
Abstract
Inflammatory bowel disease (IBD) is an important etiologic factor in the development of colorectal cancer (CRC). The risk of CRC begins to increase 8 or 10 years after the diagnosis of IBD. This type of cancer is called colitis-associated CRC (CA-CRC). The molecular pathogenesis of inflammatory epithelium might play a critical role in the development of CA-CRC. Genetic alterations detected in CA-CRC such as genetic mutations, microsatellite instability, and DNA hypermethylation are also recognized in sporadic CRC; however, there are differences in the timing and frequency of molecular events between CA-CRC and sporadic CRC. Interaction between gene–environmental factors, including inflammation, lifestyle, psychological stress, and prior appendectomy, might be associated with the etiopathology of IBD. The mucosal inflammatory mediators, such as oxidant stress, free radicals, and chemokines, may cause the genetic alterations. Understanding the molecular mechanisms of CA-CRC might be important to develop clinical efficacies for patients with IBD. This review discusses the molecular characteristics of CA-CRC, especially ulcerative colitis-associated CRC, including clinical features, signaling pathways, and interactions between genetic alterations and environment involved in inflammatory carcinogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crohn and Rosenberg reported the first case of adenocarcinoma complicating ulcerative colitis (UC) in 1925 [1]. Since then, it has been recognized that the risk of developing colorectal cancer (CRC) is increased in patients with long-term inflammatory bowel disease (IBD) such as UC and Crohn’s disease (CD) [2, 3]. Chronic inflammation plays a critical role in human carcinogenesis in some types of solid cancers [4, 5]. Colitis-associated colorectal cancer (CA-CRC) is also believed to occur by a progression from a non-neoplastic inflammatory epithelium to dysplasia to carcinoma [2]. Recent studies elucidate the molecular pathogenesis of CA-CRC, particularly in ulcerative colitis-associated CRC (UC-CRC) [6–8]. CA-CRC shows characteristic genetic changes including nucleotide mutation, chromosomal alteration, and hypermethylation in oncogenes and tumor suppressor genes. Reactive oxygen, nitrogen species, and cytokines involved in inflammatory mucosa might be associated with these genetic alterations as pathogenesis of CA-CRC. Analysis of the correlation between these molecular features and clinicopathologic features in CA-CRC might be useful to develop new biomarkers and drugs for patients with CA-CRC [9, 10]. In this paper, we present an overview of the molecular characteristics in CA-CRC, mainly in UC-CRC.
Clinical Features of Colitis-Associated CRC
An inflammatory environment is believed to play an important role for the pathogenesis of CRC in patients with chronic colitis [2]. UC-CRC accounts for about 1 % of all CRC [11]. The risk of CRC begins to increase 8 or 10 years after the diagnosis of UC [12–14]. UC-CRC patients more frequently have multiple cancerous lesions and histologically show permeating pattern of spread including mucinous or signet ring cell carcinomas [15–17]. Risk factors for CRC with UC patients include young age at diagnosis [12, 18], longer duration [19], greater anatomic extent of colonic involvement [20], the degree of inflammation [21–23], family history of CRC [24, 25], and the presence of primary sclerosing cholangitis [26]. Especially, the extent of colitis is an independent risk factor for the development of CRC. UC patients with pancolitis are at highest risk, left-sided colitis carries a moderate risk, and patients with proctitis and protosigmoiditis are at similar risk of CRC without IBD [21–23]. In addition, smoking, pseudopolyps, persisting inflammation of the colon, and backwash ileitis are also risk factor for CRC [27, 28]. Also, the relative risk of CRC in patients with CD was two- to threefold, and that for small bowel carcinoma was ten- to 12-fold [29, 30], while some studies reported that few CD patients developed cancer of the small intestine [31, 32].
Surveillance Colonoscopy
Surveillance colonoscopy is currently the most widely used method to detect dysplasia and cancer in patients with IBD [28, 33–37]. Current guidelines from the British Society of Gastroenterology (BSG) [36, 38–41], European Crohn’s and Colitis Organization (ECCO) [42], and the American Gastroenterology Association (AGA) [43] recommend colonoscopic surveillance every 1–5 years at the beginning of 8–10 years after symptom onset for IBD-colitis patients. The main aim of surveillance programs is to detect early dysplastic alterations because cancer surveillance is based on the high-risk factors that identify patients who are likely to develop cancer. The recommended guidelines of colonoscopy are as follows [36, 39–41]: (a) Screening colonoscopy should be performed when the disease is in remission. (b) Initial surveillance colonoscopy should be performed in each patient beginning 8–10 years after symptom onset, partly to reassess disease extent. (c) Regular surveillance should begin on an annual or biannual basis beginning 8–10 years of disease for patients with left-sided or extensive colitis after symptom onset. (d) Two to four random biopsy specimens should be taken every 10 cm from the entire colon, with additional samples of suspicious areas. Particularly in UC, consideration should be given to taking 4-quadrant biopsies every 5 cm in the lower sigmoid and rectum, because the frequency of CRC is higher in this region. (e) If dysplasia (of any grade) is detected, the biopsies should be reviewed by a second gastrointestinal pathologist, and if confirmed, then colectomy is usually advisable. On the other hand, surveillance of small bowel cancer is not recommended, because of its low risk of small bowel cancer in CD [44].
