
Abstract
We crossed Luyuan343, containing the Gossypium barbadense (GB) genomic component, with Lumianyan22, a high-yielding Gossypium hirsutum (GH) variety, to evaluate the introgressive exogenous genomic components that contribute to fiber quality in upland cotton. A total of 158 of 334 mapping simple sequence repeats (SSR) loci screened from 18,467 SSR primer pairs were identified as putative GB introgression loci. Twenty-four quantitative trait loci (QTLs) for fiber quality and lint percentage were detected by WinQTLCart 2.5 based on three phenotypic datasets collected over 2 years in two different locations. Of these QTLs, 20 were mapped in the introgression chromosomal regions; the GB alleles contributed to improved fiber quality at eight loci, which were located in the introgression segments of chromosome (Chr.) 3, Chr.7 and Chr.25. A total of eight digenic epistasis and nine significant QTL × environment (QE) interactions were identified by QTLNetwork-2.2. Fiber elongation was found to be most easily influenced by environment since all QTLs for this trait showed significant QE interaction with large effects. Fiber length, fiber strength, and lint percentage were scarcely affected by environment as no or minor QE interaction effects for these traits were detected. Genome-wide identification of the introgressive GB components and mapping of the fiber-related QTLs indentified in this study will be beneficial for the simultaneous marker-assistant selection of improved fiber quality and lint yield in upland cotton breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton is the leading natural fiber crop in the world, and its genome presents unique opportunities to dissect the evolution of the natural fiber, ‘lint.’ Over the past decades, the widespread use of high-speed fiber spinning machinery has increased the global demand for high-quality fiber. This calls for advances in cotton breeding programs that increase fiber quality and yield (Campbell et al. 2011). The Gossypium genus comprises approximately 50 species, including 45 diploids and five allotetraploids. There are four domesticated and cultivated species within the genus, i.e., the New World allopolyploids G. hirsutum (Upland cotton) and G. barbadense (Sea Island cotton) (2n = 52) and the Old World diploids G. arboreum and G. herbaceum (2n = 26) (Wendel et al. 2010). The two cultivated allotetraploid species have very different agronomic and fiber quality characteristics (Chee et al. 2005). Due to its higher lint yield and broad adaptation, G. hirsutum is the most widely cultivated species and is grown in more than 40 nations in both tropical and temperate regions of the world. Although G. hirsutum accounts for almost 90 % of the total world production of cotton, G. barbadense is still cultivated in several regions of Central Asia, Egypt, Sudan, India, USA, and China because of its superior fiber quality, including long, strong, and fine fibers. However, its relatively low yield has limited its importance to <10 % of the total world production (Wendel et al. 2010). Upland cotton generally has low-quality fibers, i.e., shorter or coarse fibers of relatively lower strength. The unique fiber properties of G. barbadense make it an ideal candidate for improving fiber quality for upland cotton breeding. Extensive efforts have been made on crossing and/or backcrossing G. hirsutum and G. barbadense in order to enhance the fiber quality in Upland cotton (Campbell et al. 2010, 2011; Mehboob-ur-Rahman et al. 2012). A series of introgression lines carrying desirable quantitative trait locus (QTL) alleles for fiber quality have been developed. Although there is no hybridization incompatibility between the introgression line and G. hirsutum, negative effects (unfavorable correlations due to linkage drags), such as low lint yield and poor disease-resistance, were often encountered in the breeding process for improving the fiber quality using the high-fiber quality introgression lines (Meredith 2005). It remains very difficult to resolve these complications in conventional cotton breeding programs.
DNA markers provide a useful approach for reducing unfavorable correlations by marker-assisted selection (MAS). Several QTLs for fiber quality have been identified (Draye et al. 2005; Chen et al. 2009; Shen et al. 2007; Lacape et al. 2010; Sun et al. 2011; Zhang et al. 2012); however, the majority of mapping populations are derived from interspecific hybridization of G. barbadense × G. hirsutum. Therefore, few of these QTLs can be directly applied in breeding programs. Introgression lines, which form a secondary gene pool, make it possible to transfer favorable alleles into elite cultivars. Several studies on QTL mapping to enhance fiber quality in Gossypium genus report the use of germplasm containing introgression genomic components from other species (Shen et al. 2007; Zhang et al. 2009, 2012; Sun et al. 2011). However, these studies do not provide any evidence of association between the introgression genetic components and the detected fiber quality QTLs. Recently, Zhang et al. (2011) identified QTL alleles for improved fiber quality from a wild Hawaiian cotton species Gossypium tomentosum. Also, Xu et al. (2012a) identified QTLs for fiber micronaire from a wild species G. klotzschianum introgressed into upland cotton.
In our previous study (Wang et al. 2011), we preliminarily analyzed the genetic effect of introgressive chromosomal segments from G. barbadense on fiber-related traits in upland cotton using a germplasm of superior fiber quality. After screening 4,050 simple sequence repeats (SSRs), we identified 42 loci as putative introgression alleles. Most of these were found to be clustered on the putative G. barbadense introgression chromosomal segments of chromosome (Chr.) 2, Chr.16, Chr.23, and Chr.25. Our study demonstrated that the majority of the favorable alleles for fiber quality traits were derived from the introgressive genomic components.
We undertook the current study to (1) further identify the introgression components at a genome-wide level; (2) dissect the association between QTLs responsible for fiber quality and the G. barbadense introgression chromosomal segments by constructing a longer and relatively saturated linkage map; (3) search stable QTLs which could be used in MAS for further improving the fiber quality and lint yield simultaneously in upland cotton breeding.
