and its components by using two immortalized populations of a heterotic hybrid in Gossypium hirsutum L. Mol Breed 2:297–311
Lou J, Chen L, Yue G, Lou Q, Mei H, Xiong L, Luo L (2009) QTL mapping of grain quality traits in rice. J Cereal Sci 50:145–151
Malmberg RL, Held S, Waits A, Mauricio R (2005) Epistasis for fitness-related quantitative traits in Arabidopsis thaliana grown in the field. Genetica 171:2013–2027
Mauricio R (2001) Mapping quantitative trait loci in plants: uses and caveats for evolutionary biology. Nat Rev Genet 2(5):370–381
Mei M, Syed NH, Gao W, Thaxton PM, Smith CW, Stelly DM, Chen ZJ (2004) Genetic mapping and QTL analysis of fiber-related traits in cotton (Gossypium). Theor Appl Genet 108(2):280–291
Mohan A, Kulwal P, Singh R, Kumar V, Mir R, Kumar J, Prasad M, Balyan H, Gupta P (2009) Genome-wide QTL analysis for preharvest spouting tolerance in bread wheat. Euphytica 168:319–329
Ning ZY, Chen H, Mei HX, Zhang TZ (2014) Molecular tagging of QTLs for fiber quality and yield in the upland cotton cultivar Acala-Prema. Euphytica 195:143–156. doi:10.1007/s10681-013-0990-3
Paterson AH, Brubaker C, Wendel J (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep 11:122–127
Paterson AH, Saranga Y, Menz M, Jiang CX, Wright RJ (2003) QTL analysis of genotype × environment interactions affecting cotton fiber quality. Theor Appl Genet 106(3):384–396
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin DC, Llewellyn D, Showmaker KC, Shu SQ, Udall J, Yoo MJ, Byers R, Chen W, Doron-Faigenboim A, Duke MV, Gong L, Grimwood J, Grover C, Grupp K, Hu GJ, Lee TH, Li JP, Lin LF, Liu T, Marler BS, Page JT, Roberts AW, Romanel E, Sanders WS, Szadkowski E, Tan X, Tang HB, Xu CM, Wang JP, Wang ZN, Zhang D, Zhang L, Ashrafi H, Bedon F, Bowers JE, Brubaker CL, Chee PW, Das S, Gingle AR, Haigler CH, Harker D, Hoffmann LV, Hovav R, Jones DC, Lemke C, Mansoor S, Rahman MU, Rainville LN, Rambani A, Reddy UK, Rong JK, Saranga Y, Scheffler BE, Scheffler JA, Stelly DM, Triplett BA, Van Deynze A, Waghmare VN, Vaslin MFS, Walford SA, Wright RJ, Zaki EA, Zhang TZ, Dennis ES, Mayer KFX, Peterson DG, Rokhsar DS, Wang XY, Schmutz J (2012) Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fiber. Nature 492:423–428
Qin HD, Guo WZ, Zhang YM, Zhang TZ (2008) QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor Appl Genet 117:883–894
Qin YS, Ye WX, Liu RZ, Zhang TZ, Guo WZ (2009) QTL Mapping for fiber quality properties in upland cotton (Gossypium hirsutum L.). Sci Agric Sin 42:4145–4154
Reinisch MJ, Doug J, Brubaker CL, Stelly DM, Wendel JF, Paterson AH (1994) A detailed RFLP map of cotton, Gossypium hirsutum × Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetics 138:829–847
Ren LH, Guo WZ, Zhang TZ (2002) Identification of quantitative trait loci (QTLs) affecting yield and fiber properties in chromosome 16 in cotton using substitution line. Acta Bot Sin 44:815–820
Rong JK, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, Delmonte TA, Ding X, Garza JJ, Marler BS, Park C, Pierce GJ, Rainey KM, Rastogi VK, Schulze SR, Tronlinde NL, Wendel JF, Wilkins TA, Wing RA, Wright RJ, Zhao X, Zhu L, Paterson AH (2004) A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166:389–417
Rong J, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Draye X, Saranga Y, Wright RJ, Wilkins TA, May OL, Smith CW, Gannaway JR, Wendel JF, Paterson AH (2007) Metaanalysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176:2577–2588
Said JI, Lin ZX, Zhang XL, Song MZ, Zhang JF (2013) A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom 14(1):776