Molecular Features
Genetic Alterations in Sporadic CRC
It is widely accepted that sporadic CRC result from the sequential accumulation of alterations in genes that regulate the growth of colonic epithelial cells [45, 46]. The multistep carcinogenesis concept resulted from correlative analyses between the neoplastic lesions of the colon (adenomas and carcinomas), and the genetic alterations found in association with each of the steps in the progression [47, 48]. These alterations include activating point mutations of K-ras [49, 50] and inactivation of specific tumor suppressor genes (TSGs), most notably the adenomatous polyposis coli (APC) gene on chromosome 5q21 [51], the p53 gene on 17p13 [52], and one of several candidate TSGs on chromosome 18q, most likely deleted in colon cancer (DCC)or deleted in pancreatic cancer-4 (DPC4) gene [53]. Mutational activation of K-ras has been found in more than 50 % of adenomas and CRCs [50, 54]. A typical mechanism for the inactivation of TSGs in colorectal neoplasms is the sequential inactivating mutation on one allele, followed by allelic loss, or loss of heterozygosity (LOH), of the other allele [52, 54]. Inactivation of the DNA mismatch repair (MMR) genes hMSH2 on chromosome 2p or hMLH1 on 3p leads to the mutator phenotype, which occurs in 10–15 % of CRCs [55, 56] by promoter hypermethylation.
Genetic Alterations in Ulcerative Colitis-Associated CRC and Dysplasia
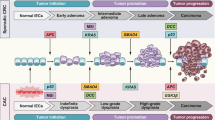
Many of the molecular changes responsible for sporadic CRC development also play a critical role in the carcinogenesis of UC-CRC. There are similarities of the genetic pathway between sporadic colon cancer and colitis-associated CRC, including MSI, DNA methylation, and mutation and eventual LOH of p53. However, distinguishing features of UC-CRC are differences in the timing and frequency of these alterations (Fig. 1). Chromosomal abnormalities are found in non-dysplastic, dysplastic, and cancerous epithelia in the UC-CRC as follows.

Molecular alteration of colitis-associated colorectal cancer and sporadic colorectal cancer. Genetic events of MSI, DNA hypermethylation, and p53 are common in malignant degeneration of both colitis-associated colorectal cancer and sporadic colorectal cancer of the genetic pathway between sporadic colon cancer and colitis-associated colorectal cancer. In contrast, frequency and sequence of APC, K-ras, and DCC/DPC4 differ between the two types. ROS reactive oxygen species, COX-2 cyclooxygenase-2, Rb retinoblastoma gene, MSI microsatellite instability, TGF transforming growth factor, APC adenomatous polyposis coli
K-ras
Most of the studies revealed a lower incidence of K-ras mutation in UC-CRC compared with that in sporadic CRC; K-ras mutation was detected in approximately 15 % of cases with inflamed mucosa (n = 100 [57]; n = 212 [58]; n = 18 [59]), and in 20–25 % of dysplasias (n = 14 [57]; n = 61 [58]; n = 8 [59]; n = 13 [60]) and of carcinomas (n = 4 [57]; n = 5 [58]; n = 9 [60]) in the IBD. The lower incidence of mutation indicated that K-ras does not seem to play a significant role in the development of UC-CRC. In contrast, mutational activation of K-ras has been reported to promote exophytic growth of intestinal neoplasms and may favor development of more differentiated intestinal type of intestinal cancer [50, 61]. UC-CRC is often raised only minimally above the level of the surrounding mucosa and grows in a more diffuse fashion than sporadic CRC [62]. Therefore, the infrequent mutational activation of K-ras might be associated with the macroscopic flat morphology and the histologic diffuse growth of UC-CRC.
Adenomatous Polyposis Coli (APC)
APC loss of function, considered to be an early event in sporadic CRC, is relatively less frequent and usually occurs as a late event in the colitis-associated dysplasia–carcinoma sequence [63–65]. Mutant APC proteins have been detected in around 3–13 % of UC-associated dysplasia- or carcinoma-bearing patients (n = 8 [63, 66]; n = 30 [63, 66]), while 26 % of sporadic cancers exhibited APC mutation (n = 42 [63, 66]). Nearly 30 % of dysplastic lesions and 50 % of cancers exhibited APC LOH (n = 21 [63, 66, 67]; n = 6 [68]). APC mutation may play a relatively unimportant role in the development of UC-associated dysplasia.
p53
The percentage of p53 mutation-containing samples is increasing with the morphological progression to carcinoma. Up to 6 % of normal cases (n = 14); 9 % in the category “indefinite for dysplasia,” 33 % with low-grade dysplasia (n = 13 [60]; n = 22 [68]), 63 % with high-grade dysplasia (n = 12 [69]), and 50–85 % of cases with cancer (n = 18 [70]; n = 10 [71]; n = 9 [60]) have been found to have a deletion of p53. p53 LOH was also observed in nearly 70 % of CRC (n = 8 [68]; n = 17 [67]) and 45 % of dysplastic lesions (n = 33 [72]; n = 19 [68]). p53 analysis might contribute to the accurate pathological diagnosis of UC-associated dysplasia [72]. Thus, the early appearance of p53 alteration might make it a clinically useful marker in the screening for UC-associated dysplasia and in the assessment of cancer risk. In contrast to the reported gatekeeper properties of p53 in sporadic CRC, which in colon adenoma is frequently altered to yield carcinoma [54], p53 might not contribute as a gatekeeper for cancer progression in UC-CRC.
Deleted in Colon Cancer (DCC) or Deleted in Pancreatic Cancer-4 (DPC4)
LOH of 18q, the site of the putative deleted in colon cancer (DCC), was observed in 12 % of eight cancers and 33 % of 30 dysplasia lesions and was not detected in non-dysplastic, inflamed epithelia [73]. Deleted in pancreatic cancer-4 (DPC4)/SMAD2 at 18q was not detected in 10 case of UC-CRC [74]. LOH of 18q is relatively a rare event and may not be important in UC-associated carcinogenesis.
Retinoblastoma Gene (Rb)
Wild-type retinoblastoma gene (Rb) suppresses neoplastic phenotypes and is frequently mutated or lost in malignant tumor [75, 76]. Rb LOH was found in about 50 % of UC-associated carcinoma or dysplasia (n = 27 [67]).