Materials and methods
Plant materials
Luyuan343 (LY343) is a high-fiber quality upland cotton germplasm developed by a natural cross between G. barbadense cv. Ashimouni with an unknown upland cotton followed by artificial selection for 13 generations (Su et al. 2000; Wang et al. 2011). It had once been cultivated in some cotton-growing region of China due to its excellent fiber quality. Lumianyan22 (LMY22) is a transgenic insect-resistant upland cotton cultivar developed in our laboratory and approved by the Chinese Ministry of Agriculture. It is now cultivated in China for its desirable lint yield performance; however, it does not have very good fiber quality. All parental lines were self-pollinated for several generations before making hybrid combinations.
Population development and phenotypic evaluation
LMY22 was crossed with LY343 at Linqing Experimental Station, Shandong Cotton Research Center (LES/SCRC) in 2006. The F 1 hybrids were planted on Hainan Island in the winter season of the same year and self-pollinated to produce F 2 seeds. The F 2 population comprising 209 individuals were grown at LES/SCRC and self-pollinated to produce the next generation in 2007. The F 2:3 family seeds were randomly divided into two groups for planting in different environments. One group was planted on Hainan Island in the winter season of 2007, and the other group was grown at LES/SCRC in the spring season of 2008. Traditional cotton cultural practices were applied at both locations. Fiber samples of F 2 individuals were harvested from the gin-out of self-pollinated bolls at LES/SCRC, and F 2:3 fiber samples were collected from the two test sites at Hainan Island and LES/SCRC.
The fiber quality parameters, including fiber length (FL, mm), fiber strength (FS, cN · Tex−1), fiber length uniformity (FU, %), fiber elongation (FE, %) and fiber micronaire (FM, fineness value) were tested by the Supervision, Inspection and Test Center of Cotton Quality, Ministry of Agriculture of China (Anyang, Henan province, P.R. China) using a high-volume precision instrument (HVI; Zellweger-Uster, Knoxville, TN). Lint percentage (LP), one of lint yield components, was evaluated following conventional cotton breeding methods.
Molecular markers and assays
Genomic DNA from the 209 F 2 plants, the mapping parents, and cv. Ashimouni (the donor G. barbadense parent of LY343, the superior introgression line) was extracted as described by Paterson et al. (1993). Most of SSR primers were downloaded from CMD (http://www.cottonmarker.org) and CottonDB (http://www.cottondb.org) which have been exploited to construct interspecific and intraspecific genetic maps for QTL mapping in tetraploid cotton (Zhang et al. 2002, 2009, 2012; Lacape et al. 2003, 2010; Rong et al. 2004, 2007; Guo et al. 2007; Shen et al. 2007; Lin et al. 2009; Sun et al. 2011). The additional SSR primer sequences were kindly provided by Dr Zhengsheng Zhang (Southwest University of P.R. China). All primers were synthesized by Invitrogen (Shanghai, China). The affirmative polymorphic primers were used to genotype F 2 plants. PCR amplification and the polyacrylamide gel electrophoresis/silver staining survey were performed according to the procedure described by Zhang et al. (2002). The putative introgression G. barbadense alleles were identified and affirmed as described in our previous report (Wang et al. 2011).
Data and QTL analysis
The linkage map was constructed using MAPMAKER/EXP 3.0 (Lander et al. 1987) with a LOD score of 6.5. The Kosambi mapping function was used to convert the recombination frequency to genetic map distance (centiMorgen, cM). The linkage groups were assigned to chromosomes based on genetic linkage map from CottonDB (http://www.cottondb.org) and the published literature (Guo et al. 2007; Zhang et al. 2012; Yu et al. 2012).
Five fiber quality traits and one of the yield components, LP, collected from F 2, F 2:3 at Linqing and Hainan were used to map QTLs. The composite interval mapping (CIM) procedure using WinQTLCart 2.5 (Wang et al. 2006) was selected to detect QTLs. The window size was set at 5 cM and the walk speed at 1 cM. LOD threshold values were estimated by 1,000 permutations to declare significant QTLs (Churchill and Doerge 1994). QTL confidence intervals (90–95 %) were set as map intervals corresponding to a decline of 1 LOD on either side of the peak. The linkage maps and QTLs were as presented by MapChart 2.2 (Voorrips 2002). QTL nomenclature follows a method developed in rice by McCouch et al. (1997). The designation begins with ‘q,’ followed by an abbreviation of the trait name, the name of chromosome, and the serial number.
To further dissect the genetic architecture of the fiber-related traits, we analyzed our data by QTLNetwork-2.2 (Yang et al. 2008). All detected QTLs and epistatic loci were fitted by a full-QTL model to estimate the main- effect QTL and epistasis and their interaction effects with environment by the MCMC (Markov Chain Monte Carlo) algorithm. The QTLs detected by QTLNetwork 2.2 to be located in the same or adjacent region of the QTLs detected by WinQTLCart 2.5 were considered to be the same QTLs.