Shao QS, Zhang FJ, Tang SY, Liu Y, Fang XM, Liu DX, Liu DJ, Zhang J, Teng ZH, Andrew HP, Zhang ZS (2014) Identifying QTL for fiber quality traits with three upland cotton (Gossypium hirsutum L.) populations. Euphytica 198:43–58
Shappley ZW, Jenkins JN, Watson CE Jr, Kahler AL, Meredith WR (1996) Establishment of molecular markers and linkage groups in two F2 populations of upland cotton. Theor Appl Genet 92:915–919
Shen XL, Guo WZ, Zhu XF, Yuan YL, John Z, Kohel RJ, Zhang TZ (2005) Molecular mapping of QTL for fiber qualities in three diverse lines in upland cotton using SSR markers. Mol Breed 15:169–181
Shen XL, Guo WZ, Lu QX, Zhu XF, Yuan YL, Zhang TZ (2007) Genetic mapping of quantitative trait loci for fiber quality and yield trait by RIL approach in upland cotton. Euphytica 155:371–380
Song XL, Zhang TZ (2009) Quantitative trait loci controlling plant architectural traits in cotton. Plant Sci 177:317–323
Sun FD, Zhang JH, Wang SF, Gong WK, Shi YZ, Liu AY, Li JW, Gong JW, Shang HH, Yuan YL (2012) QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol Breed 30:569–582
Taliercio EW, Boykin D (2007) Analysis of gene expression in cotton fiber initials. BMC Plant Biol 7:22. doi:10.1186/1471-2229-7-22
Tang FY, Xiao WJ (2014) Genetic association of within-boll yield components and boll morphological traits with fiber properties in upland cotton (Gossypium hirsutum L.). Plant Breed 133:521–529
Tanksley SD, Hewitt J (1988) Use of molecular markers in breeding for soluble solids content in tomato-a reexamination. Theor Appl Genet 75:811–823
Ulloa M, Meredith WR (2000) Genetic linkage map and QTL analysis of agronomic and fiber quality traits in an intraspecific population. J Cotton Sci 4:161–170
Ulloa M, Meredith WR, Shapply ZW, Kahler AL (2002) RFLP genetic linkage maps from F2:3 populations and a joinmap of Gossypium hirsutum L. Theor Appl Genet 104:200–208
Ulloa M, Saha S, Jenkin N, Meredith WR, McCarty JC, Stelly DM (2005) Chromosomal assignment of RFLP linkage groups harboring important QTL on an intraspecific cotton (Gossypium hirsutum L.) joinmap. J Hered 96:132–144
Van Ooijen V (2001) JoinMap R Version 3.0: Software for the calculation of genetic linkage maps. CPRO-DLO, Wageningen
Wang SC, Basten J, Zeng ZB (2005) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh
Wang BH, Wu YT, Huang NT, Zhu XF, Guo WZ, Zhang ZT (2006) QTL mapping for plant architecture traits in upland cotton using RILs and SSR markers. Acta Genet Sin 33:161–170
Wang BH, Guo WZ, Zhu XF, Wu YT, Huang NT, Zhang TZ (2007) QTL mapping of yield and yield components for elite hybrid derived-RILs in upland cotton. J Genet Genomics 34:35–45
Wang ZH, Wu XS, Ren Q, Chang XP, Li RZ, Jing RL (2010) QTL mapping for developmental behavior of plant height in wheat (Triticum aestivum L.). Euphytica 174:447–458
Wang JK, Li HH, Zhang LY, Meng L (2012) Users’ manual of QTL IciMapping Version 3.2. The Quantitative Genetics Group, Institute of Crop Science, Chinese Academy of Agricultural Sciences (CAAS), Beijing, pp 50–81
Wang FR, Xu ZZ, Sun R, Gong YC, Liu GD, Zhang JX, Wang LM, Zhang CY, Fan SJ, Zhang J (2013) Genetic dissection of the introgressive genomic components from Gossypium barbadense L. that contribute to improved fiber quality in Gossypium hirsutum L. Mol Breed 32:547–562
Wu J, Gutierrez OA, Jenkins JN, McCarty JC, Zhu J (2009) Quantitative analysis and QTL mapping for agronomic and fiber traits in an RI population of upland cotton. Euphytica 165:231–245
