
Abstract
Cholangiocarcinoma (CCA) is a primary malignancy which is often diagnosed when it is advanced and inoperable due to the lack of effective biomarkers and poor sensitivity of clinical diagnosis. Molecular profiling may provide information for improved clinical management, particularly targeted therapy. The study aimed to improve the understanding of molecular characteristics and its association with prognosis in Chinese CCA. We enrolled 41 Chinese patients with CCA, including 6 intrahepatic CCA (iCCA), 14 perihilar CCA (pCCA), and 21 distal CCA (dCCA) cases, all patients underwent radical operations and tumor samples underwent next-generation sequencing (NGS) by Foundation One Dx, which analyzed 324 genes. The patients’ genetic characteristics, clinical management, and prognosis were analyzed. The most mutated genes were TP53 (68%, 28/41), CDKN2A (37%, 15/41), and SMAD4 (29%, 12/41). The genetic mutations in dCCA, pCCA, and iCCA were significantly different. For example, NOTCH3 mutations were not found in dCCA. The gene mutations of AXL were specifically associated with lymph node metastasis in patients with CCA, whereas gene mutations of SMAD4 were specifically associated with lymphovascular invasion. Furthermore, mutations in APC, DAXX, FANCA, LTK, MAP2K4, and NOTCH1 were associated with a poor prognosis (P < 0.05). This study provides an overview of genetic alterations in Chinese patients with CCA, which will provide novel potential biomarkers for the diagnosis of CCA and may guide targeted therapeutic strategies for Chinese patients with CCA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the fact that the epidemiology of cholangiocarcinoma (CCA) varies greatly across the globe, the overall incidence is increasing [1]. It is classified as intrahepatic (iCCA) or extrahepatic (eCCA) based on its anatomical origin, with the second-order bile ducts serving as the dividing line [2]. The second most common type of primary liver cancer is intrahepatic cholangiocarcinoma (iCCA), accounting for 1% of all neoplasms [3, 4]. Furthermore, depending on whether it originates above or below the cystic duct, eCCA is classified as perihilar (pCCA) or distal (dCCA) [2]. The etiopathogenesis of these subtypes differs, with distinct risk factors [1, 2], proposed cells of origin [5, 6], and specific genomic aberrations [6, 7].
For patients in the early stages, surgical resection is currently the mainstay of curative-intent treatment; however, most patients will not benefit from radical surgery [3, 8, 9]. However, the insensitivity of CCA to radiotherapy and chemotherapy results in a poor prognosis [10, 11]. Biomarker-based targeted therapy and immunotherapy are effective treatments for some malignant tumors. There are, however, few effective biomarkers for CCA, and these need to be researched and developed to facilitate early detection and diagnosis.
The heterogeneity of CCA hinders targeted treatment for CCA subtypes that differ in their response to treatment [12]. With the development of next-generation sequencing (NGS) technology, we can identify molecular differences between subtypes. Recent research has revealed that iCCA and eCCA have distinct molecular characteristics [13, 14]. NGS studies have revealed the genomic heterogeneity of CCA subtypes, which may impact future therapy trials [13]. Although extrahepatic CCA can be divided into pCCA and dCCA, their prognosis differs. Waseem et al. reported that the mean survival of pCCA was lower than that of dCCA, but comparable to that of iCCA [15]. However, few studies have isolated pCCA and dCCA and focused on their genomic characteristics.
We enrolled 41 Chinese patients with CCA to characterize their genomic profiles according to the different subtypes. We aimed to identify potential biomarkers for prognosis and provide evidence for targeted therapy and immunotherapy.
Materials and methods
Patient information and sample collection
From 2019 to 2020, 41 CCA patients were enrolled from Peking University People’s Hospital according to the tumor locations. Informed consent was obtained from all patients and this study was approved by the Institutional Ethics Committee of Peking University People’s Hospital (2021PHB279-001). The patients were given the necessary jaundice-reducing treatment based on the results of computed tomography or magnetic resonance imaging. Surgical resection was performed once the total bilirubin level was less than 5 times normal. The tumor tissue samples were fixed in formalin for 24 h before being embedded in paraffin. Tumor tissues that have been formalin-fixed, paraffin-embedded (FFPE), and contain at least 20% tumor cells are considered tumor tissue samples and can be used for further NGS detection. For those cases, relevant demographic and clinical data from the database were extracted, including age, gender, date of diagnosis, histology type, and evaluation of treatment responses based on clinical investigator reports. Pathologists confirmed each case’s pathologic diagnosis and tumor content based on tumor tissue samples.