Cyclin-Dependent Kinase Inhibitor p16 (p16INK4a)
The cyclin-dependent kinase inhibitor p16 is a component of the Rb tumor suppressor pathway [77, 78]. LOH studies of the p16 locus at 9p showed a high rate of p16 loss in 50 % of dysplasia (n = 14) [68], and hypermethylation of the p16 promoter region is early occurring event during the process of neoplastic progression in UC as described below [79]. Alterations of p16 may be important early markers of carcinogenic progression in UC patients.
Microsatellite Instability (MSI) and DNA Repair Genes
Inactivation of the DNA MMR system leads to widespread somatic mutations at microsatellite loci. MSI tumors have been found to display microsatellite alterations not only in introns but also in coding exons. Genetic targets of this type of genomic instability include the exons in transforming growth factor β receptor type II (TGFβRII), insulin-like growth factor II receptor (IGFIIR), BAX, hMSH3, and hMSH6, all of which contain mononucleotide repeats in coding sequences [80]. The frequency of MSI in UC-associated neoplasia varies from 2.4 to 50 %, mostly 30 % [81–86]. Fujiwara et al. [81] analyzed the MSI status in fifty-seven patients with UC and found that high-frequent MSI was found in four of 11 cancer cases (36 %); five of 15 dysplasia cases (33 %); five of 11 indefinite cases (45 %); and none of 20 normal cases (0 %). The relatively high frequency of MSI in non-dysplastic, inflamed epithelia, as compared with dysplasia, suggests that MSI may be associated with the pathogenesis of IBD. A frameshift mutation of TGFβRII was significantly correlated with worsening histologic glade. High-frequent MSI was significantly associated with hMLH1 hypermethylation and loss of hMSH2 expression. The carcinogenesis process in UC-CRC was closely associated with the MSI pathway through TGFβRII mutation by a dysfunction of the MMR system [81, 82, 87]. In colorectal epithelial cells, TGFβ signal is involved mainly in the suppression of cell proliferation [88]. Microsatellite mutations of the IGFIIR gene have also been detected in UC-associated neoplasms with MSI [89]. Genetic or epigenetic alterations of mismatch repair proteins, including MLH1 promoter hypermethylation and loss of MSH2 expression, may lead to high-frequent MSI in UC-associated lesions [81, 90]. MiR-155 overexpression being particularly associated to MSI in CA-CRC [91].
Aberrant Methylation
It is well recognized that hypermethylation of CpG islands in the gene promoter regions is associated with silencing of the genes in various tumors. The density of CpG methylation increased from morphologically normal epithelia to dysplasia and carcinoma in UC [92]. The methylation of CpG islands can contribute to genomic instability and appears to be exist in the mucosa of patients with IBD carcinogenesis [81, 92]. Hypermethylation may be due to the elevated rate of cell turnover and oxidative stress characteristic of long-standing UC [92]. Promoter hypermethylation and possible silencing of the p16INK4a gene occurred in 70 % of UC-CRC and 40 % of dysplasia lesions in UC colectomy specimens (n = 89) [79], whereas it was 12.7 % of negative for dysplasia, which suggested that hypermethylation of the p16INK4a promoter region is a frequent and early occurring event during the process of neoplastic progression in UC. Methylation of p16 exon 1 was also found in the regions of normal mucosa in UC patients with dysplasia [92]. Hypermethylation of p14ARF, encoding a modulator of p53 protein levels via MDM-2, was also detected in 19 of 38 (50 %) UC-CRC, four of 12 (33 %) dysplasia lesions, and three of the 5 (60 %) non-cancerous, but only three of 40 (3.7 %) non-dysplastic lesions. Promoter hypermethylation leads to the loss of alternate reading frame product of the CDKN2A locus (p14ARF) [93]. Methylation of p14ARF is a relatively common early event in UC-associated carcinogenesis [94]. Hypermethylation is a frequent mechanism of MLH1 silencing in the subset of UC-associated dysplasias and carcinomas with high-level MSI as described above. E-cadherin promoter methylation was detected in about half of UC-CRC, while there was no difference between the UC-CRC and sporadic CRC [95]. Methylation of these genes offers potential as a biomarker for the early detection of cancer or dysplasia in UC. On the other hand, Issa et al. reported that DNA methylation alterations are uncommon in UC-CRC [96]. It will be necessary to clarify the significance of aberrant methylation in the pathogenesis of UC-CRC.
Crohn’s Disease-Associated CRC
Mutations in CARD15/NOD2 gene that activate nuclear factor NF-kB might be associated with the pathogenic mechanism of Crohn’s disease [97, 98]. Associations have also been found between Crohn’s disease and SNP in the Toll-like receptor 4 [99] or interleukin 23 receptor (IL-23R) [100]. In contrast, few studies in the pathogenesis of Crohn’s disease-associated CRC are available [101]. K-ras and p53 alterations occur early during inflammatory tumor development, while APC, DCC, and TGFβRII mutations are rare in CD-associated CRC [31, 102].
