
Abstract
The characterization of aerosol acidity has received increased attention in recent years due to its influence on atmospheric visibility, climate change and human health. Distribution of water soluble inorganic (WSI) ions in 12 different size fractions of aerosols was investigated under two different atmospheric conditions (smoke-haze and non-haze periods) in 2012 using the Micro-Orifice Uniform Deposit Impactor (MOUDI) and nano-MOUDI for the first time in Singapore. To estimate the in situ acidity ([H+]Ins) and in situ aerosol pH (pHIS), the Aerosol Inorganic Model version-IV under deliquescent mode of airborne particles was used at prevailing ambient temperature and relative humidity. The study revealed an increase in the levels of airborne particulate matter (PM) mass and concentrations of WSI ions for all size fractions during the smoke-haze period, which was caused by the trans-boundary transport of biomass burning-impacted air masses from Indonesia. A bimodal distribution was observed for concentrations of SO4 2−, NO3 −, Cl−, K+ and Na+, whereas concentrations of NH4 +, Ca2+ and Mg2+ showed a single mode distribution. The concentration of WSI ions in PM1.8 during the smoke-haze period increased by 3.8 (for SO4 2−) to 10.5 (for K+) times more than those observed during the non-haze period. The pHIS were observed to be lower during the smoke-haze period than that during the non-haze period for all size fractions of PM, indicating that atmospheric aerosols were more acidic due to the influence of biomass burning emissions. The particles in the accumulation mode were more acidic than those in the coarse mode.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Acidity is an important property of airborne particulate matter (PM) that plays a significant role in atmospheric chemistry (Pathak et al. 2004; Behera et al. 2013), in human health and ecological systems (Holma 1985; Raizenne et al. 1996; Rengarajan et al. 2011) and also in ocean biogeochemistry (Jickells et al. 2005; Nenes et al. 2011). Aerosol acidity can influence the atmospheric behavior of gaseous pollutants [e.g., sulfur dioxide (SO2), nitrous acid (HNO2), nitric acid (HNO3), hydrochloric acid (HCl), volatile organic compounds (VOCs)] and the partitioning of volatile and semi-volatile compounds between the gas and particulate phases (Chang et al. 2008; Hatch and Grassian 2008; Zhou et al. 2012). In particular, aerosol acidity can catalyze atmospheric heterogeneous reactions to enhance the formation of secondary inorganic aerosol (SIA) and secondary organic aerosol (SOA) (Surratt et al. 2007; Cao and Jang 2009; Manktelow et al. 2010). As acidic aerosols are deliquescent in nature, it is important to estimate in situ aerosol pH (pHIS), which can be defined as the pH of the aqueous phase aerosols that influence the aerosol phase chemistry and uptake of gaseous species by airborne particles (Jang et al. 2002; Zhang et al. 2007). When the acidic gases (H2SO4, HNO3 and HCl) are partially neutralized by the only alkaline gas in the atmosphere, ammonia (NH3), the resulting nature of the aerosol turns out to be acidic (Balasubramanian et al. 2003; Lin et al. 2007).
SIA is generally represented by particulate-bound sulfate (SO4 2−), nitrate (NO3 −), ammonium (NH4 +) and chloride (Cl−), and can contribute significantly to the PM mass in the atmosphere (Behera and Sharma 2010; Weijers et al. 2011). For example, SIA can account for 20–50 % of the mass of PM2.5 (PM with aerodynamic diameter (AED) ≤ 2.5 μm) (Lin and Cheng 2007; Tsai et al. 2013). In recent years, the epidemiological studies reported that ultrafine particles (UFPs; PM0.1) have more adverse health effects than coarse particles of the same mass, as UFPs have the ability to penetrate easily into the alveolar region of human lungs due to a higher number concentration and larger specific surface area (Katsouyanni et al. 2001; Kittelson 1998; Pope and Dockery 2006; Leiva et al. 2013). Therefore, it is important to characterize size-resolved particulate inorganic ions to assess their in situ acidic properties.
In atmospheric chemistry, strong acidity, ion-balanced acidity ([H+]Total) and in situ acidity ([H+]Ins) are the three parameters used to express aerosol acidity (Xue et al. 2011; Zhou et al. 2012). While the strong acidity is calculated from the laboratory results obtained from the aqueous extracts of PM, [H+]Total and [H+]Ins are estimated from the measured ion concentrations in the liquid phase of aerosols (Pathak et al. 2003; Yao et al. 2007; Zhang et al. 2007). As the estimation of [H+]Ins includes prevailing meteorological factors, it is thus a better indicator of the actual particle acidity. Therefore, [H+]Ins is preferred to [H+]Total in expressing the acidity of aerosol (Xue et al. 2011; He et al. 2012; Zhou et al. 2012; Behera et al. 2013). During the long-range transport of air masses, primary aerosol particles can undergo interactions with primary and secondary precursor gases (e.g., SO2, H2SO4, NH3, N2O5 and HNO3), resulting in significant variations of their chemical composition, size distributions and acidic characteristics (Song and Carmichael 1999; Ma et al. 2003).
Southeast Asia (SEA) experiences regional smoke-haze episodes caused by uncontrolled forest and peat fires in Indonesia, frequently. These haze episodes carry primary aerosol particles of biomass burning origin as well as SIA and SOA (Balasubramanian et al. 2003; See et al. 2006; Betha et al. 2013; Pavagadhi et al. 2013; Reid et al. 2013; Betha et al. 2014). However, the acidity of size-fractionated aerosols remains poorly unknown despite its importance in various environmental issues and human health effects. Therefore, it is essential to characterize water soluble inorganic (WSI) components (e.g., SO4 2−, NO3 −, Cl−, NH4 +, and Na+) of size-resolved aerosols and to gain insights into their acidic characteristics. Smoke-haze episodes occur not only in SEA, but also in the mega cities of China, India, USA, South America, contributing to significant effects on atmospheric visibility, cloud formation, public health and global climate (e.g., Menon et al. 2002; Nel 2005; Tan et al. 2009).
We carried out a comprehensive field study to characterize WSI ions in size-resolved aerosols (10 nm–10 μm) from September through November, 2012 under two different atmospheric conditions (smoke-haze and non-haze periods) in a tropical urban atmosphere (Singapore: latitude 1°17′N and longitude 103°50′E; Fig. 1) of SEA. This work represents the first study of its kind that investigates the acidic characteristics of size-resolved aerosols, influenced by biomass burning-impacted air masses, using the latest thermodynamic model (AIM-IV). Specifically, there are four major components in this article: (1) measurement of WSI components (NH4 +, Na+, K+, Ca2+, Mg2+, Cl−, NO3 − and SO4 2−) of size-resolved aerosols, (2) assessment of the variation of concentrations of these WSI components between smoke-haze and non-haze periods, (3) estimation of size-resolved aerosol acidity in terms of [H+]Ins and pHIS using the AIM-IV model, and (4) examination of the size-resolved [H+]Ins and pHIS during smoke-haze and non-haze periods.