DNA extraction and library preparation
Between May and November 2020, all tissue samples were sent to Foundation Medicine for analysis. NGS was performed in tumor tissue samples with at least 20% tumor cellularity and sequencing panels of 324 genes (FoundationOne® CDx) (https://www.foundationmedicine.com/genomic-testing/foundation-one-cdx), respectively. Foundation Medicine provided the tests as part of an expanded access program at no cost to patients or institutions. The NGS report was sent to the ordering physician to discuss the treatment plan with the patient. Between May, 2020 and July, 2020, all the reports and baseline characteristics were collected at Peking University People’s Hospital, and results were retrospectively analyzed.
Sequence data processing
The qualified DNA libraries were sequenced on the Illumina NovaSeq6000 platform (Illumina, San Diego, CA) and produced 150 bp paired-end reads. Base calls from the Illumina NovaSeq6000 were made to FASTQ files. An automated data management system monitored the sequencers and started the analysis pipeline when the sequencing run finished. BWA MEM was used to align sequence reads to the human genome (hg19). To reduce alignment artifacts, ABRA was used to realign reads around indels, and the Genome Analysis Toolkit was used to recalibrate base quality scores. Duplicate reads were flagged for removal, and the resulting BAM files were used to discover variants. MuTect, Pindel, and Somatic Indel Detector union calls were then subjected to automated filtering to produce a comprehensive list of somatic mutation calls that included single nucleotide variants, short and long indels. Copy number changes were detected using an in-house developed algorithm, and structural variants were detected using Delly. Germline variants were eliminated by using patient-matched blood DNA. Each alteration identified by the pipeline was manually reviewed to ensure that no false positives were reported, complex events identified separately were merged and properly represented, and mutation annotations were Human Genome Variation Society (HGVS, http://varnomen.hgvs.org) compliant [16].
The public cohort
All clinical and genomic data of 346 CCA patients were acquired from cBioPortal (http://www.cbioportal.org) [17] and the ICGC portal (http://dcc.icgc.org/releases/current/Projects). We selected the WES dataset for CCA patients only. The repositories used were BTCA-JP [18], BTCA-SG [19], TCGA-CHOL (TCGA, Pan Cancer Atlas) [20].
Statistical analysis
The Kaplan-Meier curve analysis OS and PFS was compared using the log-rank test. The Cox proportional hazards regression was applied for univariable and multivariate analysis, and available confounding factors including sex, age, subtype, and TMB level were adjusted. Student’s t-test was used to compare continuous variables, and the χ2 test or Fisher’s exact test was used to compare categorical data. All reported P values were two-tailed, and P < 0.05 was considered statistically significant. Statistical analyses were performed using R v. 4.0.3 (https://www.r-project.org).
Results
Clinical characteristics of Chinese CCA patients
A total of 41 patients with CCA, 34 (82.9%) males and 7 (17.1%) females, with a median age of 64 years (range 42–78 years) were enrolled in this study. Twenty-one of the 41 patients harbored hepatitis B virus and one harbored hepatitis C virus, 19 patients were negative. In all, 34.1% of the patients were lymph-node metastasis positive. There were 14.6% iCCA, 34.1% pCCA, and 51.3% dCCA tumors. All patients were treated surgically and 39 (95.1%) patients had R0 resection. Thirty patients received postoperative adjuvant chemotherapy, 8 did not, and 3 patients’ postoperative treatment was unknown. Intraoperative exploration showed 1 (2.4%) patient had vascular invasion, 39 (95.1%) had none, and 1 patient had no information. Histologically, 26 (63.4%) samples were well or moderately differentiated and 14 (34.2%) samples were poorly differentiated or undifferentiated. Histological information was not available for 1 (2.4%) patient. The patients’ clinical and pathological information is shown in Table 1.
Genomic alterations in Chinese CCA
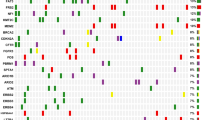
A total of 545 clinically relevant genomic alterations in 199 genes were identified in 41 patients with CCA. The alterations included 351 (64.4%) substitution/indels, 97 (17.80%) truncations, 13 (2.39%) splice site mutations, 57 (10.46%) gene amplifications, and 27 (4.95%) gene deletion. The most mutated genes were TP53 (68%, 28/41), CDKN2A (37%, 15/41), SMAD4 (29%, 12/41), ATM (22%, 9/41), MLL2 (22%, 9/41), ERBB2 (22%, 9/41), KRAS (20%, 8/41), IRS2 (17%, 7/41), and ARID1A (17%, 7/41) (Fig. 1A). The genetic mutations in dCCA, pCCA, and iCCA were significantly different. The most mutated genes in dCCA were TP53 (76%, 16/21), SMAD4 (33%, 7/21), and ATM (33%, 7/21). While in pCCA they were TP53 (64%, 9/14), CDKN2A (57%, 8/14), and SMAD4 (36%, 5/14). In iCCA they were TP53 (50%, 3/6), MLL2 (33%, 2/6), and STK11 (33%, 2/6) (Fig. 1A). However, only the NOTCH3 gene was significantly different between the three groups, as it did not mutate in the patients with dCCA (Fig. 1B).

