
Summary
Breast cancer threatens women’s health. Although there are a lot of methods to treat breast cancer, chemotherapy resistance still hinders the effectiveness of treatment. This study attempts to explore the mechanism of chemotherapy resistance from the perspective of miRNA and look for several new targets for developing new drugs. Three datasets (GSE73736, GSE71142 and GSE6434) from Gene Expression Omnibus (GEO) were used for the bioinformatics analysis. Differentially expressed miRNAs (DE-miRNAs) and differentially expressed genes (DE-genes) were obtained by using R package “limma”. DAVID tool was used to perform gene ontology annotation analysis (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for the overlapping genes. Protein-protein interaction (PPI) network was established by STRING database and visualized by software Cytoscape. Hub genes were identified by software Cytoscape. The prognostic value of hub genes was assessed through Kaplan–Meier plotter website. In total, 22 DE-miRNAs, 1932 DE-genes and top 10 hub genes were obtained. The genes were mainly enriched in cell signaling pathways like ErbB signaling pathway and PI3K / AKT/mTOR pathway. These pathways have a significant impact on the proliferation, invasion and drug resistance in cancer. MiRNA-Gene interaction may provide new insight for exploring the mechanism of chemotherapy resistance in breast cancer. Our study ultimately identified effective biomarkers and potential drug targets, which may enhance the effect of chemotherapy in patients with breast cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common malignant tumor among women and is a highly heterogeneous disease [1]. According to the hormone status and human epidermal growth factor receptor 2 (Her2) status, it can be divided into four subtypes: luminal A, luminal B, Her2-positive and triple negative breast cancer (TNBC) [2]. Due to improvements in treatment and early detection, the mortality of breast cancer declines year by year. However, as a result of resistance to chemotherapy, some patients especially those who can not benefit from other treatments still have low survival rates [3]. Therefore, it is very necessary to identify effective biomarkers and novel drug targets to improve the prognosis of breast cancer patients.
MicroRNA(miRNA) is a class of small noncoding RNAs whose length is 19–22 nucleotides. MiRNAs can regulate gene expression at the post-transcriptional level by binding to the complementary sequence in the untranslated regions (UTRs) of their target mRNAs [4]. Besides, miRNAs also can interact with long non-coding RNAs (lncRNAs), preventing miRNAs from binding to their target genes, thereby interfering with gene expression [5]. In addition to cancer, aberrant expression of miRNAs is also associated with many other human diseases, like Parkinson’s disease and Coronary Artery disease [6, 7]. In breast cancer, studies have shown that miRNAs are involved in proliferation, apoptosis, migration, invasion and drug-resistance [8,9,10,11].
In this study, we detected differentially expressed miRNAs between chemo-sensitive breast cancer tissues and chemo-resistant breast cancer tissues through the integration of two microarray profiling datasets from Gene Expression Omnibus (GEO) database. Then, we used miRNA-Genes interaction prediction database to obtain their target genes and compared them with chemo-resistant related genes from another dataset to generate a link between chemo-resistant miRNAs and genes. Finally, we applied bioinformatics analysis to identify the miRNAs and genes that play an important role in breast cancer chemotherapy resistance, which might become new biomarkers or drug targets.
Materials and methods
MiRNA microarray
The microarray datasets GSE71142, GSE73736 and GSE6434 were downloaded from the GEO database. GSE71142 and GSE73736 contained miRNA microarrays from both chemo-sensitive tissues and chemo-resistant tissues of breast cancer. GSE6434 contained the gene expression data from chemotherapy sensitive and resistant patients.
Screening for DE-miRNAs
The raw data were normalized and analyzed by R package “limma” from the Bioconductor project. The miRNAs with |log2 fold changes (FC)|>1 and P value<0.05 were considered as DE-miRNAs. The overlapping DE-miRNAs from the two datasets were finally selected.