Validation of QTLs using the F 2:8 advanced population
An advanced population of F 2:8 was developed from the same LMY22 × LY343 cross as the QTL mapping population. Individuals were randomly selected and self-pollinated from the initial F 2 population. Progenies by consecutive single seed descent (a randomly harvested selfed individual in a certain progeny is derived from a single seed of the last generation) were grown at LES/SCRC and in the following winter were planted on Hainan Island until F 2:8 lines were developed. A total of 359 lines of the F 2:8 were planted in trial plots at LES/SCRC in 2011. Fiber sample collection and phenotypic evaluation were conducted as that of F 2:3 family lines. We extracted DNA from all the 359 advanced lines for marker genotype survey. Based on the genotype of the nearest marker to the QTL, we selected homozygous lines for LMY22 and LY343 alleles to compare the differences in fiber quality traits and LP by analysis of variance using DPS software (Tang and Feng 2007) as described in our previous work (Wang et al. 2011).
Results
Molecular linkage map construction and identification of putative introgression G. barbadense genomic components
A total of 284 of the 14,417 SSR markers screened exhibited polymorphism between the mapping parents and genotyped F 2 populations. The ratio of polymorphic loci was only 1.82 %. Integrating the results from our previous study (Wang et al. 2011), we increased the total number of polymorphic loci to 366, of which 87 showed skewed Mendelian segregation. The linkage map comprises 43 linkage groups with 334 loci spanning 1,779.2 cM, covering approximately 35.36 % of the tetraploid cotton genome (Stelly 1993). Thirty-nine linkage groups were assigned to 25 chromosomes, with the exception of Chr.14, which contained only one polymorphic SSR marker.
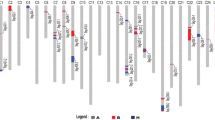
A total of 158 of the polymorphic loci assembled into the linkage groups were identified as the putative introgression G. barbadense (GB) loci, which distributed on 20 chromosomes. The introgression segments covered 646.91 cM and accounted for 12.86 % of tetraploid cotton genome. The length of the introgression segment per chromosome was found to range from 3.94 cM (Chr.26) to 94.42 cM (Chr.10) (Fig. 1).


Distribution of quantitative trait loci (QTLs) for fiber quality and lint percentage (LP) on the linkage map constructed in this paper. The putative introgression genomic components and QTLs for fiber quality and LP are shown in this figure. The linkage group bars filled with red color represent the introgression genomic regions and the corresponding markers are represented by red letters. The distorted segregation loci are shown in italics. The green, red and black bars with slash represent the QTLs detected in three, two and one datasets of F 2 in Linqing and F 2:3 in Linqing and Hainan, respectively. QTL bars and lines indicate 1 LOD and 2 LOD likelihood intervals. The names of the QTLs presented in the Fig. 1 are also shown in the tables. (Color figure online)
QTL mapping, epistasis, and their interactions with environments
The phenotypic characteristics of the F 2 population, as well as those of the F 2:3 in two environments and the mapping parents, were described in our previous study (Wang et al. 2011). Based on our three datasets, a total of 24 QTLs were detected by WinQTLCart 2.5, including 19 for fiber quality and five for LP. These QTLs were mainly targeted on nine chromosomes, and 20 of the QTLs were in the introgression chromosomal regions. The GB alleles contributed to increase fiber quality at eight loci (Fig. 1; Table 1).
A total of 18 QTLs with additive effect (A) were detected by QTLNetwork-2.2 based on the mixed liner model, among which 14 QTLs were also detected by WinQTLCart 2.5 (Table 2). Nine of the additive effect QTLs showed significant interaction with environment. Eight digenic epistastic interactions with additive × additive (AA), and/or additive × dominative (AD), dominative × additive (DA), dominative × dominance (DD) effects were identified, the majority (6/8) of which occurred between several pairs of genetic background loci not linked to any QTL. Only one epistastic interaction showed significant AAE effect (Table 3).
Fiber length
Four FL QTLs were detected by WinQTLCart 2.5, each explaining 6.98–16.36 % of the phenotypic variance. All of these QTLs were located in the introgression chromosomal regions, and the GB allele contributed to increased fiber length at three loci (qFL-C7-1, qFL-C16-1, and qFL-C25-1). Three of the QTLs with significant additive effect were detected by QTLNetwork 2.2. One epistasis interaction between qFL-C16-1 and qFL-C25-1 was identified which explained 1.35 % of the total phenotypic variance. No QTL showed significant interaction with environment.
Fiber uniformity
Only two FU QTLs were detected by WinQTLCart 2.5. The phenotypic variance explained by each individual QTL was 7.3 and 9.17 %, respectively. Both of the FU QTLs were mapped in non-introgression regions, and the favorable alleles were derived from the G. hirsutum parent (GH, LMY22). When analyzed by QTLNetwork-2.2, two QTLs with additive effect were detected, and both showed significant QTL × environment (QE) interaction. Two epistasis interactions between the genetic background loci on Chr.8 and Chr.11 with AA, DA, and DD epistastic effects were identified.
Fiber fineness (fiber micronaire)
Fiber micronaire is regarded as an indication of both fineness (linear density) and maturity (degree of cell-wall development), and a somewhat low FM value has been used as a predictor of desirable fine cotton fibers with adequate maturity, while a relatively high FM value represents thick cotton fibers (Montalvo 2005). We used the FM value for fiber fineness evaluation in our study. Three fiber fineness QTLs were detected by WinQTLCart 2.5, each explaining 10.45–12.33 % of the phenotypic variance. Two of the QTLs were mapped in the introgression chromosomal segments, and the GH parent (LMY22) conferred the favorable allele at three loci. Only one additive effect QTL (qFM-C16-1) was detected by QTLNetwork-2.2, and no significant QE interaction and epistasis effect were observed.