Mutation frequency and significantly mutated genes in Chinese CCA. (A) Mutational spectrum of patients with CCA. (B) Comparative analysis of high frequently mutated genes in pCCA (green), iCCA (blue), and dCCA (orange). The X-axis represents the most mutated genes and the Y-axis represents the mutation frequency of each gene in different CCA subtypes. The significant differences were marked with * for P < 0.05, ** for P < 0.01, and *** for P < 0.001, NA not available. (C) The tumor mutation burden (TMB) of nonsynonymous mutations (substitutions and indels) between dCCAs (presented in red), pCCAs (presented in navy blue), and iCCAs (presented in blue)
We identified the tumor mutation burden (TMB) value of 41 patients with clinically relevant genomic alterations. The median TMB value was 1.26 mutations/million bases (Mb), ranging from 0 to 22.7 mutations/Mb. There was no significant difference in TMB among the three groups (Fig. 1C).
Correlations between mutated genes and clinical characteristics
To explore potential biomarkers, we analyzed the association between mutations and clinical characteristics, such as visceral organ, nerve, and vascular invasion, lymph node metastasis, and lymphovascular invasion. Only lymph node metastasis and lymphovascular invasion were significantly associated with gene mutations. Thirteen patients had lymph node metastases and 10 patients, lymphovascular invasion. There was a significant association between AXL mutation and lymph node metastasis (P = 0.027) (Fig. 2A), and SMAD4 mutation and lymphovascular invasion (P = 0.045) (Fig. 2B).

The correlation between mutated genes and lymph node metastasis and lymphovascular invasion. (A)AXL mutation was associated with lymph node metastasis. (B)SMAD4 mutation was associated with lymphovascular invasion
Genetic mutations associated with progression‑free survival (PFS)
Thirty-six patients who had radical surgery were followed up. The median progression-free survival (PFS) was not reached. Taking PFS as a continuous variable, it was associated with APC, DAXX, FANCA, LTK, MAP2K4, and NOTCH1 mutations, and PFS. Survival curve analysis showed that patients with these mutations had a shorter PFS time (Fig. 3). To confirm this result, we performed multivariate Cox regression analysis of gender, age, TMB, and other clinical characteristics. The results showed that APC, DAXX, FANCA, LTK, and MAP2K4 (P < 0.05) mutations were still significantly associated with a shorter PFS, while NOTCH1 mutations were not (Fig. 4); the association between NOTCH1 mutations and PFS was easily neutralized by other clinical characteristics.

Correlation analysis between mutated genes and progression-free survival (PFS). Kaplan–Meier curves of the PFS in patients with (red)/without (blue) APC(A), DAXX(B), FANCA(C), LTK(D), MAP2K4(E), and NOTCH1 mutations

Multivariate cox regression analysis to confirm the correlation between PFS and genetic mutations. APC(A), DAXX(B), FANCA(C), LTK(D), MAP2K4(E), and NOTCH1 mutations (F). Forest plot showed the risk of PFS in various subgroups of patients such as gender, age, and tumor grade
Actionable target mutations of Chinese CCAs
Actionable alterations in various types of cancers were collected and summarized by the OncoKB team, and 17 clinically relevant genes with 26 potential therapies for CCA were examined [21]. Potential target drugs for KRAS mutations included asadagrasib, cobimetinib, binimetinib, and trametinib, while debio1347, infigratinib, erdaftinib, and AZD4547 were potential target drugs for FGFR mutations. Trametinib and cobimetinib were potential target drugs for NF1 mutations. In this cohort, there were 7 actionable mutated genes in 41 (17.1%) patients with CCA. The most common drug target mutations were KRAS (17.1%), BRAF (12.2%), NF1 (4.9%), and FGFR3 (4.9%) (Table 2). Cobimetinib and trametinib are the potential target drugs for both KRAS mutation and NF1 mutations, and our data showed that 21.9% (9/41) of patients with CCA harboring KRAS mutations or NF1 mutations might benefit from them.
Further, the actionable target mutations were different in the three groups. In patients with dCCA, the most common drug target mutations were KRAS (19.1%) and BRAF (9.5%). Each patient harbored NF1, FGFR3, and CDK12 (Table 2). In patients with pCCA, 3 patients harbored KRAS (21.4%) and BRAF (21.4%), and only one patient had NF1 (7.1%). In patients with iCCA, each patient harbored KRAS, FGFR2, FGFR3, and MET.