Prediction of target genes of DE-miRNAs and identification of DE-genes
The target genes of DE-miRNAs were predicted by miRWalk3 database, which predicted target genes by using its own TarPmiR algorithm or other miRNA-target prediction datasets like TargetScan, miRDB and miRTarBase. Only common target genes predicted by at least two methods were chosen as potential target genes. The common genes from both the potential target genes and differentially expressed genes in GSE6434 were considered as DE-genes.
Gene ontology annotation (GO) and KEGG pathway analysis
The online website, DAVID (http://david-d.ncifcrf.gov/), was used to perform gene ontology annotation and KEGG pathway enrichment analysis for DE-genes. All the data were visualized by R package “ggplot2”.
PPI network analysis, hub gene identification and miRNA-gene network construction
The STRING database is an online tool to analyze protein-protein interaction. We submitted the DE-genes to the STRING database and only chose the interactions with a confidence score > 0.7. The PPI network was constructed by software Cytoscape. The degree of connectivity in networks was calculated to obtain the significant nodes and hub genes (25). The miRNA-gene network was visualized by software Cytoscape.
Survival analysis of hub genes
Kaplan-Meier plotter website (https://kmplot.com/analysis/) is able to assess the effect of fifty thousand genes on survival in several cancer types including breast, ovarian, lung and gastric cancer. In order to analyze the relapse free survival (RFS), the patient samples were split into two groups according to the median level of each gene’s expression. The website can automatically compare the two patient groups by a Kaplan-Meier survival plot and calculate the hazard ratio (HR) with 95% confidence intervals and log rank P values.
Results
Identification of DE-miRNAs and their target genes
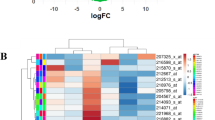
According to the previously defined threshold, 22 DE-miRNAs were finally screened out, including 10 upregulated and 12 downregulated miRNAs (Fig. 1) in chemo-resistant tissues, compared with chemo-sensitive tissues. MiRWalk3 database was employed to predict the target genes of the DE-miRNAs and finally generated 3754 potential target genes, including 1824 genes related to upregulated miRNAs and 1930 genes related to downregulated miRNAs. Then, we took the intersection of two miRNA sourced target genes and chose the common part of genes between miRNA sourced genes and GSE6434 sourced genes. Totally, 1932 genes were acquired, including 978 genes for upregulated miRNAs and 954 genes for downregulated miRNAs.

Heatmap for DE-miRNAs. a GSE71142, b GSE73736
Functional and pathway enrichment analysis
To further investigate the biological functions, KEGG pathway analysis and GO analysis including molecular function (MF), cellular component (CC) and biological process (BP) were performed through DAVID tools. The top 10 most significant Go terms and KEGG pathway terms for the target genes of upregulated DE-miRNAs were shown in Fig. 2. On the BP level, the genes were mainly enriched in positive regulation of transcription from RNA polymerase II promoter, regulation of phosphatidylinositol 3-kinase signaling and signal transduction. On the CC level, the genes were mainly enriched in cytosol, nucleoplasm and cytoplasm. On the MF level, the genes were mainly enriched in protein binding, RNA polymerase II core promoter proximal region sequence specific binding and kinase activity. The enriched KEGG pathways included ErbB signaling pathway, pathways in cancer and insulin signaling pathway.

GO and KEGG pathway analysis of the overlapping target genes from the upregulated DE-miRNAs. a Biological process, b Cellular component, c Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and d Molecular Function
We also conducted GO functional and KEGG pathway analysis for the target genes of the downregulated DE-miRNAs. The top 10 most significant Go terms and KEGG pathway terms were shown in Fig. 3. On the BP level, the genes were mainly enriched in positive regulation of transcription from RNA polymerase II promoter, negative regulation of transcription from RNA polymerase II promoter and intracellular signal transduction. On the CC level, the genes were mainly enriched in cytosol, cytoplasm and nucleoplasm. On the MF level, the genes were mainly enriched in protein binding, transcription factor binding and sequence-specific DNA binding. The enriched KEGG pathways included proteoglycans in cancer, neurotrophin signaling pathway and GnRH signaling pathway.