A map showing the study area in Singapore and its neighboring regions in SEA
Methodology
Study area and sampling campaign
Singapore, an urbanized developed country, has a very high population density (~7,700 population/km2) in Southeast Asia (http://www.singstat.gov.sg). The important sources of local air pollution in Singapore are industries (refinery, chemical industries and power plants etc.) and transportation (roads and ships). Singapore has the typical climate of a tropical environment with two monsoon seasons (northeast monsoon and southwest monsoon). Occasionally, Singapore’s air quality is affected by regional smoke-haze episodes, caused by trans-boundary movement of biomass burning-impacted air masses from Indonesia during the southwest monsoon season (July through October) (See et al. 2006; Reid et al. 2013). During the southwest monsoon season in 2012, the land clearing practices triggered forest and peat fires in Indonesia. With the prevailing south-westerly and southerly wind directions, the smoke haze was spread over SEA including Singapore and lasted for a few weeks during September–October, 2012.
The field study was conducted at the Atmospheric Research Station (latitude 1°18′N and longitude of 103°66′E) in the National University of Singapore (NUS), which is 67 m above sea level (Fig. 1). A detailed description of the characteristics of the study area and the sampling site is provided elsewhere (Balasubramanian et al. 2003; Betha et al. 2014). In brief, the usual air quality at the sampling site is mainly influenced by local traffic from the two roads (within 200 m), nearby industrial activities and a costal port 1 km away.
MOUDI (Micro-Orifice Uniform Deposition Impactor) (Model 100R, MSP Corp, USA) and Nano-MOUDI (Nano-Micro-Orifice Uniform Deposition Impactor) (Model 115, MSP Corp, USA) samplers were used for the collection of size-segregated PM samples. The size ranges of airborne particles were as follows: 5.6–10, 3.2–5.6, 1.8–3.2, 1.0–1.8, 0.56–1.0, 0.32–0.56, 0.18–0.32, 0.10–0.18, 0.056–0.100, 0.032–0.056, 0.018–0.032 and 0.010–0.018 μm. The air flow rates maintained for the sampling were 30 liter per minute (LPM) and 10 LPM for MOUDI and Nano-MOUDI, respectively. Polytetrafluoroethylene (PTFE) filters were used for the PM sampling, which were pre-conditioned and post-conditioned at T = 22 °C and RH = 33 %. The gravimetric method was applied for estimation of the PM mass concentration (μg m−3) by dividing the mass of particles (μg) collected onto filters by the total volume of air sampled (m3). A total of 132 PM samples (five sets of smoke haze and six sets of non-haze aerosols) were collected over 48 h duration for gravimetric and chemical analyses. Field and laboratory blanks (one blank in 10 sampling filters) were collected for QA-QC checks. We considered PM1.8 (0.01 μm < d p < 1.8 μm) and PM1.8–10 as fine and coarse particles, respectively, as the sampler MOUDI 110 does not have a 2.5 μm cut-point (Zhuang et al. 1999; Yao et al. 2003).
The interpretation of the PM data in this paper has been done on the basis of six major PM size categories; i.e., PM10 (total suspended PM), coarse (PM1.8–10), fine (PM1.8), accumulation mode (PM0.1–1, 0.1 μm < d p < 1.0 μm), ultrafine (PM0.01–0.1, 0.01 μm < d p < 0.10 μm), and nano particles (PM0.01–0.056, 0.01 μm < d p < 0.056 μm). The results reported in this paper are blank subtracted values.
During the measurement campaign, meteorological parameters (temperature, relative humidity (RH), wind speed, wind direction and rainfall) were acquired every 5 min by an automated meteorological station located at the sampling site. The automated meteorological station consisted of sensors (Vaisala, Model CS 500) for measurement of ambient T and RH, and anemometer and wind vane (RM Young, Model 03001) for measurements of wind speed and wind direction. During the smoke-haze period, the average meteorological parameters observed were as follows: T (28 °C), RH (76 %), wind speed (2.2 m s−1), wind direction (199°), rainfall (0 mm), pollution standard index (an indicator of ambient air quality), PSI (58), and PM2.5 concentration (56 μg m−3). The average meteorological parameters observed during the non-haze period were as follows: T (27 °C), RH (78 %), wind speed (2.1 m s−1), wind direction (99°), rainfall (0.04 mm), PSI (22), and PM2.5 concentration (21 μg m−3). The PSI and PM2.5 concentration data were obtained from the website of National Environmental Agency (NEA), Singapore (http://www.nea.gov.sg/psi/).
Sample preparation and chemical analysis
For the sample preparation before chemical analysis, half of each filter sample and blank filter was cut and extracted by 15 mL deionized water (Millipore, 18.2 MΩ) using an ultrasonic bath for 1 h at T about 60 °C. The extracted samples were filtered using 0.22 μm Teflon filters to remove insoluble matters (Karthikeyan and Balasubramanian 2006; Behera and Sharma 2010). For the analysis of cation concentrations (NH4 +, Na+, K+, Mg2+ and Ca2+) and anion concentrations (Cl−, NO3 − and SO4 2−), ion chromatography (Dionex, ICS-3000) was used. The separation and analysis of anions were achieved using IonPac AS 11HC (4 × 250 mm) analytical column, 11 HC guard column with a 10 μL sample loop, and a self-regenerating CSRS-300 suppressor, and an eluent from the solution of 2.7 mM Na2CO3/0.3 mM NaHCO3 at a flow rate of 1.5 mL/min. The cations were analyzed using IonPac CS 12A (4 × 250 mm) analytical column, CG 12A guard column with a 50 μL sample loop, a self-regenerating suppressor ultra, and an eluent from the solution of 20 mM methanesulfonic acid at a flow rate of 1 mL/min. In this analytical method, the detection limits ranged from 0.004 μg m−3 (K+) to 0.05 μg m−3 (NO3 −) and the ion recoveries ranged from 94 % (Na+) to 106 % (NO3 −). The reported concentrations of Na+, SO4 2–, K+ and Ca2+ are of non-sea-salt components estimated using the empirical formula given in earlier studies (Kennish 1994; Balasubramanian et al. 2003). There was a modification in the formula by assuming that half of the soluble Na+ in particulate samples came from sea salts.
Thermodynamic modeling and estimation of aerosol acidity
In the first step for assessment of acidic nature of aerosols, the total acidity ([H+]total) in molar concentration at each size fraction was estimated from the experimental data of ions, as given by Eq. (1):
where [SO4 2−], [NO3 −], [Cl−], [NH4 +] and [Na+] were taken from the field observations in the unit of molar concentration at each aerosol size fraction.
It should be noted that the estimation of [H+]Total ignores organic anionic species (e.g., formate, acetate, oxalate) that may be neutralized by Na+ and NH4 +. However, this approximation has been widely used for estimation of [H+]Total (Ziemba et al. 2007; Xue et al. 2011; Behera et al. 2013). There are several thermodynamic models that can estimate [H+]Ins, for example, MARS (Saxena et al. 1993), SCAPE2 (Meng et al. 1995), ISORROPIA (Nenes et al. 1998), Aerosol Inorganic Model (AIM) (Clegg et al. 1998a, b). Out of the three versions of the AIM models (AIM-II, AIM-III and AIM-IV), AIM-IV model is considered to be realistic as it includes variable meteorological parameters in the atmosphere and the simulation covers more components of cations and anions in comparison with other versions of AIM model (AIM-II and AIM-III).