Hyperprogressive disease (HPD) associated genetic alterations
In recent years, with the emergence of precision medicine therapies such as targeted therapy and immunotherapy, the treatment of biliary tract cancer (BTC) has entered a new era. The FDA approved durvalumab in combination with chemotherapy for the first-line treatment of locally advanced or metastatic BTC in 2022. Increasing clinical trial data supports the effectiveness of immunotherapy with chemotherapy in the treatment of CCA. However, some patients still did not benefit from the combination and were even at risk of hyperprogressive disease (HPD). Therefore, we analyzed gene variants associated with HPD (Table 3). There were 7 HPD-associated genetic alterations, CDKN2A loss (9, 21.9%), CDKN2B loss (9, 21.9%), CCND1 amplification (4, 9.8%), FGF3 amplification (4, 9.8%), FGF4 amplification (4, 9.8%), FGF19 amplification (4, 9.8%), and EGFR amplification (2, 4.9%). The distribution of HPD-associated genetic alterations in the three groups differed. The pCCA group showed a higher rate in HPD-associated genetic alterations (36%, 5/14), followed by dCCA (24%, 5/21), and iCCA (17%, 1/6).
Differences in genomic alterations between Chinese and western patients with CCA
In 346 public cohort CCA samples in Western patients, the most common mutations were TP53, KRAS, ARID1A, SMAD4, and APC. In 41 Chinese patients with CCA, the mutations of TP53, SMAD4, and KRAS also frequently occurred. Based on statistical analysis, the mutations of TP53, ATM, IRS2, and LTK were significantly higher in Chinese patients (P < 0.01) (Fig. 5).

Comparative analysis of high frequency mutant genes in Chinese and Western patients with CCA. The X-axis represents the most mutated genes and the Y-axis represents the mutation frequency of each gene in different CCA subtypes. The significant differences were marked with * for P < 0.05, ** for P < 0.01, and *** for P < 0.001
Discussion
Cholangiocarcinoma (CCA) is a highly lethal and aggressive cancer of the biliary epithelium [22]. Although the CCA subtypes differ in their presentation and natural history, and necessitate different approaches to diagnosis and management [23, 24], there are few reports of the differences in their molecular characteristics. We enrolled 41 patients with CCA, including 6 iCCA, 14 pCCA, and 21dCCA, and identified their mutational profiles. The greatest difference in genetic mutation occurred with NOTCH3, as none of the patients with dCCA showed NOTCH3 mutations. NOTCH is a master regulator of cell fate in the mammalian liver, and reactivation of the NOTCH pathway has been reported to varying degrees in biliary malignancies. NOTCH3 was found to be important in the development and progression of CCA [25]. O’Rourke et al. [26] discovered a network-wide imbalance of the NOTCH pathway in CCA, including a relationship between NOTCH1 and NOTCH3 and NOTCH ligands, and proposed γ-secretase modulation as a therapeutic option. However, because NOTCH3 mutations were not found in dCCA patients, the pathogenesis of dCCA and iCCA/pCCA may differ, and these genes may be specific and potential tumor biomarkers in dCCA.
Many studies on potential biomarkers for CCA have been conducted, and revealed different molecular characteristics in patients from various regions [14, 23]. Another research focused on Chinese pCCA and iCCA founded that top 10 mutated genes were TP53, KRAS, ARID1A, SMAD4, PBRM1, NF1, MACF1, GNAS, PIK3CA, and EPHA2, while our pCCA and iCCA were TP53, CDKN2A, MLL2, STK11, KRAS, NOTCH3, ERBB3, SMAD4, MET, and ERBB2. The difference could be attributed to the relatively small number of our patients, particularly those with iCCA; on the other hand, it could be attributed to the fact that MACF1 and EPHA2 were not included in the FoundationOne® CDx gene list. However, CDKN2A deletion one of the genes with significant copy number alteration, indicating that CDKN2A deletion was important for patients with pCCA and iCCA [27]. Our findings, when combined with gene mutation data of Japanese liver cancer displaying biliary phenotype (LCB, which means iCCA and combined hepatocellular cholangiocarcinoma) [28] and Thai iCCA [29] revealed that KRAS was important for Asian iCCA patients. Furthermore, the top mutated gene in Chinese and Thai iCCA patients was TP53. The gene mutations of the iCCA in our study were also significantly different from those of patients in Japan and Thailand. For example, the top mutated genes in iCCA were TP53, MLL2, STK11, KRAS, IRS2, ERBB3, NOTCH3, and MET. In Japan, they were primarily TERT promoter, KRAS, IDH family (IDH1 or IDH2), PBRM1, ARID2, BAP1, PCLO, ODZ1, and XIRP2. The most common mutations in Thailand’s iCCA were TP53, ARID1A, KRAS, SMAD4, APC, KMT2C, GNAS, LRP1B, CSMD3, and ERBB2. The differences could be attributed to regional differences (Japan, Thailand, and China), differences in sequencing technology (WGS vs. targeted panel sequencing), and large sample numbers. Our study and another Thailand’s CCA genetic data [19] revealed that TP53, ERBB2, and ARID1A were the main driver genes. Additionally, the most frequently mutated genes in our CCA patients included SMAD4, ATM, MLL2, KRAS, IRS2, and CDKN2A, whereas in Thailand, the most commonly mutated genes contained FGFR, BAP1, and IDH1/2. Recently, Lowery et al. [24] reported the genomic profiling of patients with CCA, including 152 iCCA and 43 eCCA cases, from Caucasian (89.2%, 174/195), Asian (7.1%, 14/195), and African American (3.6%, 7/195) patients. IDH1, TP53, ARID1A, BAP1, KRAS, PBKM1, SMAD4, and ATM were the most common mutations. In addition, Tian et al. [14] reported that mutations of TP53, KRAS, and SMAD4 were significantly higher in Chinese patients, whereas the mutations of IDH1 and BAP1 were significantly lower than in Western patients. We detected common high-frequency mutations (> 10%) of TP53, KRAS, SMAD4, TERT, and ARID1A in Chinese CCA patients; compared with Western patients, TP53, ATM, IRS2, and LTK mutations were significantly higher (P < 0.001).