GO and KEGG pathway analysis of the overlapping target genes from the downregulated DE-miRNAs. a Biological process, b Cellular component, c Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and d Molecular Function
PPI network analysis, hub gene identification and miRNA-gene network construction
As we all know, proteins usually perform their functions by interacting with other proteins. Thus, we used STRING database to analyze the PPI network of the DE-genes of the both upregulated and downregulated miRNAs. Software Cytoscape was used to identify the top 25 hub genes according to degree level (Fig. 4). For the upregulated miRNAs, the top 10 hub genes were MAPK1, CTNNB1, PIK3CA, PIK3R1, MAPK3, KRAS, APP, HSP90AA1, PTEN and MAPK8. Among these genes, MAPK1 showed the highest node degree(degree=64). For the downregulated miRNAs, the top 10 hub genes were PRKACA, CDC42, MAPK1, MAPK8, KRAS, POLR2E, SMAD3, STAT3, PTPN11 and PTEN. Among these genes, PRKACA showed the highest node degree(degree=54). As shown in Fig. 5, miRNA-hub genes network was constructed by software Cytoscape. Hub genes of upregulated miRNAs could be regulated by miR-200c-3p, miR-214-3p, miR-33b-3p, miR-3927-3p, miR-4277, miR-4422 and miR-4771. Among them, miR-214-3p was predicted to target the most hub genes (n=12), indicating that this miRNA may play an important role in chemotherapy resistance in breast cancer. The hub genes of downregulated miRNAs could be regulated by miR-30c-2-3p, miR-586, miR-587, miR-592, miR-1178-3p, miR-2681-3p, miR-3116, miR-3162-5p, miR-4282 and miR-4472. Among them, miR-587 was predicted to target the most hub genes (n=9). Besides, miR-4472 and miR-4422 could respectively regulate 6 and 7 hub genes suggesting these four miRNAs may have great effect on chemotherapy resistance in breast cancer.

The PPI network of top 25 hub genes. a Hub genes targeted by upregulated miRNAs, b Hub genes targeted by downregulated miRNAs

miRNA-hub genes interaction networks. a The network of hub genes and upregulated miRNAs b The network of hub genes and downregulated miRNAs
Prognostic value of hub genes
The prognostic value of 16 unrepeated hub genes was obtained from Kaplan–Meier plotter website. As shown in Fig. 6, 13 out of 16 genes had statistical significance including SMAD3, PTPN11, KRAS. High expression of PIK3R1, MAPK3, KRAS, MAPK8, PRKACA, CDC42, SMAD3, STAT3, PTPN11 and lower expression of PIK3CA, HSP90AA1, PTEN, POLR2E were associated with a better RFS of breast cancer patients (p<0.05).