To be specific, AIM-IV considers the state of thermodynamic equilibrium system of H+–NH4 +–Na+–SO4 2−–NO3 −–Cl−–H2O with temperature (T) ranging from 263.15 to 330 K and prevailing ambient RH in the atmosphere (Friese and Ebel 2010; Behera et al. 2013). AIM-IV model predicts thermodynamic properties and concentrations of aqueous phase ions under equilibrium conditions. The development of AIM model involves derivation of several mathematical equations on chemical thermodynamics in a complex system, and the model solved those equations by computer programming through FORTRAN. The validation of predicted aqueous phase thermodynamic parameters and concentrations of aqueous phase ionic species has been reported by Clegg and co-workers (e.g., Clegg et al. 1998a, b; Friese and Ebel 2010). For example, AIM model outputs for prediction of water activities (which are equal to equilibrium RH) were validated using an extensive experimental data set derived from aqueous solutions and mixtures. Several research groups (e.g., Yao et al. 2006, 2007; Zhang et al. 2007; Cheng et al. 2011; Xue et al. 2011; Zhou et al. 2012) have used AIM models in the past to predict thermodynamic properties of aerosol ionic species in aqueous phase and to estimate the in situ aerosol pH, pHIS.
In view of the above considerations under variable ambient temperatures and RH, we used AIM-IV model (http://www.aim.env.uea.ac.uk/aim) in this study to estimate [H+]Ins, pHIS and the deliquescent properties of aerosols. In the modeling exercise, the following input parameters were used: T, RH, molar concentrations of H+ ([H+]Total: Eq. (1)), NH4 +, Na+, SO4 2−, NO3 − and Cl−. A deliquescent mode of airborne particles was assumed to exist as an aqueous solution at ambient RH. The outputs of the model were as follows: equilibrium composition of the aerosols, including solid and liquid partitioning per m3 of air. When the molar concentrations of cations (NH4 + and Na+) participating in the formation of secondary inorganic aerosols (SIA) exceed those of anions (SO4 2−, NO3 − and Cl−), the concentration of [H+]Total would be negative indicating a complete neutralization of anions by cations. Under these cases, data sets were discarded for the acidity analysis as part of the quality control protocol adopted in the study.
In situ pH, pHIS of size-fractionated aerosols was estimated using Eq. (2):
where \(a_{{{\text{H}}^{ + } }}\) is the activity of aqueous phase H+ in the particle in the unit of mol L−1, \(\gamma_{{{\text{H}}^{ + } }}\) is the activity coefficient of aqueous phase H+, [H+]Ins is the concentration of free aqueous phase H+ in the unit of mol per m−3 of air, ρ is the aerosol aqueous phase density in g cm−3, and m is the air concentration in the aqueous phase in the unit of g m−3. The parameters, ρ,\(\gamma_{{{\text{H}}^{ + } }}\), [H+]Ins and the concentration of aerosol water content ([H2O]), were derived from AIM-IV model. The parameter, m, was taken as the sum of concentrations of all ionic solutes in the aqueous phase plus [H2O].
The values of [H+]Ins predicted by AIM-IV depend on many factors including, aqueous phase concentrations of respective ionic species, activity coefficients, and partial pressure in the thermodynamic equilibrium (Friese and Ebel 2010; Behera et al. 2013). When ambient RH is lower than the deliquescence point (DRH), the particle is considered by AIM to exist as a pure solid phase and thus there was no output for the [H+]Ins of aerosol (Cheng et al. 2011; Zhou et al. 2012; Behera et al. 2013). In addition, the model cannot predict the [H+]Ins for fully neutralized aerosols (i.e., ([H+]Total ≥ 1).
In this study, we used [H+]Ins and pHIS in the deliquescent particles as indicators of aerosol acidity. In addition, ammonium-to-sulfate ratio, i.e. [NH4 +]/[SO4 2−] by mole, of ambient aerosols was also used as an indicator of the extent of neutralization of acidic sulfate in the atmosphere, as the concentrations of these two species can be easily obtained from the field measurements. This ratio is an overall parameter indicating the basic and acidic contents of the particles, but does not describe a complete atmospheric loading of aerosol acidity (He et al. 2012; Zhou et al. 2012).
The uncertainty associated with measurements of secondary inorganic aerosols (SIA) used in the calculation was estimated based on the propagation of errors due to filter sampling of ambient aerosols and subsequent chemical analysis by ion chromatography. Overall, the measurement uncertainties of SIA were determined as less than 6 % for particles of 0.18 μm < d p < 10.0 μm and less than 9 % for particles of 0.01 μm < d p < 0.18 μm. As the estimation of [H+]total was involved with the addition and subtraction of five parameters (SIA), absolute uncertainties of [SO4 2−], [NO3 −], [Cl−], [NH4 +] and [Na+] were used for estimating a propagated uncertainty. The propagated uncertainties for estimation of [H+]total were found to be less than 13 % for particles of 0.18 μm < d p < 10.0 μm and less than 20 % for particles of 0.01 μm < d p < 0.18 μm. As the prediction of [H+]Ins in AIM-IV model required the input parameters of [H+]total, [SO4 2−], [NO3 −], [Cl−], [NH4 +] and [Na+], the propagated uncertainty for prediction of [H+]Ins was estimated from the absolute uncertainties of respective empirical parameters on the basis of addition/subtraction operations. The propagated uncertainties for prediction of [H+]Ins were found to be less than 19 % for particles of 0.18 μm < d p < 10.0 μm and less than 29 % for particles of 0.01 μm < d p < 0.18 μm. The estimation of pHIS involved a logarithm function of four variables (\(\gamma_{{{\text{H}}^{ + } }}\), [H+]Ins, ρ and m). The propagated uncertainties of pHIS were estimated based on multiplication and division operations. The uncertainties of predicted values of \(\gamma_{{{\text{H}}^{ + } }}\), ρ and m from AIM-IV model were assumed to be the same as those of [H+]Ins. Hence, the propagated uncertainties of pHIS estimation were found to be less than 18 % for particles of 0.18 μm < d p < 10.0 μm and less than 25 % for particles of 0.01 μm < d p < 0.18 μm.
Results and discussions
Overall results on mass of aerosols
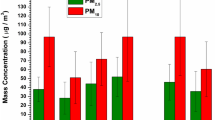
The mass concentrations of size-fractionated PM and the overall particle-size distributions are shown in Fig. 2 for each sampling set during the measurement campaign. The levels of PM10 concentrations ranged from 54.1 to 83.2 and 19.2 to 26.3 μg m−3 while those of PM1.8 concentrations varied from 43.1 to 59.4 and 12.9 to 18.1 μg m−3 during smoke-haze and non-haze days, respectively. The PM1.8 concentrations exceeded the 24-h WHO guideline of 25 μg m−3 for PM2.5 during the smoke-haze period. Overall, the observed concentrations of PM10 and PM1.8 during the smoke-haze period were 2–4 times higher than those during the non-haze period.

The mass concentrations of size-fractionated aerosols during both smoke-haze and non-haze periods
The ratios of PM1.8/PM10 observed during both these time periods were examined to assess the relative contribution of fine (aerodynamic diameter (d p) ≤ 1.8 μm) versus coarse particles (d p ≥ 1.8 μm) to the total PM10 mass. The average concentration ratio of PM1.8/PM10 during the smoke-haze period was observed to be 0.71 while it was 0.63 during the non-haze period. The trends in variations of these ratios under two different atmospheric conditions are similar to the studies that were conducted at other parts of the world (Guangzhou, Shanghai and Beijing) (Tan et al. 2009; Du et al. 2011; Sun et al. 2013). Among the different fractions of PM1.8, the mass concentration of particles in the size range of 0.1–1.0 μm (PM0.1–1; the accumulation mode) increased in particular by a factor of 3.6 during the smoke-haze period compared to that during the non-haze period. The particles in the accumulation mode are normally formed by nucleation of precursor gases along with primary emissions (Kerminen et al. 1995, 2001). Therefore, the significant increase in the proportion of particles in the accumulation mode during the smoke-haze period is indicative of the influence of the long-range transport of biomass smoke aerosols and/or formation of new particles in the atmosphere under favorable atmospheric conditions on the particulate pollution in Singapore.