Dysplasia in UC
Dysplasia arising on the grounds of UC may precede the development of carcinoma [62]. Classification of polypoid mucosa of UC is important with respect to clinical treatment for dysplasia. The dysplasia found in IBD is categorized as follows: low-grade dysplasia (LGD), high-grade dysplasia (HGD), dysplasia-associated lesion or mass (DALM), adenoma-like mass (ALM), and adenoma-like DALM. UC with HGD usually leads to a total colectomy because of the high incidence of adenocarcinoma [41, 103, 104]. When HGD in flat mucosa was the initial discovery, surgery or polypectomy is done [41]. In contrast, the management of LGD is controversial [105]. There is evidence that an unrecognized synchronous CRC may already be present in up to 20 % of individuals who undergo colectomy for LGD [8, 103]. In contrast, some studies have shown that patients with LGD have a lower rate of CRC than previously thought [106]. Dysplasia found in DALM is believed to be the origin CRC [107, 108]. DALM has been reported to be associated with CRC in up to 46 % of CD specimens and 62.1 % of UC specimens, supporting the requirement for surgical resection [109, 110]. In contrast, ALM, a lesion found in an area without inflammation, is tend to be treated by standard polypectomy. A strong correlation between p53 mutations and the histologic progression from LGD to invasive carcinoma in patients with IBD has been shown [111]. DALM or areas without any macroscopically visible mucosal alteration are considered to be the origin CRC [107, 108]. Mutations of p53 occur more frequently in DALM. Rb LOH was detected in 25 % of UC patients with DALM, or dysplasia [67]. The guidelines for surveillance colonoscopy also state that particular attention should be paid to DALM [37], because the occurrence of DALM is frequently associated with synchronic or metachronic CRC. Therefore, patients with DALM are recommended to undergo prophylactic proctocolectomy with ileoanal pouch. Recently, raised dysplastic lesions or DALMs with the appearance of sporadic adenomas have been termed adenoma-like DALM. UC-associated non-adenoma-like DALMs have a different molecular genotype than UC-related adenoma-like DALMs and non-CRC sporadic adenomas [112]. Serrated adenomas are polypoid lesions present in the colon that are characterized by saw-toothed or serrated crypts with dysplasia, and the serrated neoplasia pathway was recently proposed in CRC [61]. Bossard et al. [113] found that serrated lesions, such as hyperplastic polyps and sessile serrated polyps/adenomas, accounted for approximately 7 % of premalignant lesions in the inflamed mucosa in patients with IBD. The serrated lesions contained BRAF mutations. It has been reported that LOH of APC, chromosome 3 (chromosome 3p), p53 locus, and K-ras mutations were present in 0 % (0/11), 20 % (2/10), 0 % (0/11), and 37 % (4/11) of sporadic hyperplastic polyps [61]. In contrast, Odze et al. [114] reported that LOH of APC, chromosome 3p, p53, and K-ras mutations were present in 21, 40, 27, and 19 % of UC-associated hyperplastic polyps. UC-associated hyperplastic polyps are more likely to have an LOH event on at APC and p53 locus, compared with sporadic hyperplastic polyps. Hyperplastic polyps are generally regarded as non-neoplastic lesions; however, UC-associated hyperplastic may evolve through a different genetic pathway than sporadic hyperplastic polyps.
The Interactions Between Environmental Component and Molecular Alterations in the Inflammatory Bowel Disease
IBD and cancer are complex disease processes driven by multiple interacting genes in concert with environmental influences [115]. A gene–environment interaction might contribute to the progressive process of cancer with genetic and epigenetic dysfunction in multiple systems including DNA repair and immune functions [116]. The etiopathology of IBD might be also associated with the gene × environment interactions [117–119]. Some of environmental factors, including inflammation, lifestyle, psychological stress, and prior appendectomy, have been suggested to be associated with IBD [120, 121]. Mucosal inflammatory mediators such as oxidative stress, nitric oxide, cytokines, receptors on the epithelial cells, COX-2, and luminal microbiota might be mainly responsible for the molecular alterations in the development of CA-CRC.
Inflammation
Oxidative Stress
One of major mechanisms, which link inflammation to pro-neoplastic genetic alterations, is oxidative stress [122–124]. IBD has been considered to be an “oxyradical overload” disease, in which chronic inflammation increases the risk of cancer [125]. Oxidative stress is mainly produced by inflammatory immune cells such as macrophages and granulocytes and includes the generation of various reactive oxygen and nitrogen species such as reactive oxygen species (ROS) and nitric oxide synthase (NOS) that pose a constant mutational challenge for the intestinal epithelium [126]. This results in DNA breaks, DNA adducts, and damage to cellular lipids and proteins [126]. Oxidative stress secondary to chronic inflammation also plays a pivotal role in IBD-associated colorectal carcinogenesis [124, 125, 127]. ROS, reactive nitrogen free radicals, releases a cascade of inflammatory mediators such as inflammatory cytokines including interleukin (IL)-1, IL-6, tumor necrosis factor-α (TNFα), and interferon-γ (IFN-γ) [8, 128, 129]. These cytokines bind DNA, RNA, proteins, or lipid [130], supposed to cause gene alterations, genetic instability, and aberrant methylation. Telomere damage [131, 132] in UC has been linked with the development of dysplasia [133, 134]. Lipid peroxidation occurs when ROS and NOS interact with cell membranes, causing DNA adducts leading to transition mutations [135] and frequently involving the p53 TSG [136]. In addition, these free radicals inhibit DNA repair proteins and are believed to be initiators of MSI [137]. The oxidative stress also increases the mutation of mitochondrial DNA and possibly correlates with the pathogenesis of UC-CRC [138]. The therapeutic effects of 5-aminosalicylic acid (5-ASA) have been attributed to antioxidant, iron-chelating, and radical scavenging effects [28, 139–144].
Nitric Oxide (NO)
Serum nitrite levels, a measure of NOS activity, are increased in active UC and CD patients and correlate with their disease activity [145–147]. NOS is induced in the inflamed human colonic epithelium and is associated with the formation of peroxynitrite and the nitration of cellular proteins [148]. High activity of the inducible NOS contributes to early onset IBD, which may contribute to colon carcinogenesis [149]. It has been suggested that ursodesoxycholic acid acts antioxidative and thereby reduces mutational stress by NOS [150, 151], and NO level suggested to be a useful biomarker of treatment response in IBD [152].