Fiber elongation
Seven QTLs for FE were detected by WinQTLCart 2.5. The phenotypic variance explained by individual QTLs ranged from 5.29 to 20.66 %. All of the QTLs were mapped in the introgression regions, and the GB allele contributed to increased elongation at three loci (qFE-C3-1, qFE-C3-2, and qFE-C3-3). Five FE QTLs with additive effect were detected by QTLNetwork-2.2, and four of the QTLs were located on the same marker intervals or adjacent region as the QTLs detected by WinQTLCart 2.5. All five QTLs showed significant QE interaction effects in one or more environments, and the QE effects were remarkably larger than the additive effect. One epistasis interaction between the genetic background loci on Chr.7 and Chr.6 with AD effect was detected, and significant AAE interaction was also identified. Our results suggest that fiber elongation was markedly influenced by environment.
Fiber strength
Three QTLs for FS were detected by WinQTLCart 2.5, explaining 7.3–27.09 % of the phenotypic variance. These QTLs were all mapped in the introgression segments, and the GB allele contributed to increase fiber strength at two loci on Chr.7. One of the QTLs, qFS-C7-1, was detected in all the three datasets and explained 13.74, 26.21, and 25.58 % of the phenotype variance in each population, respectively. When analyzed by QTLNetwork-2.2, two additional QTLs (qFS-C8-1 and qFS-C4-1) besides qFS-C7-1 were detected with additive effect. qFS-C7-1 showed QE interaction with a minor effect (\( h_{\text{ae}}^{2} = 1.98 \)). Two epistasis interactions were identified, one between qFS-C7-1 and background locus on Chr.7 and the other between two background loci on Chr.16 and Chr.17.
Lint percentage
We detected five QTLs for LP by WinQTLCart 2.5, explaining 6.54–15.52 % of the phenotypic variance. The favorable alleles of the five QTLs were all derived from the GH parent. qLP-C17-1 could be detected in all three datasets and explained 15.52, 14.88, and 7.97 % of the phenotype variance, respectively. This QTL was flanked by an expressed sequence tag (EST)—SSR marker, HAU1396, at a distance ranging from 0.1 (F 2 and F 2:3 at Linqing) to 0.9 cM (F 2:3 at Hainan). When analyzed by QTLNetwork 2.2, four of the QTLs (except for qLP-C23-1) were also detected, and only qLP-C7-1 showed significant QE interaction. Two epistasis interactions were identified between genetic background loci, one between loci on Chr.16 and Chr.2 and the other between loci on Chr.16 and Chr.17.
Confirmation of the QTLs in advanced breeding population
One of the challenges faced by MAS during breeding programs is the stability of a QTL across different generations. In order to confirm the stability of the identified QTLs in early generations, we performed an association analysis based on the genotype of the nearest marker to each QTL using F8 advanced population from the same cross. Of the 24 QTLs, 19 showed significant differences between homozygous genotype of LMY22 and LY343, including four QTLs for FL, one for FU, six for FE, three for FS, and five for LP. QTLs for fiber fineness were not found in the advanced population (Table 4).
We identified four QTLs responsible for FL with a significant difference in the advanced generation. The introgression alleles at three QTLs (qFL-C3-1, qFL-C7-1, qFL-C25-1) increased FL by 0.35–1.08 mm, while at qFL-C16-1, the GH allele increased FL by 0.79 mm. All three QTLs responsible for FS were detected in the advanced F8 generation and showed significant difference. The introgression line allele increased FS by 2.71 and 2.73 cN · Tex−1 for the two QTLs (qFS-C7-1, qFS-C7-2) located on Chr.7. Five QTLs responsible for LP identified in F 2 and F 2:3 populations were also detected in the F8 generation. The GH alleles conferred an increase in LP by 1.13 (qLP-C7-1) to 2.03 % (qLP-C15-1).
Discussion
Identification of introgressive exogenous genomic components at the genome-wide level
Reinisch et al. (1994) constructed the first allotetrploid cotton genetic map comprising 705 restriction fragment length polymorphism marker loci assembled into 41 linkage groups and covered 4,675 cM by using an interspecific (G. hirsutum × G. barbadense) mapping population. Rong et al. (2004) subsequently constructed a sequence-tagged site genetic map composed of 2,584 loci at 1.72-cM intervals in 26 linkage groups and covered 4,447.9 cM using the same population. Since then several tetraploid cotton maps have been constructed using various types of molecular markers among which PCR-based markers, such as SSRs, are in common use (Guo et al. 2007; Yu et al. 2012). These interspecific tetraploid maps are close to the overall map length of tetraploid cotton and cover all 26 chromosomes in the allotetraploid cotton genome. Several other relatively well-covered intraspecific G. hirsutum maps have been constructed by SSRs (Zhang et al. 2009, 2012) and other PCR-based markers (Lin et al. 2009). It is worthwhile to note that all the markers exploited for interspecific mapping could be applied to intraspecific mapping in G. hirsutum.
In an attempt to identify introgressive exogenous genomic components at a genome-wide level, we constructed intraspecific maps using the markers previously exploited in interspecific mapping. A total of 18,467 SSR primer pairs, including 4,050 SSRs reported in our previous study (Wang et al. 2011), were applied to screen for polymorphism between the mapping parents LMY22 and LY343. Of the 334 mapping SSR loci, 158 were identified as putative introgression G. barbadense loci. All of the introgression loci distributed on 41 introgression segments that were assigned to 20 chromosomes. The total introgression segments covered 646.91 cM and accounted for about 12.86 % of the tetraploid cotton genome. Identification of genome-wide introgression components by screening SSRs covering most of the tetraploid cotton genome greatly facilitated our analysis of the association between fiber quality QTLs and the G. barbadense introgression chromosomal segments.