Kaplan–Meier survival curves of 16 hub genes. a MAPK1, b CTNNB1, c PIK3CA, d PIK3R1, e MAPK3, f KRAS, g APP, h HSP90AA1, i PTEN, j MAPK8, k PRKACA, l CDC42, m POLR2E, n SMAD3, o STAT3, p PTPN11
Discussion
Although significant progress has been achieved in targeted therapy, immunotherapy and endocrine therapy, breast cancer chemotherapy resistance is still the main reason for limiting patients’ survival rates [12]. Yusuf Baran et.al pointed out that there are many reasons for chemotherapy resistance and the main reason that limits the success of chemotherapy is the development of multidrug resistance [13]. Wang et.al showed that changing the expression of miRNAs can effectively re-sensitise cancer cells to cisplatin regimen in certain types of cancers including breast, gastric, ovarian and prostate [14]. Here, we explored the possible biomarkers and drug targets of breast cancer chemotherapy resistance from the perspective of miRNA. In this study, we used the method of bioinformatics to download data from public databases and screened out the disordered miRNAs associated with breast cancer chemotherapy resistance and their corresponding target genes. We found that mir-3927-3p had the largest difference multiple in the upregulated miRNAs, but probably because mir-3927-3p was found late, it has not been studied yet in the aspect of tumor proliferation or drug resistance. Mir-619 had the largest difference multiple in the downregulated miRNAs. Study has shown that the expression of mir-619 decreased in drug resistant head and neck tumors [15]. As a sponge to lncRNA and mRNA, mir-619 can not only promote tumor angiogenesis but also the proliferation and invasion of colorectal cancer [16, 17]. In our study, the genes interacting with miR-214-3p were the most, so we can speculate that miR-214-3p may play a big role in chemotherapy resistance of breast cancer. Study showed that miR-214-3p can enhance cells’ proliferation by directly binding to the 3’UTR of FOXP2 and down-regulating its expression in breast cancer [18]. MiR-214-3p also can regulate the viability, invasion, migration and Epithelial-Mesenchymal Transition (EMT) of TNBC cells by targeting to ST6GAL1 [19]. Besides, overexpression of miR-214-3p in NCCIT cells may lead to cisplatin resistance [20]. Therefore, what roles these miRNAs play in chemotherapy resistance is worth studying in depth.
Subsequently, we performed functional enrichment analysis and pathway analysis of target genes of dysregulated miRNAs. Consistent with the predictions, we found that these genes were mainly concentrated in the cell signal transduction, and the signaling pathways were concentrated in the cell proliferation and invasion-related pathways, such as Ras signaling pathway, mTOR signaling pathway and ErbB signaling pathway. In the study of hub genes, we also found the similar situation. Most of the identified hub genes were key genes in the signal transduction pathways, such as MAPK1, CTNNB1 (encoding β-catenin), KRAS, PTEN, SMAD3, BRCA1, etc. They mainly focus on Ras/MAPK pathway, PI3K / AKT/mTOR pathway, Wnt / β-catenin/mTOR pathway and SMAD / TGF- β pathway. Ras/MAPK pathway is a classical pathway in tumor. About 15% TNBC patients were Ras/MAPK altered at the genomic level and Ras/MAPK activity can suppress expression of MHC-I and MHC-II, which suggests cancer cells can circumvent antigen presentation pathways by activating the MAPK pathway [21]. It is believed that Ras/MAPK pathway is involved in chemotherapy resistance in breast cancer. Steelman et.al pointed out that PI3K/AKT/mTOR and Ras/ MAPK signaling pathways can regulate HER2 expression, while HER2 overexpression can also activate both the PI3K/Akt/mTOR and Ras/ MAPK pathways [22]. New evidence showed that activation of PI3K/AKT/mTOR pathway can partly cause the drug resistance in HER2-positive breast cancer with brain metastasis [23]. In addition, TGF-β/SMAD3 pathway also plays an important role in the induction and maintenance of resistance to anti-HER2 drugs and inhibiting SMAD3 can attenuate resistance to anti-HER2 drugs in HER2-positive breast cancer cells [24]. All these studies indicate that our research is credible and the interactions between miRNAs and genes may have a great effect on the chemotherapy resistance in breast cancer.
In summary, our study identified several miRNAs and hub genes involved in chemotherapy resistance in breast cancer. According to the functional analysis and survival analysis, these genes and miRNAs may serve as potential biomarkers or drug targets contributing to improve the survival rates of breast cancer patients with chemotherapy resistance. In the next plan, we will continue to explore the underlying mechanisms of breast cancer chemotherapy resistance in more depth.
Data availability
All data is available under reasonable request.