There have been many studies of the association between genetic mutations and patients’ clinical characteristics. Tian et al. [14] assessed gender-associated specific genomic characteristics in Chinese patients with CCA. They identified CCND1, FBXW7FGF3/4/19, PIK3C1, NF1 and STK11 as mutations specific to male patients, while mutations in ERBB2, AXIN2, CREBBP, ERBB3, MTOR and MYB were specific to female patients. Feng et al. [30] identified that patients with KRAS mutations may have a higher risk of vascular invasion and poor prognosis. However, little is known about the genomic characteristics associated with lymph node metastasis and lymphovascular invasion in patients with CCA. The present study identified an association between AXL mutation and lymph node metastasis (P = 0.027), and SMAD4 mutation and lymphovascular invasion (P = 0.045). AXL is a receptor tyrosine kinase involved in many pathological conditions, including carcinogenesis. It has been previously reported to be significantly associated with lymph node metastasis in CCA [31], and in most melanoma lymph node metastases [32]. Its expression was associated with differentiation grade, TNM stage, lymph node, and distant metastasis in solid malignancies [33]. The genetic mutations may affect the expression or function of AXL, which is then associated with lymph node metastasis in CCA. SMAD4 is found to be mutated in many cancers. It acts as a tumor suppressor regulating the TGF-β signaling pathway. The expression of SMAD4 was associated with the histological grade, clinical stage, and metastasis of ICC (P < 0.05) [34]. A history of biliary surgery and mutations in SMAD4 were independent protective factors for metastasis in patients with advanced CCA [35]. The association of SMAD4 with lymphovascular invasion in CCA was first identified in the present study, and the specific molecular mechanisms need to be studied further.
We investigated the possible link between genetic alterations and prognosis and found worse outcomes for patients with certain genetic mutations although statistically, this was of borderline significance. Our results showed that APC, DAXX, FANCA, LTK, and MAP2K4 mutations were significantly associated with a shorter PFS (P < 0.05). The adenomatous polyposis coli (APC) gene is the most frequently mutated in colon cancer and is responsible for familial adenomatous polyposis. Mutations in the APC gene play a critical role in the adenoma-carcinoma pathway of colorectal tumorigenesis [36, 37]. In addition, downregulated APC expression was associated with a poor prognosis in patients with esophageal squamous cell carcinoma [38]. Loss of heterozygosity was displayed close to the APC gene in iCCA [39]. It was first identified that the mutations of APC were associated with CCA prognosis. The death domain-associated protein encoded by the DAXX gene is involved in transcriptional regulation, cell apoptosis, carcinogenesis, and anti-virus infection. DAXX is a transcriptional repressor that regulates p53 chromatin binding and DNA damage response [40]. It has been linked to a number of cancers, and its role in tumorigenesis and development is complex. By activating the ERK signaling pathway, it promotes ovarian cancer ascites, cell proliferation, and migration [41]. It also promotes prostate cancer tumorigenicity by suppressing autophagy [42], and inhibits cancer stemness and epithelial-mesenchymal transition in gastric cancer [43]. Its role in the development of CCA remains unknown. FANCA mutations exhibit a clinical impact in Fanconi anemia, which is characterized by congenital abnormalities, bone marrow failure, and cancer predisposition. It is rarely reported in CCA; however, there is one case report of a patient with stage IV CCA who developed fatal myelotoxicity after receiving palliative chemotherapy with cisplatin and gemcitabine [44]. Leukocyte tyrosine kinase (LTK) that has been found to be overexpressed in human leukemia. Its function is largely unknown, but it is very similar to anaplastic lymphoma kinase, which is frequently mutated in human cancer [45]. Mitogen-activated protein kinase 4 (MAP2K4) belongs to the family of mitogen-activated protein kinase (MAPK) activators. MAPK signaling is important for cell proliferation, differentiation, transcriptional regulation, and development. It promotes human prostate cancer metastasis [46], breast cancer pathogenesis [47], and functions as a tumor suppressor in lung adenocarcinoma [48]. However, the specific function and mechanism of MAP2K4 in CCA have not been clarified.