Xia Z, Zhang X, Liu YY, Jia ZF, Zhao HH, Li CQ, Wang QL (2014) Major gene identification and quantitative trait locus mapping for yield-related traits in upland cotton (Gossypium hirsutum L.). J Integr Agric 13(2): 299–309
Xing YZ, Tan YF, Hua JP, Sun XL, Xu CG, Zhang QF (2002) Characterization of the main effects, epistatic effects and their environmental interactions of QTL on the genetic basis of yield traits in rice. Theor Appl Genet 105:248–257
Xu SZ, Jia ZY (2007) Genome-wide analysis of epistatic effects for quantitative traits in barley. Genetics 175:1955–1963
Xu YB, Zhu LH (1994) Molecular Quantitative Genetics. Chinese Agriculture Press, Beijing, China, pp12
Xu ZY, Kohel RJ, Song GL, Cho JM, Alabady M, Yu J, Koo P, Chu J, Yu SX, Wilkins TA, Zhu YX, Yu JZ (2008) Gene rich islands for fiber development in the cotton genome. Genomics 92:173–183
Xu DL, Cai YL, Lv XG, Dai GQ, Wang GQ, Wang JG, Sun HY, Tan HN (2009) QTL mapping for plant-tape traits in maize. J Maize Sci 17:27–31
Yu SB, Li JX, Tan YF, Gao YJ, Li XH, Zhang QF, Saghai Maroof MA (1997) Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 94(17):9226–9231
Yu JW, Yu SX, Liu C, Wu W, Fan SL, Song MZ, Lin ZX, Zhang XL, Zhang JF (2007) High-density linkage map of cultivated allotetraploid cotton based on SSR, TRAP, SRAP and AFLP markers. J Integr Plant Biol 49:716–724
Yu Y, Yuan DJ, Liang SG, Li XM, Wang XQ, Lin ZX, Zhang XL (2011) Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genom 12(1):15
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Zhang J, Wu YT, Guo WZ, Zhang TZ (2000) Fast screening of SSR markers in cotton with PAGE/silver staining. Cotton Sci Sinica 12:267–269
Zhang WP, Shen XY, Wu P, Hu B, Liao CY (2001) QTLs and epistasis for seminal root length under a different water supply in rice. Theor Appl Genet 103:118–125
Zhang J, Guo WZ, Zhang TZ (2002) Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. × Gossypium barbadense L.) with a haploid population. Theor Appl Genet 105:1166–1174
Zhang ZS, Xiao YH, Luo M, Li XB, Luo XY, Hou L, Li DM, Pei Y (2005) Construction of a genetic linkage map and QTL analysis of fiber-related traits in upland cotton (Gossypium hirsutum L.). Euphytica 144:91–99
Zhang ZS, Hu MC, Zhang J, Liu DJ, Zheng J, Zhang K, Wang W, Wan Q (2009) Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol Breed 24(1): 49–61
Zhang J, Chen X, Zhang K, Liu DJ, Wei XQ, Zhang ZS (2010) QTL mapping of yield traits with composite cross population in upland cotton (Gossypium hirsutum L.). J Agric Biol 18:476–481
Zhang F, Jiang JF, Chen SM, Chen FD, Fang WM (2012a) Mapping single-locus and epistatic quantitative trait loci for plant architectural traits in chrysanthemum. Mol Breed 30:1027–1036
Zhang K, Zhang J, Ma J, Tang S, Liu D, Teng Z, Liu D, Zhang Z (2012b) Genetic mapping and quantitative trait locus analysis of fiber quality traits using a three-parent composite population in upland cotton (Gossypium hirsutum L.). Mol Breed 29:335–348
Zhang T, Qian N, Zhu X, Chen H, Wang S, Mei H, Zhang Y (2013) Variations and transmission of QTL alleles for yield and fiber qualities in upland cotton cultivars developed in China. PLoS ONE 8:e57220
Acknowledgments
This program was financially supported in part by National Key Technology Support Program of China during the twelfth Five-year Plan Period (2013BAD01B03-04) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, S., Wang, T., Liu, Q. et al. Quantitative trait locus analysis of boll-related traits in an intraspecific population of Gossypium hirsutum . Euphytica 203, 121–144 (2015). https://doi.org/10.1007/s10681-014-1281-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1281-3