References
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70(1):7–30. https://doi.org/10.3322/caac.21590
DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, Jemal A, Siegel RL (2019) Breast cancer statistics, 2019. CA Cancer J Clin 69(6):438–451. https://doi.org/10.3322/caac.21583
Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J, Cardoso F (2019) Breast cancer. Nat Rev Dis Primers 5(1):66. https://doi.org/10.1038/s41572-019-0111-2
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297. https://doi.org/10.1016/s0092-8674(04)00045-5
Tay Y, Rinn J, Pandolfi PP (2014) The multilayered complexity of ceRNA crosstalk and competition. Nature 505(7483):344–352. https://doi.org/10.1038/nature12986
Hu YB, Zhang YF, Wang H, Ren RJ, Cui HL, Huang WY, Cheng Q, Chen HZ, Wang G (2019) miR-425 deficiency promotes necroptosis and dopaminergic neurodegeneration in Parkinson's disease. Cell Death Dis 10(8):589. https://doi.org/10.1038/s41419-019-1809-5
Dégano IR, Camps-Vilaró A, Subirana I, García-Mateo N, Cidad P, Muñoz-Aguayo D, Puigdecanet E, Nonell L, Vila J, Crepaldi FM, de Gonzalo-Calvo D, Llorente-Cortés V, Pérez-García MT, Elosua R, Fitó M, Marrugat J (2020) Association of Circulating microRNAs with coronary artery disease and usefulness for reclassification of healthy individuals: the REGICOR study. J Clin Med 9(5):1402. https://doi.org/10.3390/jcm9051402
Liu Y, Yang Y, Du J, Lin D, Li F (2020) MiR-3613-3p from carcinoma-associated fibroblasts exosomes promoted breast cancer cell proliferation and metastasis by regulating SOCS2 expression. IUBMB Life 72:1705–1714. https://doi.org/10.1002/iub.2292
Lei B, Wang D, Zhang M, Deng Y, Jiang H, Li Y (2020) miR-615-3p promotes the epithelial-mesenchymal transition and metastasis of breast cancer by targeting PICK1/TGFBRI axis. J Exp Clin Cancer Res 39(1):71. https://doi.org/10.1186/s13046-020-01571-5
Zhou Q, Guo J, Huang W, Yu X, Xu C, Long X (2020) Linc-ROR promotes the progression of breast cancer and decreases the sensitivity to rapamycin through miR-194-3p targeting MECP2. Mol Oncol 14:2231–2250. https://doi.org/10.1002/1878-0261.12700
Yi D, Wang R, Shi X, Xu L, Yilihamu Y, Sang J (2020) METTL14 promotes the migration and invasion of breast cancer cells by modulating N6-methyladenosine and hsa-miR-146a-5p expression. Oncol Rep 43(5):1375–1386. https://doi.org/10.3892/or.2020.7515
Kachalaki S, Ebrahimi M, Mohamed Khosroshahi L, Mohammadinejad S, Baradaran B (2016) Cancer chemoresistance; biochemical and molecular aspects: a brief overview. Eur J Pharm Sci 89:20–30. https://doi.org/10.1016/j.ejps.2016.03.025
Kartal-Yandim M, Adan-Gokbulut A, Baran Y (2016) Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol 36(4):716–726. https://doi.org/10.3109/07388551.2015.1015957
Wang S, Li MY, Liu Y, Vlantis AC, Chan JY, Xue L, Hu BG, Yang S, Chen MX, Zhou S, Guo W, Zeng X, Qiu S, van Hasselt CA, Tong MC, Chen GG (2020) The role of microRNA in Cisplatin resistance or sensitivity. Expert Opin Ther Targets 24:885–897. https://doi.org/10.1080/14728222.2020.1785431
Zheng Y, Song A, Zhou Y, Zhong Y, Zhang W, Wang C, Ding X, Du Y, Zhang W, Li G, Wu H, Wu Y, Song X (2020) Identification of extracellular vesicles-transported miRNAs in Erlotinib-resistant head and neck squamous cell carcinoma. J Cell Commun Signal 14:389–402. https://doi.org/10.1007/s12079-020-00546-7