Recently, several immune checkpoint inhibitor (ICI) therapies have been developed for treating advanced-stage cancers, although they currently benefit only a minority of patients. TMB, a novel predictive biomarker, can predict the clinical response to ICI and identify patients who may benefit from this therapy [49]. An increased TMB indicates increased levels of tumor antigen, which is beneficial for activating the body’s immune response. Previous studies of TMB have found a strong link between increased TMB and improved response to ICI therapy. However, patients with CCA showed a relatively low TMB value; there were only two patients whose TMB was higher than 10. In addition, there was no significant difference between the TMB in the iCCA, pCCA, and dCCA groups. The importance of hyperprogressive disease (HPD) in ICI therapy cannot be overstated. However, few studies have explored HPD in CCA. In our study, we analyzed HPD-associated genetic mutations in Chinese CCA patients. Our results showed that there were 11 (27%) patients who harbored HPD-associated genetic mutations, and many patients had more than one. The pCCA group showed the highest ratio in HPD-associated genetic mutations. Therefore, immunotherapy in patients with CCA should be seriously considered.
Conclusion
In conclusion, we had investigated the comprehensive genomic features of 41 Chinese patients with CCA. TP53 mutations were significantly higher in Chinese patients than in Western patients with CCA. This suggests that TP53 may be more suitable as a predictive and diagnostic biomarker in Chinese patients with CCA. Furthermore, gene mutations that were associated with clinical characteristics, such as lymph node metastasis and lymphovascular invasion, were detected. Overall, these data provide an insight into the diagnosis and prognosis of Chinese patients with CCA. The results may guide future therapeutic strategies, including targeted and immune therapies.
Data Availability
The datasets used in the study are available from the corresponding author on reasonable request.
References
Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P et al (2016) ;13(5):261 – 80. https://doi.org/10.1038/nrgastro.2016.51, PMID: 27095655. https://doi.org/10.1038/nrgastro.2016.51
Blechacz B, Komuta M, Roskams T, Gores GJ (2011) Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 8(9):512–522. https://doi.org/10.1038/nrgastro.2011.131
Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ (2018) Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 15(2):95–111. https://doi.org/10.1038/nrclinonc.2017.157
Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Akinyemiju TF, Al Lami FH, Alam T, Alizadeh-Navaei R, Allen C et al (2018) Global, Regional, and National Cancer incidence, mortality, years of Life Lost, Years lived with disability, and disability-adjusted life-years for 29 Cancer groups, 1990 to 2016: a systematic analysis for the global burden of Disease Study. JAMA Oncol 4(11):1553–1568. https://doi.org/10.1001/jamaoncol.2018.2706
Rizvi S, Gores GJ (2013) Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 145(6):1215–1229. https://doi.org/10.1053/j.gastro.2013.10.013
Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM (2016) Molecular Pathogenesis and targeted therapies for Intrahepatic Cholangiocarcinoma. Clin Cancer Res 22(2):291–300. https://doi.org/10.1158/1078-0432.CCR-14-3296
Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M et al (2015) Genomic spectra of biliary tract cancer. Nat Genet 47(9):1003–1010. https://doi.org/10.1038/ng.3375
Weber SM, Ribero D, O’Reilly EM, Kokudo N, Miyazaki M, Pawlik TM (2015) Intrahepatic cholangiocarcinoma: expert consensus statement. HPB (Oxford) 17(8):669–680. https://doi.org/10.1111/hpb.12441
Jarnagin WR, Fong Y, DeMatteo RP, Gonen M, Burke EC, Bodniewicz BSJ et al (2001) Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann Surg 234(4):507–517 discussion 517-9. https://doi.org/10.1097/00000658-200110000-00010
Hidalgo E, Asthana S, Nishio H, Wyatt J, Toogood GJ, Prasad KR et al (2008) Surgery for hilar cholangiocarcinoma: the Leeds experience. Eur J Surg Oncol 34(7):787–794. https://doi.org/10.1016/j.ejso.2007.10.005
Yoo T, Park SJ, Han SS, Kim SH, Lee SD, Kim TH et al (2018) Proximal resection margins: more prognostic than distal resection margins in patients undergoing Hilar Cholangiocarcinoma Resection. Cancer Res Treat 50(4):1106–1113. https://doi.org/10.4143/crt.2017.320