Figure 3 shows size distributions of PM mass concentrations observed during both smoke-haze and non-haze periods. The coarse mode peak appeared at 1.8–3.2 μm during both time periods. However, the fine mode peak appeared at 0.56–1.0 μm during the smoke-haze period while it was at 0.18–0.32 μm during the non-haze period. The reason for the shift in mass median aerodynamic diameter to larger particle size during the smoke-haze period (from 0.87 to 0.95 μm) could be due to the coagulation of freshly emitted smoke haze particles during their long-range transport (See et al. 2006; Betha et al. 2013).

Particle mass size distributions during smoke-haze and non-haze periods
Size distributions of particulate WSI ions
The ratios of concentrations of WSI ions (NH4 +, Na+, K+, Ca2+, Mg2+, SO4 2−, NO3 − and Cl−) during the smoke-haze period to the non-haze period in different size-modes are presented in Table 1. For all size-modes, it was observed that the contributions of WSI ions to the PM mass were significantly higher during the smoke-haze period than those during the non-haze period. For example, the contribution of WSI ions to the PM1.8 mass ranged from 26 to 51 % (average 38.3 ± 9.7 %) during the smoke-haze period and from 14 to 29 % (average 21.1 ± 4.8 %) during the non-haze period. The observation on the contribution of WSI ions to the PM1.8 mass from this study is comparable to a previous study conducted in Beijing, China (Wang et al. 2006) that showed an increase in the contribution of WSI ions to the PM2.5 mass from 32 to 53 % between the non-haze and smoke-haze periods. A similar observation was also made in a field study conducted in Guangzhou, China (Tan et al. 2009); there was an increase from 24.7 to 44.9 % in the contribution of WSI ions to the PM2.5 mass from the non-haze to the smoke-haze period.
Among all WSI ions, SO4 2−, NO3 − and NH4 + were the major PM constituents that accounted for an average of 84 % of all WSI ions in PM1.8 (Table S1: supplementary section). The ratios of average concentrations between the smoke-haze and non-haze periods were more than 3 for SO4 2−, NO3 − and NH4 + in PM1.8, suggesting that these ions might have played a significant role in the formation of smoke haze. SO4 2− concentrations were the highest among all WSI ions in all modes during both smoke-haze and non-haze periods. However, the ratio of SO4 2−concentration during the smoke-haze episode to that during the non-haze period was lower than that of NO3 − (3.8 for SO4 2− and 7.4 for NO3 −). The reason for such a significant increase in the NO3 − concentration could be due to higher emission of NOx than SO2 from biomass burning (Balasubramanian et al. 2003; Behera and Balasubramanian 2014). In addition, the atmospheric concentration of the key photochemical oxidant, H2O2, could be greatly reduced under high NOx conditions, and further reduced the possibility of SO4 2− formation via its multiphase reactions (Poppe et al. 1993; Behera and Sharma 2012; Sun et al. 2013).
The trends observed in the concentrations of major WSI ions in PM1.8 are summarized as: SO4 2− > NO3 − > NH4 + > K+ > Cl− > Ca2+ during the smoke-haze period, while it was SO4 2− > NO3 − > NH4 + > Ca2+ > K+ > Cl− during the non-haze period. This difference in the trend clearly indicates that higher concentrations of K+ were emitted from biomass burning during the smoke-haze period. Another notable observation was that the concentration ratio of fine to coarse mode of NO3 − was much lower than those of SO4 2− and NH4 + (2.3 for NO3 −, 4.7 for SO4 2− and 3.4 for NH4 +). This trend indicated that SO4 2− and NH4 + particles were more populated in the fine mode compared to NO3 − particles. Cl−, Na+ and K+ also showed similar behaviors to that of SO4 2− and NH4 +. In the case of Ca2+ and Mg2+, the concentrations in the coarse mode were dominant over the fine mode. Overall, it can be inferred that the major ions, SO4 2− and NH4 +, were mostly formed with fine mode dominance, whereas NO3 − was formed in both fine and coarse modes. The results were similar to those from studies conducted elsewhere (Pierson and Brachaczek 1988; Zhuang et al. 1999).
The size distributions of the mass concentrations of WSI ions are shown in Fig. 4 during both smoke-haze and non-haze periods. The concentrations of K+, Cl−, NO3 − and SO4 2− showed bimodal distributions in a manner similar to that of PM mass. The bimodal distributions for the concentrations of these ions were also reported by several other studies (Yao et al. 2003; Wang et al. 2006; Tsai et al. 2012, 2013; Sun et al. 2013). The fine mode peak of these ions appeared at 0.56–1.0 μm during the smoke-haze period and at 0.18–0.32 μm during the non-haze period. However, the coarse mode peak occurred with the same size range at 1.8–3.2 μm during both smoke-haze and non-haze periods. In the case of Na+ and NH4 +, significant peaks were observed in the fine mode at 0.56–1.0 μm and 0.18–0.32 μm during the smoke-haze and non-haze periods, respectively. Insignificant peaks were also observed in the coarse mode for both Na+ and NH4 +. The significant peak of NH4 + in the fine mode could be due to its formation mechanism in the accumulation mode during the gas-to-particle conversion process involving the precursors gases (Zhuang et al. 1999; Yu et al. 2011). Ca2+ and Mg2+ exhibited single peaks in the coarse mode at 1.8–3.2 μm during both smoke-haze and non-haze periods. This can be attributed to re-suspension of road dust and eroded dust from soils and crustal rocks (Sun et al. 2006; Wang et al. 2006; Wall et al. 1988).

Mass-size distributions of particulate WSI ions during smoke-haze and non-haze period
The fine mode peaks of NO3 − and SO4 2− at 0.18–0.32 μm during the non-haze period can be attributed to the transformation of SO2 and NOx via heterogeneous reactions (Yao et al. 2003; Sun et al. 2013). However, the peaks at 0.56–1.0 μm during the smoke-haze period can be attributed to their formation through in-cloud, or aqueous phase chemical reactions (Meng and Seinfeld 1994; Kerminen and Wexler 1995). The dominance of SO4 2− and NH4 + concentrations in the fine mode with the particle size <1.8 μm could be due to kinetics of their formations. Behera and Sharma (2012) observed that the tendency of NH3 to undergo reactions with H2SO4 to form (NH4)2SO4 was more than that with HNO3 to form NH4NO3 due to the higher reaction rate of formation. The concentration of NO3 − in the coarse mode could be due to the possibility of heterogeneous reactions of HNO3 vapor with NaCl in sea salts, or crustal materials resulting in the formation of NaNO3 and Ca(NO3)2 (Pierson and Brachaczek 1988; Zhuang et al. 1999). Previous studies also confirmed that the NO3 − in the fine mode existed in the form of NH4NO3 and NO3 − in the coarse mode in the form of NH4NO3, as well as sodium nitrate (NaNO3) (Lee et al. 1999; Behera and Sharma 2010). The fine mode peak of Cl− at 0.18–0.32 μm could be due to the formation of NH4Cl, whereas the coarse mode peak at 1.8–3.2 μm could be due to the influence of sea salt (Tsai et al. 2011).