Genetic effects of the putative introgression chromosomal segments from G. barbadense on fiber-related traits
Exploitation and utilization of favorable exogenous alleles in related species can facilitate the broadening of the genetic base of popular crop cultivars and cause dramatic improvements in various desirable traits (Tanksley and McCouch 1997; Zamir 2001, 2008). Marker-guided introgression of exogenous genomic components into cultivated crop varieties for potential application in MAS has been reported in several crops, such as tomato (Fernie et al. 2006; Korff et al. 2006, 2008; Zamir 2008; Barone et al. 2009; Chapman et al. 2012), rice (Steele et al. 2006; Xu et al. 2012b), wheat (Pestsova et al. 2006), maize (Zheng 2008), and oat (Yu and Herrmann 2006).
Cultivated G. hirsutum, which dominates the world’s cotton fiber production, lacks a genetic base for fiber quality improvement. However, other species in Gossypium genus, especially the cultivated tetraploid species G. barbadense, provide an ideal gene pool for superior fiber quality (Wendel et al. 2010; Mehboob-ur-Rahman et al. 2012). Numerous QTLs for fiber quality traits have been mapped using molecular markers, but few have been applied in breeding selection since most of the studies selected G. barbadense varieties as one of the mapping parents. Superior fiber quality germplasm lines introgressed by other Gossypium spp. have been used as a parent to map fiber-related QTLs (Shen et al. 2007; Lin et al. 2009; Zhang et al. 2009, 2012; Sun et al. 2011). However, these studies do not provide any evidence of association between the introgressive genetic components and the detected QTLs. Recently, Zhang et al. (2011) identified QTL alleles for improved fiber quality from a wild Hawaiian cotton, G. tomentosum, and Xu et al. (2012a) identified QTLs for FM on Chr.7 from a wild species, G. klotzschianum, that were introgressed into Upland cotton.
In this study, a total of 24 QTLs for fiber quality and LP were identified by WinQTLCart 2.5, of which 20 were located in the introgression segments of G. barbadense. The GB allele contributed to the improvement of only fiber quality traits at eight loci, including three for FL, two for FS, and three for fiber elongation. While for fiber uniformity, fineness, and LP, the favorable alleles were derived from the GH parent. qFL-C7-1 and qFS-C7-1, which could be detected in more than two environments and explained relatively large phenotypic variances, were located in the same introgression chromosomal segment on Chr.7. qFL-C25-1, which was detected in two environments with a large genetic effect, was located in the introgression region of Chr.25. These introgression segments might carry important genes controlling the desirable superior fiber quality. Further studies targeting these regions would be beneficial to further our understanding of the genetic mechanism of superior fiber formation.
QTLs for fiber quality are frequently clustered on a chromosomal region
Classical quantitative genetic studies have revealed that there is a strong correlation among various traits that determine fiber quality (Kloth 1998). In our previous study (Wang et al. 2011), we reported that there is a positive correlation between FL and FU and FS, while there is a strong negative correlation between fiber quality and LP in the F 2 and F 2:3 population from the cross of LMY22 × LY343. Recent QTL mapping studies have shown that QTLs for fiber quality or yield traits are clustered on the same chromosomal region (Chen et al. 2009; Lacape et al. 2010; Sun et al. 2011; Zhang et al. 2012). In this study, QTL-rich regions were found on several chromosomes (four QTLs on Chr.3, five on Chr.7, four on Chr.23; see Fig. 1). Some regions consisted of QTLs for fiber quality (Fig. 1, Chr.3), while other QTL-rich regions were related not only to fiber quality but also to LP (Chr.7, Chr.23). A QTL-rich region on Chr.7 was associated with FL, FS, fiber elongation, and LP, and the direction of genetic effects of QTLs for FL and strength traits was consistent, but the direction was opposite for the two fiber quality traits and LP. These results further explain the positive phenotypic correlation between FL and FS, and the negative correlation between fiber quality and LP. Knowledge of the co-location of various QTLs on a definite chromosomal region would be helpful when the aim is to improve different fiber quality parameters simultaneously.
Highly heritable and stable QTLs could be candidates for MAS in Upland cotton breeding programs
The heritability of fiber quality and yield components is moderate to high, suggesting that these traits could be manipulated by MAS (Paterson et al. 2003). Our study shows that the heritability of QTLs with additive effect for FL, FS, and LP (25.31, 23.2, and 17.62 %, respectively) was higher than that for other fiber properties (8.49, 3.67, and 12.43 % for FU, FM, and FE, respectively) (Table 2). It also shows that the QTLs for these traits had no or only a minor QE interaction effect, indicating that these traits were scarcely affected by environment and MAS would be useful in improving these traits. However, FU and FE were markedly influenced by environments as all of the QTLs for these two traits showed significant QE interaction, especially those for FE whose heritability of QE interaction effect was much larger than that of the additive effect. Although the QTL for FM showed no QE interaction, the heritability of the additive effect was only 3.67 %. Therefore, these QTLs for FU, FE, and FM should be carefully chosen for in MAS manipulation.