Cytokines
The chronic inflammatory changes in IBD are associated with increased levels of inflammatory cytokines from immune cells. It is now becoming clear that cytokines and growth factors released during inflammation may influence the carcinogenesis process [153]. IL-6 and IL-23, which play significant roles in the induction and maintenance of gut inflammation in IBD, have been recently shown to influence the development and growth of CA-CRC [153–157]. Also, cytokines activate receptors on intestinal epithelial cells that activate oncogenic transcription factors such as nuclear factor-kappaB (NF-κB) and Stat3 in the development of UC-CRC [156, 158]. TNFα increased gene mutations, gene amplification, micronuclei formation, and chromosomal instability [159], and a close relationship between the polymorphism of TNFα-308 G>A and the gene instability in UC-CRC [160]. NF-κB regulates the expression of various cytokines, modulates the inflammatory processes in IBD [161], and controls apoptosis, cell cycle progression and proliferation, and cell differentiation [162, 163]. NFκB is activated not only in sites of inflammation, but also in many solid tumors [164]. 5-ASA, the NFκB pathway inhibitor, is the first line agent for anti-inflammatory therapy [142, 144]. Toll-like receptors (TLR) play an important role in the interaction between the intestinal microflora and the mucosal immune defense via NFκB activation [165]. The potential association between TLR4 and chitinase 3-like 1 signaling has been reported, which seems to contribute to the proliferation, migration, and neoplastic progression of colonic epithelial cells under inflammatory conditions [166, 167].
Cyclooxygenase-2 (COX-2)
COX-2 is only induced by inflammation. COX-2 is triggered by inflammatory stimuli such as IL-1, IFN-γ, and TNFα and develops neoplastic changes [168–170]. Overexpression of COX-2 in epithelial, mesenchymal, and inflammatory cells results in the production of prostaglandins (PGs). PGE2 induced by COX-2 transactivates PPARδ through β-catenin and P13K/Akt signaling, which promotes cell survival and tumor growth [171]. PPARδ acts as a focal point of crosstalk between the PGs and Wnt/β-catenin pathways, which results in a shift from cell death to cell survival and consequently increased tumor growth [171]. Wnt/β-catenin signalings with downstream events including c-Myc and Cyclin-D1 represent the connection between IBD and increased risk of developing CRC [172]. Selective COX-2 inhibitor and PGE2 receptor inhibitor exert the cancer chemopreventive effects through the suppression of cell proliferation [173, 174].
Luminal Microbiota
Many studies have found a link between alterations in the commensal bacteria of the gut, termed the microbiota, and the pathogeneses of IBD [120, 175, 176]. Diet, such as Western diet and vegetarianism, and genetic factors might influence the changes in the microbiota composition [177]. The intestinal microbiota makes a significant contribution to the development of not only colitis, but also neoplasia by production of toxic and genotoxic bacterial [125]. Mice colonized with enterotoxigenic B. Fragilis exhibit colonic Th17 inflammatory infiltrates that are involved in induction of colon tumors by activating Stat3 [178]. Dysbiosis of gut microflora may also cause alterations in the immune response and increase risk of cancer [179, 180]. Ghadimi et al. [181] described that the commensal bacteria inhibited the production of the IBD-causing cytokines, IL-17 and IL-23, thereby reducing histone acetylation and enhanced DNA methylation, suggesting that an imbalanced intestinal microbiota might be associated with the increased risk of CRC development in IBD. The microbiota might regulate the expression of heat shock protein (Hsp) that is associated with the immune system by folding, refolding, translocation, and degradation of intracellular proteins under normal and stress conditions. Studies conducted on patients affected by IBD showed a decrease in Hsp60, Hsp10, and Hsp70 in epithelium and lamina propria after a combined therapy of 5ASA [182].
Lifestyle Factors
The epigenetic changes are influenced by lifestyle factors, such as diet, smoking, and physical inactivity [183]. These factors might drive changes in gene expression and increase cancer risk. Poullis et al. [184] found a significant positive relationship between risk factors for CRC and increasing age, obesity, and physical inactivity, and an inverse relationship with fiber intake and vegetable consumption. Huxley et al. reported that the risk of CRC based on 103 cohort studies was significantly associated with alcohol, smoking, diabetes, obesity and high meat intakes, and physical activity.
Dietary
Amre et al. [185] hypothesized that interactions between dietary substrates (fats, vegetables, and fruits) and DNA variants in the xenobiotic metabolizing enzymes would modify risk of IBD. Slattery et al. suggest that diet may be involved in disease pathways represented by p53 loss [186], Ki-ras mutations [187], and MSI [188]. Some studies suggest that vitamin D is associated with IBD [189, 190]. Vegetable consumption has long been hypothesized to be protective against CRC. Hutter et al. [191] reported that vegetable consumption is closely associated with CRC by a gene–environment interaction across studies. A meta-analysis reported that vegetable consumption of fruit and vegetable intake showed a significant inverse association with CRC risk [192]. Chen et al. reported that high intake of red and processed meat is associated with significant increased risk of CRC [193]. In contrast, several studies have failed to find a relationship [194, 195]. The ability of fruits and vegetables confers clearly that the ROS might be responsible for the mechanism [196]. Slattery et al. [197] suggest that alcohol contributes to rectal cancer risk.
Smoking
Smoking habit is an important environmental factor in UC [176, 198, 199]. Wang et al. examined the predictive value of combining the 133 UC risk loci with genetic interactions using genome-wide association studies and identified interactions between genes (HLA-DQA1, CALM3, TRIB1, and IL-2/IL-21) and smoking in the discovery cohort [119], while the exact mechanisms by which smoking influences the development of IBD are unknown. Slattery et al. suggest that smoking statistically significantly contributes to MSI in colon tumors [200] and that significant interactions were observed between MLH1 polymorphisms and smoking [201].
Psychological Stress
Psychosocial stress increases the likelihood of developing IBD and multiple types of malignant neoplasms [202]. Peters et al. suggested that chronic psychosocial stress increases the risk of inflammation-related CRC using azoxymethane/dextran sodium sulfate CRC mouse model, and colonic liver receptor homolog-1, COX-2, tumor necrosis factor, forkhead box P3 mRNA as well as colonic ß-catenin were also increased in CSC [203]. Acute psychologic stress induces systemic and mucosal proinflammatory responses, which could contribute to exacerbations of UC [204]. The effects of stress on inflammation in IBD are likely to be mediated through changes in hypothalamic–pituitary–adrenal function, alterations in bacterial-mucosal floral interactions, activation of mucosal mast cells, and peripheral release of corticotrophin releasing factor [202]. Soderholm et al. [205] reported that chronic psychological stress can be an initiating factor in intestinal inflammation by impairing mucosal defenses against luminal bacteria and highlight the importance of mast cells in this process. In contrast, Timmer et al. [206] reported that there was no evidence for efficacy of psychological therapy in adult patients with IBD in general.