The occurrence of peak of K+ at 0.56–1.0 μm during the smoke-haze period could be due to its direct emissions from biomass burning sources in Indonesia. Ryu et al. (2004) also observed an increase in the levels of K+ during the biomass burning period. Andreae et al. (1998) reported that K+ is the major electrolyte in cell cytoplasm and that it can be released in large amounts as K-rich PM in the submicron size fraction during the burning of plants. Therefore, K+ is a useful tracer for pyrogenic aerosols derived from vegetation fires (See et al. 2006; Tsai et al. 2012; Betha et al. 2013).
Influence of biomass burning on the characteristics of WSI ions
To assess the influence of biomass burning emissions on the chemical characteristics of PM, we estimated correlation coefficients between K+ and other WSI ions during both smoke-haze and non-haze periods. K+ has been used as a tracer of biomass burning and PM10 was chosen in this study, as a bimodal size distribution was observed for K+ (described in the previous section). Sigma Plot 12.5 English was used to construct a correlation matrix between WSI ions and the PM10 mass. Table 2 shows the results of correlation analysis. K+ showed a significant correlation with the PM10 mass and major ions (SO4 2−, NO3 − and NH4 +) during the smoke-haze period, suggesting a strong influence of the trans-boundary transport of biomass burning emissions from Indonesia on the chemical composition of aerosols collected at the receptor.
To confirm the origin of air masses received at the receptor, a back-trajectory analysis was performed during both smoke-haze and non-haze periods (Fig. 5). For this purpose, the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model was employed by using the archived data of the Global Data Assimilation System of NOAA (National Oceanic and Atmospheric Administration) (Draxler and Rolph 2013). The simulation was done for duration of 5 days starting at 14:00 h (local time) and at three different altitudes (500, 1,000 and 1,500 m above the ground level) for each sampling day. The results from HYSPLIT showed that the biomass burning-impacted air masses that originated from biomass fires (hotspots) in Indonesia reached Singapore during the entire smoke-haze period. However, the oceanic (clean) air masses derived from the northeast direction were intermixed with the local urban emissions during the non-haze period. Thus, the increased PM loading with enrichment in the concentration of WSI ions in the atmosphere during the smoke haze period was due to combination of biomass burning-impacted air masses from Indonesia and local urban emissions within Singapore.

Backward trajectory of air mass during and biomass burning hotspots on two representative days of smoke-haze and non-haze periods from the outputs of HYSPLIT modeling (NOAA, USA)
To examine the formation of smoke haze in Singapore, the sources responsible for it and the environmental implication of the haze, we conducted a series of field studies to investigate the physical, chemical and optical characteristics of PM (e.g., See et al. 2006, 2007; Betha et al. 2013; Engling et al. 2014). Some important findings derived from our previous studies on examining the sources of smoke haze in Singapore are described below.
The details of peat fire emissions and their impact on the air quality of downwind sites were reported in See et al. (2007). In this study, ambient PM samples were collected simultaneously at three different sites located at varying distances from the peat fires, representing a rural site affected by the local peat combustion, a semirural site, and an urban site situated downwind of the peat fires. From the results of source apportionment of PM, it was confirmed that peat fires had significant contribution to PM mass at both semi-urban and urban sites located downwind of the hotspots.
In another study (See et al. 2006), PM samples were collected in Singapore from March 2001 to March 2002 under varying weather conditions (haze and clear) to investigate physical, chemical and optical characteristics. This study revealed that the average mass concentrations of PM2.5 and most chemical components increased on hazy days. The long-range transport of biomass burning-impacted air masses from Indonesia to Singapore under favorable conditions was confirmed by the backward air trajectories. Scanning electron microscope (SEM) images of “aged” (long-range transported) particles and “freshly emitted” particles of local origin collected during smoke-haze and the non-haze periods were examined to assess their surface morphology and atmospheric behavior (See et al. 2006). From the SEM images, it was clear that aged aerosol particles were formed through hygroscopic growth and agglomeration during the long-range transport.
In our most recent study (Engling et al. 2014), the contribution of long-range transport of biomass-impacted smoke haze to the local air quality in Singapore was investigated through a combination of source apportionment techniques and backward air trajectory analyses during two different conditions (haze and non-haze). This study revealed an enhancement in concentrations of organic molecular tracers of biomass burning (such as levoglucosan) by more than two orders of magnitude and the diagnostic ratios of individual organic compounds indicated that biomass burning emissions and their subsequent transport caused a regional smoke haze. The source apportionment results also showed that the contribution of peat fires was as much as 75 % to the PM mass during the smoke-haze period. From the findings of all these studies, it can be confirmed that the occurrence of smoke haze in Singapore is due to mixing of long-range transported biomass and peat burning emissions from Indonesia with the local anthropogenic emissions.
Size distributed in situ aerosol acidity and pH
The size distributions of the molar ratios of [NH4 +]/[SO4 2−], pHIS, [H]Total and [H]Ins estimated during both smoke-haze and non-haze periods are shown in Fig. 6. To examine the difference in the values of these parameters between smoke-haze and non-haze periods, a paired t test was performed using Sigma Plot 12.5 English. From the results of the t test, it was found that all parameters except [NH4 +]/[SO4 2−] showed a significant difference between the two sets of data at 95 % level of confidence for all size fractions of PM. The levels of pHIS varied from 0.08 at 0.01–0.018 μm to 0.32 at 0.056–0.1 μm during the smoke-haze period and from 0.16 at 0.01–0.018 μm to 0.59 at 0.056–0.1 μm during the non-haze period. The average of pHIS at all size fractions was 0.21 during the smoke-haze period and 0.36 during the non-haze period. Thus, aerosols during the smoke-haze period were more acidic than that during the non-haze period.

Size distributions of acidity characteristics during smoke-haze and non-haze period
The levels of [H]Total ranged from 0.06 nmol m−3 at 0.032–0.056 μm to 7.61 nmol m−3 at 0.56–1.0 μm during the smoke-haze period and from 0.05 nmol m−3 at 0.032–0.056 μm to 3.32 nmol m−3 at 0.56–1.0 μm during the non-haze period. The trends in the levels of [H]Ins were similar to those of [H]Total that varied from 0.02 nmol m−3 at 0.032–0.056 μm to 1.86 nmol m−3 at 0.56–1.0 μm during the smoke-haze period and from 0.01 nmol m−3 at 0.032–0.056 μm to 1.02 nmol m−3 at 0.56–1.0 μm during the non-haze period. As it can be inferred from Fig. 6, the difference between [H+]Total and [H+]Ins indicated that the particle-size-dependent [H+]Total (the aerosol acidity based on ambient SO4 2−, NO3 −, Cl−, NH4 + and Na+) is different from that of [H+]Ins. The difference between the pattern of [H+]Total and that of [H+]Ins can be attributed to the influence of other in situ parameters on the levels of [H+]Ins, such as aerosol water content, and hygroscopic properties of SIA components.
The model outputs revealed that most of the SO4 2− existed as bisulfate (HSO4 −) in the aqueous phase equilibrium of aerosols. To examine the role of HSO4 − in [H+]Ins, a statistical correlation between [HSO4 −] and [H+]Ins was studies for all size fractions. It was observed that a strong and significant correlation existed between [HSO4 −] and [H+]Ins (R 2 = 0.74 with P < 0.001 during smoke-haze and R 2 = 0.69 with P < 0.001 during non-haze period). Therefore, it appears that HSO4 − makes an important contribution to the aerosol acidity by releasing more free H+ ions to the aqueous phase. The hydrolysis of HSO4 − depends on aerosol water contents and the state of the aqueous phase equilibrium system of aerosols. However, high water contents can sometimes dilute the proton concentrations in acidic aerosols, leading to the decrease of acidity in the aqueous phase (Cheng et al. 2011; Behera et al. 2013). Fig S1 shows the size distributed [HSO4 −] during smoke-haze and non-haze periods.