The environment-stable QTL is very important for MAS during breeding programs. Five QTLs (qFL-C7-1, qFL-C16-1, qFL-C25-1, qFS-C7-1, and qLP-C17-1) could be detected in more than two datasets with no or only minor QE interaction. These five QTLs could be confirmed in F8 population. The QTL for FS, qFS-C7-1, was detected in all the datasets and could explain large phenotype variance. The region near the qFS-C7-1 locus was also associated with FL, FE, and LP and was located within the introgression G. barbadense chromosomal region. Sun et al. (2011) identified an important QTL responsible for FS on Chr.7 using an introgression line; this QTL could be detected in different generations and different environments. Zhang et al. (2009, 2012) also identified an important region on Chr.7 associated with five fiber quality QTLs using Yumian 1 as mapping parent; this region introgressed with G. barbadense chromosomal fragments, suggesting that this introgression region in Chr.7 (A7) might carry important genes controlling fiber quality traits. Another important QTL for LP (qLP-C17-1) was also detected across all datasets and explained phenotypic variation, ranging from 7.79 to 15.52 %. Since the region near the qLP-C17-1 locus was not associated with fiber quality, stacking this QTL for improving yield trait was not expected to penalize any fiber quality parameters. It should be noted that qLP-C17-1 was tightly linked with an EST-SSR HAU1396 (Fig. 1; Table 1). BLAST results indicate that this EST is orthologous to Arabidopsis REVOLUTA, which is related to the band of interfascicular fiber characteristic of Arabidopsis. These QTLs could be candidates for MAS in an Upland cotton breeding program.
QTLs for fiber quality and LP mapping in non-homologous regions could facilitate pyramiding favorable alleles by MAS for simultaneous improvement of cotton fiber quality and lint yield
Nineteen QTLs detected in our study were found to be responsible for fiber quality and five for LP. These QTLs were mainly located on nine chromosomes, three of which were in the A-subgenome and six in the D-subgenome (Fig. 1; Table 1). The phenotypic variation explained by any individual QTL ranged from 5.29 to 27.09 %, and the number of QTLs detected for each trait was more than two. In analyzing the distribution of these QTLs carefully, we found that the majority of the QTLs for the same traits were mapped in non-homologous regions, except for qFL-C7-1 and qFL-C16-1, which were mapped in the homologous chromosomal pair of Chr.7 (A07) and Chr.16 (D07). This result is similar to that reported previously by Rong et al. (2007) that the A- and D sub-genomes of the tetraploid cotton contributed QTLs for lint fiber development at largely non-homologous locations. This suggests that variations in sub-genome contributions to QTLs may depend on polyploid lineages (Fonceka et al. 2012) and could be explained by high variation in the expression of the A- and D-subgenomes (Adams et al. 2003; Chaudhary et al. 2009; Flagel and Wendel 2010). Classical genetic (May 1999) and recent QTL mapping studies have shown that fiber quality traits are controlled by multiple genes with different genetic effects. As a result, transgressive segregation of phenotypic values of fiber quality and yield is a normal phenomenon in mapping and breeding populations. It is reasonable to assume that the transgressive segregated individuals in the population might contain more favorable alleles than any one of the two parents. Thus, we can pyramid the favorable alleles or QTLs for a definite phenotype when a particular species harbors several QTLs for one trait, which are then mapped in the non-homeologous locations. For example, we can stack qFL-C3-1, qFL-C7-1, qFL-C16-1, and qFL-C25-1 up via MAS for pyramiding the favorable alleles for FL to improve the FL trait in breeding programs. Our study demonstrates that the G. barbadense alleles (qFL-C3-1, qFL-C7-1, and qFL-C25-1) and the G. hirsutum allele (qFL-C16-1) contribute to improvement of FL and yield in the hybrid populations.
Conventional breeding programs based on phenotypic selection have improved fiber quality in Upload cotton to a certain extent. However, the complicated inheritance and unfavorable correlations between fiber quality and lint yield have generally limited breeding efficiency. Combining powerful molecular tools and conventional breeding methods will provide effective approaches to developing cotton cultivars with improved fiber quality. We can pyramid favorable alleles of QTLs for mapping fiber quality and LP in non-homologous regions by MAS, with the aim of simultaneously improving cotton fiber quality and lint yield. However, to reduce the negative effect of the unfavorable correlation of the introgression alleles, we should develop larger segregation populations for fine QTL mapping and for further MAS in breeding programs. Therefore, exploitation of additional markers (new SSRs and SNPs) for mapping the target introgression chromosomal region would enable high-resolution dissection of the QTLs for fiber quality and lint yield performance. This in turn would enhance the possibility of being able to effect simultaneous improvement of both fiber quality and lint yield by MAS in Upland cotton.