Appendectomy
Prior appendectomy for appendicitis has been linked to a lower risk of UC [207], particularly among children experiencing appendicitis before 20 years of age [208, 209], while the effect of appendectomy on UC disease course remains inconclusive [210]. The appendix may act as a reservoir of enteric bacteria and may be involved in antigen sampling that regulates the immunologic response to host microflora [209]. Andersson et al. [211] suggest that appendicitis is mediated by T-helper 1 cells, which may explain the inverse associations between appendicitis and UC.
Future Perspectives
To clarify the pathogenesis of CRC in IBD, the resolution of complex gene–environmental interactions might be important.
New Biomarkers
Biomarkers for early detection of CRC in IBD are desired. Analysis of the correlation between these genetic features and clinicopathologic features might be useful to determine new biomarkers that can help in the early detection and predictive values of CRC in patients with IBD. The combination of the endoscopic and molecular screening approaches may be useful tools for the surveillance of patients with IBD.
Chemoprevention
The inflammatory stresses, such as ROS and some free radicals, have been considered to cause genetic damages to UC epithelium. The control of long-term inflammation and mucosal damage over time might be a potentially important strategy for reducing CRC risk in UC patients. Development of a new anti-inflammatory reagent might be useful to prevent and treat UC-CRC. Further clinical studies would be needed to develop useful drugs and validate potential modalities for the prevention of UC-associated carcinogenesis.
Crohn’s Disease
Genetic alteration of CRC with CD remains unclear in comparison with that of UC. Further studies are needed to evaluate similarities and differences in genetic alterations of UC-CRC and CD-associated neoplasia.
References
Crohn BRH. The sigmoidoscopic picture of chronic ulcerative colitis (non-specific). Am J Med Sci. 1925;170:220–228.
Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229–244.
Hartnett L, Egan LJ. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 2012;33:723–731.
Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444.
Fujii S, Fujimori T, Kashida H. Ulcerative colitis-associated neoplasia. Pathol Int. 2002;52:195–203.
Foersch S, Neurath MF. Colitis-associated neoplasia: molecular basis and clinical translation. Cell Mol Life Sci. 2014;71:3523–3535.
Seril DN, Liao J, Yang GY, Yang CS. Oxidative stress and ulcerative colitis-associated carcinogenesis: studies in humans and animal models. Carcinogenesis. 2003;24:353–362.
Talero E, Sanchez-Fidalgo S, Villegas I, de la Lastra CA, Illanes M, Motilva V. Role of different inflammatory and tumor biomarkers in the development of ulcerative colitis-associated carcinogenesis. Inflamm Bowel Dis. 2011;17:696–710.
Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009;29:2727–2737.
Gyde S, Prior P, Dew MJ, Saunders V, Waterhouse JA, Allan RN. Mortality in ulcerative colitis. Gastroenterology. 1982;83:36–43.
Ekbom A, Adami HO, Helmick CG, Jonzon A, Zack MM. Perinatal risk factors for inflammatory bowel disease: a case-control study. Am J Epidemiol. 1990;132:1111–1119.
Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91:854–862.
Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535.
Dobbins WO 3rd. Dysplasia and malignancy in inflammatory bowel disease. Annu Rev Med. 1984;35:33–48.
Isbell G, Levin B. Ulcerative colitis and colon cancer. Gastroenterol Clin N Am. 1988;17:773–791.
Chambers WM, Warren BF, Jewell DP, Mortensen NJ. Cancer surveillance in ulcerative colitis. Br J Surg. 2005;92:928–936.
Jess T, Simonsen J, Jorgensen KT, Pedersen BV, Nielsen NM, Frisch M: Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology. 2012;143:375–381 e371; quiz e313–e374.
Brackmann S, Andersen SN, Aamodt G, et al. Relationship between clinical parameters and the colitis-colorectal cancer interval in a cohort of patients with colorectal cancer in inflammatory bowel disease. Scand J Gastroenterol. 2009;44:46–55.
Xie J, Itzkowitz SH. Cancer in inflammatory bowel disease. World J Gastroenterol. 2008;14:378–389.
Jess T, Rungoe C, Peyrin-Biroulet L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol. 2012;10:639–645.
Soderlund S, Brandt L, Lapidus A et al. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology. 2009;136:1561–1567; quiz 1818–1569.
Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–1233.
Askling J, Dickman PW, Karlen P, et al. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. Gastroenterology. 2001;120:1356–1362.
Nuako KW, Ahlquist DA, Mahoney DW, Schaid DJ, Siems DM, Lindor NM. Familial predisposition for colorectal cancer in chronic ulcerative colitis: a case-control study. Gastroenterology. 1998;115:1079–1083.
Tsaitas C, Semertzidou A, Sinakos E. Update on inflammatory bowel disease in patients with primary sclerosing cholangitis. World J Hepatol. 2014;6:178–187.
Pinczowski D, Ekbom A, Baron J, Yuen J, Adami HO. Risk factors for colorectal cancer in patients with ulcerative colitis: a case-control study. Gastroenterology. 1994;107:117–120.
Velayos FS, Loftus EV Jr, Jess T, et al. Predictive and protective factors associated with colorectal cancer in ulcerative colitis: a case-control study. Gastroenterology. 2006;130:1941–1949.
Solem CA, Harmsen WS, Zinsmeister AR, Loftus EV Jr. Small intestinal adenocarcinoma in Crohn’s disease: a case-control study. Inflamm Bowel Dis. 2004;10:32–35.
Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther. 2006;23:1097–1104.