Kim DH, Park S, Kim H, Choi YJ, Kim SY, Sung KJ, Sung YH, Choi CM, Yun M, Yi YS, Lee CW, Kim SY, Lee JC, Rho JK (2020) Tumor-derived exosomal miR-619-5p promotes tumor angiogenesis and metastasis through the inhibition of RCAN1.4. Cancer Lett 475:2–13. https://doi.org/10.1016/j.canlet.2020.01.023
Chen G, Gu Y, Han P, Li Z, Zhao JL, Gao MZ (2019) Long noncoding RNA SBF2-AS1 promotes colorectal cancer proliferation and invasion by inhibiting miR-619-5p activity and facilitating HDAC3 expression. J Cell Physiol 234(10):18688–18696. https://doi.org/10.1002/jcp.28509
Qin CX, Yang XQ, Jin GC, Zhan ZY (2019) LncRNA TSLNC8 inhibits proliferation of breast cancer cell through the miR-214-3p/FOXP2 axis. Eur Rev Med Pharmacol Sci 23(19):8440–8448. https://doi.org/10.26355/eurrev_201910_19156
Tao Y, Zhao Z, Ma J, Dong L, Liang Y, Li S, Mao Y, Li Y, Zhang Y (2019) MiR-214-3p regulates the viability, invasion, migration and EMT of TNBC cells by targeting ST6GAL1. Cytotechnology 71(6):1155–1165. https://doi.org/10.1007/s10616-019-00352-z
Hsieh TH, Liu YR, Chang TY, Liang ML, Chen HH, Wang HW, Yen Y, Wong TT (2018) Global DNA methylation analysis reveals miR-214-3p contributes to cisplatin resistance in pediatric intracranial nongerminomatous malignant germ cell tumors. Neuro-oncology 20(4):519–530. https://doi.org/10.1093/neuonc/nox186
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV, Sánchez V, Sanders ME, Cook RS, Pilkinton MA, Mallal SA, Wang K, Miller VA, Stephens PJ, Yelensky R, Doimi FD, Gómez H, Ryzhov SV, Darcy PK, Arteaga CL, Balko JM (2016) RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res 22(6):1499–1509. https://doi.org/10.1158/1078-0432.Ccr-15-1125
Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA (2011) Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 3(3):192–222. https://doi.org/10.18632/aging.100296
Kabraji S, Ni J, Lin NU, Xie S, Winer EP, Zhao JJ (2018) Drug resistance in HER2-positive breast cancer brain metastases: blame the barrier or the brain? Clin Cancer Res 24(8):1795–1804. https://doi.org/10.1158/1078-0432.Ccr-17-3351
Chihara Y, Shimoda M, Hori A, Ohara A, Naoi Y, Ikeda JI, Kagara N, Tanei T, Shimomura A, Shimazu K, Kim SJ, Noguchi S (2017) A small-molecule inhibitor of SMAD3 attenuates resistance to anti-HER2 drugs in HER2-positive breast cancer cells. Breast Cancer Res Treat 166(1):55–68. https://doi.org/10.1007/s10549-017-4382-6
Acknowledgements
We thank every member of the research group for their cooperation. We are also grateful to the creators and maintainers of the public databases for providing resources for our research.
Author information
Authors and Affiliations
Contributions
Bo Chen designed and guided this research. Yujie Zhao, Nanxi Peng and Zuo Tao contributed to collection of data. Ming Wu performed data analysis and wrote the first draft of the manuscript. Bo Chen commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
All authors consent to the publication of this study.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, M., Zhao, Y., Peng, N. et al. Identification of chemoresistance-associated microRNAs and hub genes in breast cancer using bioinformatics analysis. Invest New Drugs 39, 705–712 (2021). https://doi.org/10.1007/s10637-020-01059-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-01059-1