Abbreviations
- SSR:
-
Simple sequence repeat
- QTL:
-
Quantitative trait locus
- QE:
-
QTL × environment
- MAS:
-
Marker-assisted selection
- GB:
-
Gossypium barbadense
- GH:
-
Gossypium hirsutum
References
Adams K, Cronn R, Percifield R, Wendel J (2003) Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc Natl Acad Sci USA 100:4649–4654
Barone A, Di Matteo A, Carputo D, Frusciante L (2009) High-throughput genomics enhances tomato breeding efficiency. Curr Genomics 10(1):1–9
Campbell BT, Saha S, Percy R, Frelichowski J, Jenkins JN, Park W, Mayee CD, Gotmare V, Dessauw D, Giband M, Du X, Jia Y, Constable G, Dillon S, Abdurakhmonov IY, Abdukarimov A, Rizaeva SM, Abdullaev A, Barroso PAV, Pádua JG, Hoffmann LV, Podolnaya L (2010) Status of the global cotton germplasm resources. Crop Sci 50(4):1161–1179
Campbell BT, Chee PW, Lubbers E, Bowman DT, Meredith WR, Johnson J, Fraser DE (2011) Genetic improvement of the Pee Dee cotton germplasm collection following seventy years of plant breeding. Crop Sci 51(3):955–968
Chapman NH, Bonnet J, Grivet L, Lynn J, Graham N, Smith R, Sun G, Walley PG, Poole M, Causse M, King GJ, Baxter C, Seymour GB (2012) High-resolution mapping of a fruit firmness-related quantitative trait locus in tomato reveals epistatic interactions associated with a complex combinatorial locus. Plant Physiol 159(4):1644–1657
Chaudhary B, Flagel L, Stupar RM, Udall JA, Verma N, Springer NM, Wendel JF (2009) Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (Gossypium). Genetics 182(2):503–517
Chee P, Draye X, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum and Gossypium barbadense (cotton) by a backcross-self approach: I. Fiber elongation. Theor Appl Genet 111(4):757–763
Chen H, Qian N, Guo W, Song Q, Li B, Deng F, Dong C, Zhang T (2009) Using three overlapped RILs to dissect genetically clustered QTL for fiber strength on Chro.D8 in Upland cotton. Theor Appl Genet 119(4):605–612
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138(3):963–971
Draye X, Chee P, Jiang CX, Decanini L, Delmonte TA, Bredhauer R, Smith CW, Paterson AH (2005) Molecular dissection of interspecific variation between Gossypium hirsutum and G. barbadense (cotton) by a backcross-self approach: II. Fiber fineness. Theor Appl Genet 111(4):764–771
Fernie AR, Tadmor Y, Zamir D (2006) Natural genetic variation for improving crop quality. Curr Opin Plant Biol 9(2):196–202
Flagel L, Wendel J (2010) Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol 186:184–193
Fonceka D, Tossim H-A, Rivallan R, Vignes H, Faye I, Ndoye O, Moretzsohn M, Bertioli D, Glaszmann J-C, Courtois B, Rami J-F (2012) Fostered and left behind alleles in peanut: interspecific QTL mapping reveals footprints of domestication and useful natural variation for breeding. BMC Plant Biol 12(1):26
Guo W, Cai C, Wang C, Han Z, Song X, Wang K, Niu X, Lu K, Shi B (2007) A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium. Genetics 176(1):527–541
Kloth R (1998) Analysis of commonality for traits of cotton fiber. J Cotton Sci 2:17–22
Korff M, Wang H, Léon J, Pillen K (2006) AB-QTL analysis in spring barley: II. Detection of favourable exotic alleles for agronomic traits introgressed from wild barley (H. vulgare ssp. spontaneum). Theor Appl Genet 112(7):1221–1231
Korff M, Wang H, Léon J, Pillen K (2008) AB-QTL analysis in spring barley: III. Identification of exotic alleles for the improvement of malting quality in spring barley (H. vulgare ssp. spontaneum). Mol Breed 21(1):81–93
Lacape JM, Nguyen TB, Thibivilliers S, Bojinov B, Courtois B, Cantrell RG, Burr B, Hau B (2003) A combined RFLP-SSR-AFLP map of tetraploid cotton based on a Gossypium hirsutum x Gossypium barbadense backcross population. Genome 46(4):612–626
Lacape JM, Llewellyn D, Jacobs J, Arioli T, Becker D, Calhoun S, Al-Ghazi Y, Liu S, Palaï O, Georges S (2010) Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum x G. barbadense RIL population. BMC Plant Biol 10(1):132
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newberg LA (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1(2):174–181
Lin Z, Zhang Y, Zhang X, Guo X (2009) A high-density integrative linkage map for Gossypium hirsutum. Euphytica 166(1):35–45
May OL (1999) Genetic variation in fiber quality. Cotton fibers, developmental biology, quality improvement, and textile processing. Food Products Press, New York, pp 183–229
McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M, Morishima H, Kinoshita T (1997) Report on QTL nomenclature. Rice Genet Newsl 14:111–131
Mehboob-ur-Rahman M-u-R, Shaheen T, Tabbasam N, Iqbal M, Ashraf M, Zafar Y, Paterson A (2012) Cotton genetic resources. A review. Agron Sustain Dev 32(2):419–432
Meredith WR (2005) Minimum number of genes controlling cotton fiber strength in a backcross population. Crop Sci 45(3):1114–1119
Montalvo JG Jr (2005) Relationships between micronaire, fineness, and maturity. Part I. Fundamentals. J Cotton Sci 9:81–88
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep 11(2):122–127
Paterson AH, Saranga Y, Menz M, Jiang CX, Wright RJ (2003) QTL analysis of genotype x environment interactions affecting cotton fiber quality. Theor Appl Genet 106(3):384–396
Pestsova EG, Borner A, Roder MS (2006) Development and QTL assessment of Triticum aestivum-Aegilops tauschii introgression lines. Theor Appl Genet 112(4):634–647