Rashid A, Hamilton SR. Genetic alterations in sporadic and Crohn’s-associated adenocarcinomas of the small intestine. Gastroenterology. 1997;113:127–135.
Jess T, Winther KV, Munkholm P, Langholz E, Binder V. Intestinal and extra-intestinal cancer in Crohn’s disease: follow-up of a population-based cohort in Copenhagen County, Denmark. Aliment Pharmacol Ther. 2004;19:287–293.
Karlen P, Kornfeld D, Brostrom O, Lofberg R, Persson PG, Ekbom A. Is colonoscopic surveillance reducing colorectal cancer mortality in ulcerative colitis? A population based case control study. Gut. 1998;42:711–714.
Riddell RH, Goldman H, Ransohoff DF, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum Pathol. 1983;14:931–968.
Choi PM, Nugent FW, Schoetz DJ Jr, Silverman ML, Haggitt RC. Colonoscopic surveillance reduces mortality from colorectal cancer in ulcerative colitis. Gastroenterology. 1993;105:418–424.
Carter MJ, Lobo AJ, Travis SP. Ibd Section BSoG: guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53:V1–V16.
Eaden JA, Mayberry JF. Guidelines for screening and surveillance of asymptomatic colorectal cancer in patients with inflammatory bowel disease. Gut. 2002;51:V10–V12.
Cairns SR, Scholefield JH, Steele RJ, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002). Gut. 2010;59:666–689.
Kornbluth A, Sachar DB, Practice Parameters Committee of the American College of G. Ulcerative colitis practice guidelines in adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105:501–523; quiz 524.
Biancone L, Michetti P, Travis S, et al. European evidence-based Consensus on the management of ulcerative colitis: special situations. J Crohns Colitis. 2008;2:63–92.
Itzkowitz SH, Present DH. Crohn’s, Colitis Foundation of America Colon Cancer in IBDSG: consensus conference: colorectal cancer screening and surveillance in inflammatory bowel disease. Inflamm Bowel Dis. 2005;11:314–321.
Van Assche G, Dignass A, Bokemeyer B, et al. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 3: special situations. J Crohns Colitis. 2013;7:1–33.
Farraye FA, Odze RD, Eaden J, et al. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:738–745.
Magro F, Langner C, Driessen A, et al. European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis. 2013;7:827–851.
Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532.
Kern SE, Fearon ER, Tersmette KW, et al. Clinical and pathological associations with allelic loss in colorectal carcinoma [corrected]. JAMA. 1989;261:3099–3103.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767.
Chen R, Rabinovitch PS, Crispin DA, et al. DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Am J Pathol. 2003;162:665–672.
Bos JL, Fearon ER, Hamilton SR, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297.
Yashiro M, Carethers JM, Laghi L, et al. Genetic pathways in the evolution of morphologically distinct colorectal neoplasms. Cancer Res. 2001;61:2676–2683.
Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237.
Baker SJ, Preisinger AC, Jessup JM, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7722.
Fearon ER, Cho KR, Nigro JM, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247:49–56.
Boland CR, Sato J, Appelman HD, Bresalier RS, Feinberg AP. Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression. Nat Med. 1995;1:902–909.
Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561.
Andersen SN, Lovig T, Clausen OP, Bakka A, Fausa O, Rognum TO. Villous, hypermucinous mucosa in long standing ulcerative colitis shows high frequency of K-ras mutations. Gut. 1999;45:686–692.
Holzmann K, Klump B, Borchard F, et al. Comparative analysis of histology, DNA content, p53 and Ki-ras mutations in colectomy specimens with long-standing ulcerative colitis. Int J Cancer. 1998;76:1–6.
Chen J, Compton C, Cheng E, Fromowitz F, Viola MV. c-Ki-ras mutations in dysplastic fields and cancers in ulcerative colitis. Gastroenterology. 1992;102:1983–1987.
Chaubert P, Benhattar J, Saraga E, Costa J. K-ras mutations and p53 alterations in neoplastic and nonneoplastic lesions associated with longstanding ulcerative colitis. Am J Pathol. 1994;144:767–775.
Yashiro M, Laghi L, Saito K, et al. Serrated adenomas have a pattern of genetic alterations that distinguishes them from other colorectal polyps. Cancer Epidemiol Biomark Prev. 2005;14:2253–2256.
Bressenot A, Cahn V, Danese S, Peyrin-Biroulet L. Microscopic features of colorectal neoplasia in inflammatory bowel diseases. World J Gastroenterol. 2014;20:3164–3172.
Umetani N, Sasaki S, Watanabe T, et al. Genetic alterations in ulcerative colitis-associated neoplasia focusing on APC, K-ras gene and microsatellite instability. Jpn J Cancer Res. 1999;90:1081–1087.
Matkowskyj KA, Chen ZE, Rao MS, Yang GY. Dysplastic lesions in inflammatory bowel disease: molecular pathogenesis to morphology. Arch Pathol Lab Med. 2013;137:338–350.
Redston MS, Papadopoulos N, Caldas C, Kinzler KW, Kern SE. Common occurrence of APC and K-ras gene mutations in the spectrum of colitis-associated neoplasias. Gastroenterology. 1995;108:383–392.
Aust DE, Terdiman JP, Willenbucher RF, et al. The APC/beta-catenin pathway in ulcerative colitis-related colorectal carcinomas: a mutational analysis. Cancer. 2002;94:1421–1427.
Greenwald BD, Harpaz N, Yin J, et al. Loss of heterozygosity affecting the p53, Rb, and mcc/apc tumor suppressor gene loci in dysplastic and cancerous ulcerative colitis. Cancer Res. 1992;52:741–745.
Fogt F, Vortmeyer AO, Goldman H, Giordano TJ, Merino MJ, Zhuang Z. Comparison of genetic alterations in colonic adenoma and ulcerative colitis-associated dysplasia and carcinoma. Hum Pathol. 1998;29:131–136.