From the analysis of size distributions of pHIS, it became clear that there was no specific trend in the levels of pHIS as a function of particle sizes during both smoke-haze and non-haze periods. However, the behavior of the levels of pHIS could be explained on the basis of different modes of particles. The level of pHIS was the lowest in the nanoparticles (0.01–0.056 μm) and increased with an increase in the size of particles during both smoke-haze and non-haze periods. In the accumulation mode (0.1–1.0 μm) and the coarse mode (1.8–10.0 μm), the levels of pHIS decreased with an increase in the size of particles during both smoke-haze and non-haze periods. The trends of pHIS observed at different size fractions indicated that pHIS not only depends on the [H+]Ins, but also on the hygroscopic growth of SIA components in the equilibrium. For example, if the mass of all ions in aqueous phase [i.e., m in Eq. (2)] increases, then with same [H+]Ins values, pH would increase, indicating that more hygroscopic growth of aerosols can reduce their acidic characteristics. Therefore, the prediction of pHIS is a complex problem as it depends on many inter-related parameters with different characteristics. However, it was confirmed that pHIS was mainly dependent upon the hygroscopic properties of SIA components. Fig. S1 shows the size-distributed aerosol water content ([H2O]), which is an indicator of hygroscopic growth of aerosols. From this figure, it can be observed that the particle-size fractions in the nanometer range had less water content than the fractions in the accumulation and coarse modes, suggesting that more hygroscopic growth of aerosols took place in the accumulation mode in which larger concentrations of SIA components were populated.
There was no systematic variation in the molar ratio of [NH4 +]/[SO4 2−] with the particle size (Fig. 6), implying that the variation of the in situ aerosol acidity cannot be explained on the basis of the values of [NH4 +]/[SO4 2−] only. However, the lower pHIS with higher values of [NH4 +]/[SO4 2−] in some size fractions could be due to higher levels of NO3 − and Cl− that did not have sufficient NH4 + for neutralization as can be seen from Eq. (1). Although the levels of NH4 + increased significantly in all size fractions of PM during the smoke-haze period, the levels of the acidic components (Cl−, NO3 − and SO4 2−) also increased significantly. Therefore, it could be inferred that the smoke-haze period experienced proportionally higher levels of acidic components than the basic components (NH4 + and Na+) that could lead to lower levels of pHIS.
Summary and conclusions
The present study investigated the size-resolved characterization (10 nm–10 μm) of water soluble inorganic ions (NH4 +, Na+, K+, Ca2+, Mg2+, Cl−, NO3 − and SO4 2−) in atmospheric aerosols during a recent smoke-haze episode that occurred in Singapore. The concentrations of these ions were used together in the thermodynamic model, AIM-IV, to assess the influence of the smoke-haze episodes on aerosol acidity. The peaks for PM mass in the fine mode occurred at 0.56–1.0 μm during the smoke-haze period and 0.32–0.56 μm during the non-haze period. The peaks of PM mass in the coarse mode occurred at 3.2–5.6 μm during both smoke-haze and non-haze periods. The significant increase in PM mass in the accumulation mode during the smoke-haze period could be due to the influence of long-range transported biomass-impacted aerosols from Indonesia on formation of new particles under favorable atmospheric conditions in Singapore. The ionic characterization showed that the peak concentrations of Cl−, NO3 − and SO4 2− in size-fractionated aerosols occurred at 0.32–0.56 μm during the smoke-haze period and at 0.56–1.0 μm during the non-haze period. A significant increase (>8 times) in the levels of particulate-bound K+ during the smoke-haze period indicated that the contribution of trans-boundary transport of biomass burning-impacted air masses from Indonesia to the enhancement in the concentration of all WSI ions during the smoke-haze period was paramount. The atmospheric nanoparticles were observed to be more acidic than other modes of PM. The average of pHIS at all size fractions was 0.21 during the smoke-haze period and 0.36 during the non-haze period. The overall conclusion is that the atmospheric aerosols characterized in this study were more acidic during the smoke-haze period compared to those during the non-haze period. The increased acidity of aerosols possibly played important roles in several atmospheric phenomena including the formation of secondary particles in the atmosphere.
References
Andreae, M. O., Andreae, T. W., Annegarn, H., Beer, J., Cachier, H., Le Canut, P., et al. (1998). Airborne studies of aerosol emissions from savanna fires in southern Africa: 2. Aerosol chemical composition. Journal of Geophysical Research: Atmospheres, 103(D24), 32119–32128.
Balasubramanian, R., Qian, W. B., Decesari, S., Facchini, M. C., & Fuzzi, S. (2003). Comprehensive characterization of PM2.5 aerosols in Singapore. Journal of Geophysical Research: Atmospheres, 108(D16), 4523. doi:10.1029/2002JD002517.
Behera, S. N., & Balasubramanian, R. (2014). Influence of biomass burning on temporal and diurnal variations of acidic gases, particulate nitrate and sulfate in a tropical urban atmosphere. Advances in Meteorology. doi:10.1155/2014/828491.
Behera, S. N., Betha, R., Liu, P., & Balasubramanian, R. (2013). A study of diurnal variations of PM2.5 acidity and related chemical species using a new thermodynamic equilibrium model. Science of the Total Environment, 452, 286–295.
Behera, S. N., & Sharma, M. (2010). Investigating the potential role of ammonia in ion chemistry of fine particulate matter formation for an urban environment. Science of the Total Environment, 408(17), 3569–3575.
Behera, S. N., & Sharma, M. (2012). Transformation of atmospheric ammonia and acid gases into components of PM2. 5: An environmental chamber study. Environmental Science and Pollution Research, 19(4), 1187–1197.
Betha, R., Behera, S. N., & Balasubramanian, R. (2014). 2013 Southeast Asian smoke haze: Fractionation of particulate-bound elements and associated health risk. Environmental Science and Technology, 48(8), 4327–4335.
Betha, R., Pradani, M., Lestari, P., Joshi, U. M., Reid, J. S., & Balasubramanian, R. (2013). Chemical speciation of trace metals emitted from Indonesian peat fires for health risk assessment. Atmospheric Research, 122, 571–578.
Cao, G., & Jang, M. (2009). An SOA model for toluene oxidation in the presence of inorganic aerosols. Environmental Science and Technology, 44(2), 727–733.
Chang, L. P., Tsai, J. H., Chang, K. L., & Lin, J. J. (2008). Water-soluble inorganic ions in airborne particulates from the nano to coarse mode: A case study of aerosol episodes in southern region of Taiwan. Environmental Geochemistry and Health, 30(3), 291–303.
Cheng, S. H., Yang, L. X., Zhou, X. H., Xue, L. K., Gao, X. M., Zhou, Y., & Wang, W. X. (2011). Size-fractionated water-soluble ions, situ pH and water content in aerosol on hazy days and the influences on visibility impairment in Jinan, China. Atmospheric Environment, 45(27), 4631–4640.