Reinisch AJ, Dong JM, Brubaker CL, Stelly DM, Wendel JF, Paterson AH (1994) A detailed RFLP map of cotton, Gossypium hirsutum x Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetics 138(3):829–847
Rong J, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, Delmonte TA, Ding X, Garza JJ, Marler BS, Park C-H, Pierce GJ, Rainey KM, Rastogi VK, Schulze SR, Trolinder NL, Wendel JF, Wilkins TA, Williams-Coplin TD, Wing RA, Wright RJ, Zhao X, Zhu L, Paterson AH (2004) A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166(1):389–417
Rong J, Feltus F, Waghmare V, Pierce G, Chee P, Draye X, Saranga Y, Wright R, Wilkins T, May O, Smith C, Gannaway J, Wendel J, Paterson A (2007) Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176:2577–2588
Shen X, Guo W, Lu Q, Zhu X, Yuan Y, Zhang T (2007) Genetic mapping of quantitative trait loci for fiber quality and yield trait by RIL approach in Upland cotton. Euphytica 155(3):371–380
Steele KA, Price AH, Shashidhar HE, Witcombe JR (2006) Marker-assisted selection to introgress rice QTLs controlling root traits into an Indian upland rice variety. Theor Appl Genet 112(2):208–221
Stelly DM (1993) Interfacing cytogenetics with the cotton genome mapping effort. Pro Beltwide Cotton Conf 3:1545–1550
Su X, Gao G, Shi X, Zhu D (2000) Breeding of a terrestrial long-staple cotton—Luyuan 343. Acta Agric Nucleatae Sinica 14(3):180–183
Sun FD, Zhang JH, Wang SF, Gong WK, Shi YZ, Liu AY, Li JW, Gong JW, Shang HH, Yuan YL (2011) QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol Breed 30(1):1–14
Tang QY, Feng MG (2007) DPS data processing system: experimental design, statistical analysis, and data mining. Science Press, Beijing
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277(5329):1063–1066
Voorrips R (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93(1):77–78
Wang S, Basten CJ, Zeng ZB (2006) Windows QTL Cartographer 2.5 Department of Statistics, North Carolina State University, Raleigh. Available at: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Wang F, Gong Y, Zhang C, Liu G, Wang L, Xu Z, Zhang J (2011) Genetic effects of introgression genomic components from Sea Island cotton (Gossypium barbadense L.) on fiber related traits in upland cotton (G. hirsutum L.). Euphytica 181(1):41–53
Wendel JF, Brubaker CL, Seelanan T (2010) The origin and evolution of Gossypium. In: Stewart JM, Oosterhuis DM, Heitholt JJ, Mauney JR (eds) Physiology of cotton. Springer, Dordrecht, pp 1–18
Xu P, Zhu J, Zhang X, Ni W, Xu Y, Shen X (2012a) Molecular mapping and identification of QTLs for fiber micronaire on chromosome 7 from Gossypium klotzschianum. Acta Agron Sin 38(03):447–453
Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, Li J, He W, Zhang G, Zheng X, Zhang F, Li Y, Yu C, Kristiansen K, Zhang X, Wang J, Wright M, McCouch S, Nielsen R, Wang J, Wang W (2012b) Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotech 30(1):105–111
Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24(5):721–723
Yu J, Herrmann M (2006) Inheritance and mapping of a powdery mildew resistance gene introgressed from Avena macrostachya in cultivated oat. Theor Appl Genet 113(3):429–437
Yu JZ, Kohel RJ, Fang DD, Cho J, Van Deynze A, Ulloa M, Hoffman SM, Pepper AE, Stelly DM, Jenkins JN, Saha S, Kumpatla SP, Shah MR, Hugie WV, Percy RG (2012) A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3 Genes Genomes Genet 2(1):43–58
Zamir D (2001) Improving plant breeding with exotic genetic libraries. Nat Rev Genet 2(12):983–989
Zamir D (2008) Plant breeders go back to nature. Nat Genet 40(3):269–270
Zhang J, Guo W, Zhang T (2002) Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. × Gossypium barbadense L.) with a haploid population. Theor Appl Genet 105(8):1166–1174
Zhang ZS, Hu MC, Zhang J, Liu DJ, Zheng J, Zhang K, Wang W, Wan Q (2009) Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol Breed 24(1):49–61
Zhang Z, Rong J, Waghmare V, Chee P, May OL, Wright R, Gannaway J, Paterson A (2011) QTL alleles for improved fiber quality from a wild Hawaiian cotton. Gossypium tomentosum. Theor Appl Genet 123(7):1075–1088
Zhang K, Zhang J, Ma J, Tang S, Liu D, Teng Z, Liu D, Zhang Z (2012) Genetic mapping and quantitative trait locus analysis of fiber quality traits using a three-parent composite population in upland cotton (Gossypium hirsutum L.). Mol Breed 29(2):335–348
Zheng P (2008) A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat Genet 40:367–372
Acknowledgments
This work was financially supported in part by grants from the National Science Foundation in China (31171598), National Major Project on Transgenic Breeding in China (2013ZX08005-003), The National High-tech Program of China (2012AA101108-04-02), and Agro-Bio Resources Innovation and Utilization Project of Seed-Industrialized Development Program in Shandong Province.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Furong Wang and Zhenzhen Xu contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wang, F., Xu, Z., Sun, R. et al. Genetic dissection of the introgressive genomic components from Gossypium barbadense L. that contribute to improved fiber quality in Gossypium hirsutum L.. Mol Breeding 32, 547–562 (2013). https://doi.org/10.1007/s11032-013-9888-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-013-9888-y