Huggett MT, Passant H, Hurt C, Pereira SP, Bridgewater J, Mukherjee S (2014) Outcome and patterns of care in advanced biliary tract carcinoma (ABC): experience from two tertiary institutions in the United Kingdom. Tumori 100(2):219–224. https://doi.org/10.1177/030089161410000217
Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J et al (2014) Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS ONE 9(12):e115383. https://doi.org/10.1371/journal.pone.0115383
Tian W, Hu W, Shi X, Liu P, Ma X, Zhao W et al (2020) Comprehensive genomic profile of cholangiocarcinomas in China. Oncol Lett 19(4):3101–3110. https://doi.org/10.3892/ol.2020.11429
Waseem D, Tushar P (2017) Intrahepatic, perihilar and distal cholangiocarcinoma: management and outcomes. Ann Hepatol 16(1):133–139. https://doi.org/10.5604/16652681.1226927
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR et al (2017) Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23(6):703–713. https://doi.org/10.1038/nm.4333
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1. https://doi.org/10.1126/scisignal.2004088
Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M et al (2015) Genomic spectra of biliary tract cancer. Nat Genet 47(9):1003–1010. https://doi.org/10.1038/ng.3375
Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N et al (2017) Whole-genome and epigenomic landscapes of etiologically distinct subtypes of Cholangiocarcinoma. Cancer Discov 7(10):1116–1135. https://doi.org/10.1158/2159-8290.CD-17-0368
Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E et al (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of Cancer. Cell 173(2):291–304e6. https://doi.org/10.1016/j.cell.2018.03.022
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J et al OncoKB: a Precision Oncology Knowledge Base. JCO Precis Oncol 2017;2017:PO.17.00011. https://doi.org/10.1200/PO.17.00011
Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR et al (2020) Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 17(9):557–588. https://doi.org/10.1038/s41575-020-0310-z
Xue L, Guo C, Zhang K, Jiang H, Pang F, Dou Y et al (2019) Comprehensive molecular profiling of extrahepatic cholangiocarcinoma in chinese population and potential targets for clinical practice. Hepatobiliary Surg Nutr 8(6):615–622. https://doi.org/10.21037/hbsn.2019.08.05
Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M et al (2018) Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 24(17):4154–4161. https://doi.org/10.1158/1078-0432.CCR-18-0078
Guest RV, Boulter L, Dwyer BJ, Kendall TJ, Man TY, Minnis-Lyons SE et al (2016) Notch3 drives development and progression of cholangiocarcinoma. Proc Natl Acad Sci U S A 113(43):12250–12255. https://doi.org/10.1073/pnas.1600067113
O’Rourke CJ, Matter MS, Nepal C, Caetano-Oliveira R, Ton PT, Factor VM et al (2020) Identification of a pan-gamma-secretase inhibitor response signature for Notch-Driven Cholangiocarcinoma. Hepatology 71(1):196–213. https://doi.org/10.1002/hep.30816
Zhang Y, Ma Z, Li C, Wang C, Jiang W, Chang J et al (2022) The genomic landscape of cholangiocarcinoma reveals the disruption of post-transcriptional modifiers. Nat Commun 13(1):3061. https://doi.org/10.1038/s41467-022-30708-7
Fujimoto A, Furuta M, Shiraishi Y, Gotoh K, Kawakami Y, Arihiro K et al (2015) Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun 6:6120. https://doi.org/10.1038/ncomms7120
Chaisaingmongkol J, Budhu A, Dang H, Rabibhadana S, Pupacdi B, Kwon SM et al (2017) Common molecular subtypes among asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 32(1):57–70e3. https://doi.org/10.1016/j.ccell.2017.05.009
Feng F, Wu X, Shi X, Gao Q, Wu Y, Yu Y et al (2021) Comprehensive analysis of genomic alterations of chinese hilar cholangiocarcinoma patients. Int J Clin Oncol 26(4):717–727. https://doi.org/10.1007/s10147-020-01846-z
Khamko R, Wasenang W, Daduang J, Settasatian C, Limpaiboon T (2022) Combined OPCML and AXL expression as a prognostic marker and OPCML enhances AXL inhibitor in Cholangiocarcinoma. In Vivo 36(3):1168–1177. https://doi.org/10.21873/invivo.12816
Nyakas M, Fleten KG, Haugen MH, Engedal N, Sveen C, Farstad IN et al (2022) AXL inhibition improves BRAF-targeted treatment in melanoma. Sci Rep 12(1):5076. https://doi.org/10.1038/s41598-022-09078-z
Zhang S, Xu XS, Yang JX, Guo JH, Chao TF, Tong Y (2018) The prognostic role of Gas6/Axl axis in solid malignancies: a meta-analysis and literature review. Onco Targets Ther 11:509–519. https://doi.org/10.2147/OTT.S150952
Yan XQ, Zhang W, Zhang BX, Liang HF, Zhang WG, Chen XP (2013) Inactivation of Smad4 is a prognostic factor in intrahepatic cholangiocarcinoma. Chin Med J (Engl) 126(16):3039–3043. https://doi.org/10.3760/cma.j.issn.0366-6999.20121235