Clegg, S. L., Brimblecombe, P., & Wexler, A. S. (1998a). Thermodynamic model of the system H+–NH4 +–Na+–SO4 2−–NO3 −–Cl−–H2O at 298.15 K. The Journal of Physical Chemistry A, 102(12), 2155–2171.
Clegg, S. L., Brimblecombe, P., & Wexler, A. S. (1998b). Thermodynamic model of the system H+–NH4 +–SO4 2−–NO3–H2O at tropospheric temperatures. The Journal of Physical Chemistry A, 102(12), 2137–2154.
Draxler, R. R., & Rolph, G.D. (2013). HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) model access via NOAA ARL READY. Website http://www.arl.noaa.gov/HYSPLIT.php. College Park, MD: NOAA Air Resources Laboratory.
Du, H., Kong, L., Cheng, T., Chen, J., Du, J., Li, L., et al. (2011). Insights into summertime haze pollution events over Shanghai based on online water-soluble ionic composition of aerosols. Atmospheric Environment, 45(29), 5131–5137.
Engling, G., He, J., Betha, R., & Balasubramanian, R. (2014). Assessing the regional impact of Indonesian biomass burning emissions based on organic molecular tracers and chemical mass balance modeling. Atmospheric Chemistry and Physics, 14, 8043–8054.
Friese, E., & Ebel, A. (2010). Temperature dependent thermodynamic model of the system H+–NH4 +–Na+–SO4 2−–NO3 −–Cl−–H2O. The Journal of Physical Chemistry A, 114(43), 11595–11631.
Hatch, C. D., & Grassian, V. H. (2008). 10th anniversary review: Applications of analytical techniques in laboratory studies of the chemical and climatic impacts of mineral dust aerosol in the Earth’s atmosphere. Journal of Environmental Monitoring, 10(8), 919–934.
He, K., Zhao, Q., Ma, Y., Duan, F., Yang, F., Shi, Z., et al. (2012). Spatial and seasonal variability of PM 2.5 acidity at two Chinese megacities: Insights into the formation of secondary inorganic aerosols. Atmospheric Chemistry and Physics, 12(3), 1377–1395.
Holma, B. (1985). Influence of buffer capacity and pH-dependent rheological properties of respiratory mucus on health effects due to acidic pollution. Science of the Total Environment, 41(2), 101–123.
Jang, M., Czoschke, N. M., Lee, S., & Kamens, R. M. (2002). Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science, 298(5594), 814–817.
Jickells, T. D., An, Z. S., Andersen, K. K., Baker, A. R., Bergametti, G., Brooks, N., et al. (2005). Global iron connections between desert dust, ocean biogeochemistry, and climate. Science, 308(5718), 67–71.
Karthikeyan, S., & Balasubramanian, R. (2006). Determination of water-soluble inorganic and organic species in atmospheric fine particulate matter. Microchemical Journal, 82(1), 49–55.
Katsouyanni, K., Touloumi, G., Samoli, E., Gryparis, A., Le Tertre, A., Monopolis, Y., et al. (2001). Confounding and effect modification in the short-term effects of ambient particles on total mortality: Results from 29 European cities within the APHEA2 project. Epidemiology, 12(5), 521–531.
Kennish, M. J. (1994). Practical handbook of marine science. Boca Raton, FL: C.R.C. Press.
Kerminen, V. M., Hillamo, R., Teinilä, K., Pakkanen, T., Allegrini, I., & Sparapani, R. (2001). Ion balances of size-resolved tropospheric aerosol samples: Implications for the acidity and atmospheric processing of aerosols. Atmospheric Environment, 35(31), 5255–5265.
Kerminen, V. M., & Wexler, A. S. (1995). Growth laws for atmospheric aerosol particles: An examination of the bimodality of the accumulation mode. Atmospheric Environment, 29(22), 3263–3275.
Kittelson, D. B. (1998). Engines and nanoparticles: A review. Journal of Aerosol Science, 29(5), 575–588.
Lee, H. S., Kang, C. M., Kang, B. W., & Kim, H. K. (1999). Seasonal variations of acidic air pollutants in Seoul, South Korea. Atmospheric Environment, 33(19), 3143–3152.
Leiva, G. M. A., Santibañez, D. A., Ibarra, E. S., Matus, C. P., & Seguel, R. (2013). A five-year study of particulate matter (PM2.5) and cerebrovascular diseases. Environmental Pollution, 181, 1–6.
Lin, C. C., Chen, S. J., Huang, K. L., Lee, W. J., Lin, W. Y., Liao, C. J., & Chiu, C. H. (2007). Water-soluble ions in nano/ultrafine/fine/coarse particles collected near a busy road and at a rural site. Environmental Pollution, 145(2), 562–570.
Lin, Y. C., & Cheng, M. T. (2007). Evaluation of formation rates of NO2 to gaseous and particulate nitrate in the urban atmosphere. Atmospheric Environment, 41(9), 1903–1910.
Ma, J., Tang, J., Li, S. M., & Jacobson, M. Z. (2003). Size distributions of ionic aerosols measured at Waliguan observatory: Implication for nitrate gas‐to‐particle transfer processes in the free troposphere. Journal of Geophysical Research: Atmospheres, 108(D17). doi:10.1029/2002JD003356.
Manktelow, P. T., Carslaw, K. S., Mann, G. W., & Spracklen, D. V. (2010). The impact of dust on sulfate aerosol, CN and CCN during an East Asian dust storm. Atmospheric Chemistry and Physics, 10(2), 365–382.
Meng, Z., & Seinfeld, J. H. (1994). On the source of the submicrometer droplet mode of urban and regional aerosols. Aerosol Science and Technology, 20(3), 253–265.
Meng, Z., Seinfeld, J. H., Saxena, P., & Kim, Y. P. (1995). Atmospheric gas-aerosol equilibrium: IV. Thermodynamics of carbonates. Aerosol Science and Technology, 23(2), 131–154.
Menon, S., Hansen, J., Nazarenko, L., & Luo, Y. (2002). Climate effects of black carbon aerosols in China and India. Science, 297(5590), 2250–2253.
Nel, A. (2005). Air pollution-related illness: Effects of particles. Science, 308(5723), 804–806.
Nenes, A., Krom, M. D., Mihalopoulos, N., Van Cappellen, P., Shi, Z., Bougiatioti, A., et al. (2011). Atmospheric acidification of mineral aerosols: A source of bioavailable phosphorus for the oceans. Atmospheric Chemistry and Physics, 11(13), 6265–6272.
Nenes, A., Pandis, S. N., & Pilinis, C. (1998). ISORROPIA: A new thermodynamic equilibrium model for multiphase multicomponent inorganic aerosols. Aquatic Geochemistry, 4(1), 123–152.
Pathak, R. K., Louie, P. K. K., & Chan, C. K. (2004). Characteristics of aerosol acidity in Hong Kong. Atmospheric Environment, 38(19), 2965–2974.
Pathak, R. K., Yao, X., Lau, A. K., & Chan, C. K. (2003). Acidity and concentrations of ionic species of PM2.5 in Hong Kong. Atmospheric Environment, 37(8), 1113–1124.
Pavagadhi, S., Betha, R., Venkatesan, S., Balasubramanian, R., & Hande, M. P. (2013). Physicochemical and toxicological characteristics of urban aerosols during a recent Indonesian biomass burning episode. Environmental Science and Pollution Research, 20(4), 2569–2578.
Pierson, W. R., & Brachaczek, W. W. (1988). Coarse-and fine-particle atmospheric nitrate and HNO3(g) in Claremont, California, during the 1985 nitrogen species methods comparison study. Atmospheric Environment, 22(8), 1665–1668.