Song H, Huang Y, Jiang X (2022) Mutation spectrum associated with metastasis of advanced cholangiocarcinoma. J Int Med Res 50(6):3000605221102080. https://doi.org/10.1177/03000605221102080
Zhang Y, Liu X, Li A, Tang X (2022) A pan-cancer analysis on the carcinogenic effect of human adenomatous polyposis coli. PLoS ONE 17(3):e0265655. https://doi.org/10.1371/journal.pone.0265655
Aghabozorgi AS, Bahreyni A, Soleimani A, Bahrami A, Khazaei M, Ferns GA et al (2019) Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 157:64–71. https://doi.org/10.1016/j.biochi.2018.11.003
Wang W, Shao F, Yang X, Wang J, Zhu R, Yang Y et al (2021) METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N6-methyladenosine-dependent YTHDF binding. Nat Commun 12(1):3803. https://doi.org/10.1038/s41467-021-23501-5
Koch E, Fiedler W, Tannapfel A, Ballhausen WG (2003) Alteration of the fragile histidine triad gene in intrahepatic cholangiocarcinoma. Eur J Gastroenterol Hepatol 15(8):907–913. https://doi.org/10.1097/00042737-200308000-00012
Gulve N, Su C, Deng Z, Soldan SS, Vladimirova O, Wickramasinghe J et al (2022) DAXX-ATRX regulation of p53 chromatin binding and DNA damage response. Nat Commun 13(1):5033. https://doi.org/10.1038/s41467-022-32680-8
Liu SB, Lin XP, Xu Y, Shen ZF, Pan WW (2018) DAXX promotes ovarian cancer ascites cell proliferation and migration by activating the ERK signaling pathway. J Ovarian Res 11(1):90. https://doi.org/10.1186/s13048-018-0462-4
Puto LA, Brognard J, Hunter T, Transcriptional Repressor DAXX (2015) Promotes prostate Cancer tumorigenicity via suppression of Autophagy. J Biol Chem 290(25):15406–15420. https://doi.org/10.1074/jbc.M115.658765
Wu C, Ding H, Wang S, Li Y, Liu SB, Wang X et al (2020) DAXX inhibits cancer stemness and epithelial-mesenchymal transition in gastric cancer. Br J Cancer 122(10):1477–1485. https://doi.org/10.1038/s41416-020-0800-3
Engel NW, Schliffke S, Schüller U, Frenzel C, Bokemeyer C, Kubisch C et al (2019) Fatal myelotoxicity following palliative Chemotherapy with Cisplatin and Gemcitabine in a patient with stage IV Cholangiocarcinoma Linked to Post Mortem diagnosis of Fanconi Anemia. Front Oncol 9:420. https://doi.org/10.3389/fonc.2019.00420
Roll JD, Reuther GW (2012) ALK-activating homologous mutations in LTK induce cellular transformation. PLoS ONE 7(2):e31733. https://doi.org/10.1371/journal.pone.0031733
Pavese JM, Ogden IM, Voll EA, Huang X, Xu L, Jovanovic B et al (2014) Mitogen-activated protein kinase kinase 4 (MAP2K4) promotes human prostate cancer metastasis. PLoS ONE 9(7):e102289. https://doi.org/10.1371/journal.pone.0102289
Liu S, Huang J, Zhang Y, Liu Y, Zuo S, Li R (2019) MAP2K4 interacts with vimentin to activate the PI3K/AKT pathway and promotes breast cancer pathogenesis. Aging 11(22):10697–10710. https://doi.org/10.18632/aging.102485
Ahn YH, Yang Y, Gibbons DL, Creighton CJ, Yang F, Wistuba II et al (2011) Map2k4 functions as a tumor suppressor in lung adenocarcinoma and inhibits tumor cell invasion by decreasing peroxisome proliferator-activated receptor γ2 expression. Mol Cell Biol 31(21):4270–4285. https://doi.org/10.1128/MCB.05562-11
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY et al (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51(2):202–206. https://doi.org/10.1038/s41588-018-0312-8
Funding
This study was supported by Beijing Municipal Natural Science Foundation (7202220).
Author information
Authors and Affiliations
Contributions
Drafting of the manuscript: Changkun Zhang. Data collection and analysis: Changkun Zhang and Qin Zhang. Figure preparation and verification of the original data: Xia You. Preparation of the tables and verification of the original data: Changkun Zhang and Xia You. Revision of the manuscript for important intellectual content: Changkun Zhang and Dong Wang. Design of the study and review of the final manuscript: Dong Wang.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Institutional Ethics Committee of Peking University People’s Hospital (2021PHB279-001).
Consent to participate
Informed consent was waived given the retrospective nature of the study.
Consent to publish
Not applicable.
Competing interests
The authors have no conflict of interests related to this publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, C., You, X., Zhang, Q. et al. Molecular profiling and prognostic analysis in Chinese cholangiocarcinoma: an observational, retrospective single-center study. Invest New Drugs 42, 24–34 (2024). https://doi.org/10.1007/s10637-023-01394-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-023-01394-z