Pope, C. A., & Dockery, D. W. (2006). Health effects of fine particulate air pollution: Lines that connect. Journal of the Air and Waste Management Association, 56(6), 709–742.
Poppe, D., Wallasch, M., & Zimmermann, J. (1993). The dependence of the concentration of OH on its precursors under moderately polluted conditions: A model study. Journal of Atmospheric Chemistry, 16(1), 61–78.
Raizenne, M., Neas, L. M., Damokosh, A. I., Dockery, D. W., Spengler, J. D., Koutrakis, P., et al. (1996). Health effects of acid aerosols on North American children: Pulmonary function. Environmental Health Perspectives, 104(5), 506.
Reid, J. S., Hyer, E. J., Johnson, R. S., Holben, B. N., Yokelson, R. J., Zhang, J., et al. (2013). Observing and understanding the Southeast Asian aerosol system by remote sensing: An initial review and analysis for the Seven Southeast Asian Studies (7SEAS) program. Atmospheric Research, 122, 403–468.
Rengarajan, R., Sudheer, A. K., & Sarin, M. M. (2011). Aerosol acidity and secondary organic aerosol formation during wintertime over urban environment in western India. Atmospheric Environment, 45(11), 1940–1945.
Ryu, S. Y., Kim, J. E., Zhuanshi, H., Kim, Y. J., & Kang, G. U. (2004). Chemical composition of post-harvest biomass burning aerosols in Gwangju, Korea. Journal of the Air and Waste Management Association, 54(9), 1124–1137.
Saxena, P., Mueller, P. K., Kim, Y. P., Seinfeld, J. H., & Koutrakis, P. (1993). Coupling thermodynamic theory with measurements to characterize acidity of atmospheric particles. Aerosol Science and Technology, 19(3), 279–293.
See, S. W., Balasubramanian, R., Rianawati, E., Karthikeyan, S., & Streets, D. G. (2007). Characterization and source apportionment of particulate matter 2.5 μm in Sumatra, Indonesia, during a recent peat fire episode. Environmental Science and Technology, 41(3488–3494), 2007.
See, S. W., Balasubramanian, R., & Wang, W. (2006). A study of the physical, chemical, and optical properties of ambient aerosol particles in Southeast Asia during hazy and nonhazy days. Journal of Geophysical Research: Atmospheres, 111(D10). doi:10.1029/2005JD006180.
Song, C. H., & Carmichael, G. R. (1999). The aging process of naturally emitted aerosol (sea-salt and mineral aerosol) during long range transport. Atmospheric Environment, 33(14), 2203–2218.
Sun, Z., Mu, Y., Liu, Y., & Shao, L. (2013). A comparison study on airborne particles during haze days and non-haze days in Beijing. Science of the Total Environment, 456, 1–8.
Sun, Y., Zhuang, G., Tang, A., Wang, Y., & An, Z. (2006). Chemical characteristics of PM2.5 and PM10 in haze-fog episodes in Beijing. Environmental Science and Technology, 40(10), 3148–3155.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., & Seinfeld, J. H. (2007). Effect of acidity on secondary organic aerosol formation from isoprene. Environmental Science and Technology, 41(15), 5363–5369.
Tan, J. H., Duan, J. C., Chen, D. H., Wang, X. H., Guo, S. J., Bi, X. H., et al. (2009). Chemical characteristics of haze during summer and winter in Guangzhou. Atmospheric Research, 94(2), 238–245.
Tsai, J. H., Chang, L. P., & Chiang, H. L. (2013). Size mass distribution of water-soluble ionic species and gas conversion to sulfate and nitrate in particulate matter in Southern Taiwan. Environmental Science and Pollution Research, 20(7), 4587–4602.
Tsai, J. H., Lin, J. H., Yao, Y. C., & Chiang, H. L. (2012). Size distribution and water soluble ions of ambient particulate matter on episode and non-episode days in Southern Taiwan. Aerosol and Air Quality Research, 12, 263–274.
Tsai, H. H., Yuan, C. S., Hung, C. H., & Lin, C. (2011). Physicochemical properties of PM2. 5 and PM2.5–10 at Inland and offshore sites over Southeastern Coastal Region of Taiwan Strait. Aerosol and Air Quality Research, 11, 664–678.
Wall, S. M., John, W., & Ondo, J. L. (1988). Measurement of aerosol size distributions for nitrate and major ionic species. Atmospheric Environment, 22(8), 1649–1656.
Wang, Y., Zhuang, G., Sun, Y., & An, Z. (2006). The variation of characteristics and formation mechanisms of aerosols in dust, haze, and clear days in Beijing. Atmospheric Environment, 40(34), 6579–6591.
Weijers, E. P., Schaap, M., Nguyen, L., Matthijsen, J., Denier Van Der Gon, H. A. C., Ten Brink, H. M., & Hoogerbrugge, R. (2011). Anthropogenic and natural constituents in particulate matter in the Netherlands. Atmospheric Chemistry and Physics, 11(5), 2281–2294.
Xue, J., Lau, A. K., & Yu, J. Z. (2011). A study of acidity on PM2.5 in Hong Kong using online ionic chemical composition measurements. Atmospheric Environment, 45(39), 7081–7088.
Yao, X., Lau, A. P., Fang, M., Chan, C. K., & Hu, M. (2003). Size distributions and formation of ionic species in atmospheric particulate pollutants in Beijing, China: 1—inorganic ions. Atmospheric Environment, 37(21), 2991–3000.
Yao, X., Ling, T. Y., Fang, M., & Chan, C. K. (2007). Size dependence of in situ pH in submicron atmospheric particles in Hong Kong. Atmospheric Environment, 41(2), 382–393.
Yu, C. Y., Dong, C., Wang, X. F., Yang, L. X., & Wang, W. X. (2011). Size distributions of water-soluble inorganic ions of atmospheric aerosol particles in autumn in Jinan. Journal of Environmental Science (China), 31(4), 561–5677.
Zhang, Q., Jimenez, J. L., Worsnop, D. R., & Canagaratna, M. (2007). A case study of urban particle acidity and its influence on secondary organic aerosol. Environmental Science and Technology, 41(9), 3213–3219.
Zhou, Y., Xue, L., Wang, T., Gao, X., Wang, Z., Wang, X., et al. (2012). Characterization of aerosol acidity at a high mountain site in central eastern China. Atmospheric Environment, 51, 11–20.
Zhuang, H., Chan, C. K., Fang, M., & Wexler, A. S. (1999). Size distributions of particulate sulfate, nitrate, and ammonium at a coastal site in Hong Kong. Atmospheric Environment, 33(6), 843–853.
Ziemba, L. D., Fischer, E., Griffin, R. J., & Talbot, R. W. (2007). Aerosol acidity in rural New England: Temporal trends and source region analysis. Journal of Geophysical Research: Atmospheres, 112(D10S22). doi:10.1029/2006JD007605.
Acknowledgments
This research programme is funded by the National Research Foundation (NRF), Prime Minister’s Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE) programme. The authors are grateful to NRF for the financial support from Grant No. R-706-002-101-281.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Behera, S.N., Cheng, J. & Balasubramanian, R. In situ acidity and pH of size-fractionated aerosols during a recent smoke-haze episode in Southeast Asia. Environ Geochem Health 37, 843–859 (2015). https://doi.org/10.1007/s10653-014-9660-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10653-014-9660-1