Fogt F, Zhuang Z, Poremba C, Dockhorn-Dworniczak B, Vortmeyer A. Comparison of p53 immunoexpression with allelic loss of p53 in ulcerative colitis-associated dysplasia and carcinoma. Oncol Rep. 1998;5:477–480.
Yin J, Harpaz N, Tong Y, et al. p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology. 1993;104:1633–1639.
Burmer GC, Crispin DA, Kolli VR, et al. Frequent loss of a p53 allele in carcinomas and their precursors in ulcerative colitis. Cancer Commun. 1991;3:167–172.
Walsh SV, Loda M, Torres CM, Antonioli D, Odze RD. P53 and beta catenin expression in chronic ulcerative colitis–associated polypoid dysplasia and sporadic adenomas: an immunohistochemical study. Am J Surg Pathol. 1999;23:963–969.
Mikami T, Mitomi H, Hara A, et al. Decreased expression of CD44, alpha-catenin, and deleted colon carcinoma and altered expression of beta-catenin in ulcerative colitis-associated dysplasia and carcinoma, as compared with sporadic colon neoplasms. Cancer. 2000;89:733–740.
Lei J, Zou TT, Shi YQ, et al. Infrequent DPC4 gene mutation in esophageal cancer, gastric cancer and ulcerative colitis-associated neoplasms. Oncogene. 1996;13:2459–2462.
Zheng L, Lee WH. The retinoblastoma gene: a prototypic and multifunctional tumor suppressor. Exp Cell Res. 2001;264:2–18.
Paggi MG, Baldi A, Bonetto F, Giordano A. Retinoblastoma protein family in cell cycle and cancer: a review. J Cell Biochem. 1996;62:418–430.
Medema RH, Herrera RE, Lam F, Weinberg RA. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci USA. 1995;92:6289–6293.
Wiman KG. The retinoblastoma gene: role in cell cycle control and cell differentiation. FASEB J. 1993;7:841–845.
Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942–3945.
Arnold CN, Goel A, Blum HE, Boland CR. Molecular pathogenesis of colorectal cancer: implications for molecular diagnosis. Cancer. 2005;104:2035–2047.
Fujiwara I, Yashiro M, Kubo N, Maeda K, Hirakawa K. Ulcerative colitis-associated colorectal cancer is frequently associated with the microsatellite instability pathway. Dis Colon Rectum. 2008;51:1387–1394.
Cawkwell L, Sutherland F, Murgatroyd H, et al. Defective hMSH2/hMLH1 protein expression is seen infrequently in ulcerative colitis associated colorectal cancers. Gut. 2000;46:367–369.
Brentnall TA, Crispin DA, Bronner MP, et al. Microsatellite instability in nonneoplastic mucosa from patients with chronic ulcerative colitis. Cancer Res. 1996;56:1237–1240.
Heinen CD, Noffsinger AE, Belli J, et al. Regenerative lesions in ulcerative colitis are characterized by microsatellite mutation. Genes Chromosomes Cancer. 1997;19:170–175.
Suzuki H, Harpaz N, Tarmin L, et al. Microsatellite instability in ulcerative colitis-associated colorectal dysplasias and cancers. Cancer Res. 1994;54:4841–4844.
Kern SE, Redston M, Seymour AB, et al. Molecular genetic profiles of colitis-associated neoplasms. Gastroenterology. 1994;107:420–428.
Souza RF, Lei J, Yin J, et al. A transforming growth factor beta 1 receptor type II mutation in ulcerative colitis-associated neoplasms. Gastroenterology. 1997;112:40–45.
Yashiro M, Hirakawa K, Boland CR. Mutations in TGFbeta-RII and BAX mediate tumor progression in the later stages of colorectal cancer with microsatellite instability. BMC Cancer. 2010;10:303.
Souza RF, Appel R, Yin J, et al. Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet. 1996;14:255–257.
Fleisher AS, Esteller M, Harpaz N, et al. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000;60:4864–4868.
Svrcek M, El-Murr N, Wanherdrick K, et al. Overexpression of microRNAs-155 and 21 targeting mismatch repair proteins in inflammatory bowel diseases. Carcinogenesis. 2013;34:828–834.
Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577.
Sato F, Harpaz N, Shibata D, et al. Hypermethylation of the p14(ARF) gene in ulcerative colitis-associated colorectal carcinogenesis. Cancer Res. 2002;62:1148–1151.
Moriyama T, Matsumoto T, Nakamura S, et al. Hypermethylation of p14 (ARF) may be predictive of colitic cancer in patients with ulcerative colitis. Dis Colon Rectum. 2007;50:1384–1392.
Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 2001;48:367–371.
Konishi K, Shen L, Wang S, Meltzer SJ, Harpaz N, Issa JP. Rare CpG island methylator phenotype in ulcerative colitis-associated neoplasias. Gastroenterology. 2007;132:1254–1260.
Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606.
Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603.
De Jager PL, Franchimont D, Waliszewska A, et al. The role of the Toll receptor pathway in susceptibility to inflammatory bowel diseases. Genes Immun. 2007;8:387–397.
Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604.
Freeman HJ. Colorectal cancer risk in Crohn’s disease. World J Gastroenterol. 2008;14:1810–1811.
Sachar DB. Cancer in Crohn’s disease: dispelling the myths. Gut. 1994;35:1507–1508.
Bernstein CN, Shanahan F, Weinstein WM. Are we telling patients the truth about surveillance colonoscopy in ulcerative colitis? Lancet. 1994;343:71–74.
Kornbluth A, Sachar DB. Practice Parameters Committee of the American College of G: ulcerative colitis practice guidelines in adults (update): American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2004;99:1371–1385.
Pekow JR, Hetzel JT, Rothe JA, et al. Outcome after surveillance of low-grade and indefinite dysplasia in patients with ulcerative colitis. Inflamm Bowel Dis. 2010;16:1352–1356.
Befrits R, Ljung T, Jaramillo E, Rubio C. Low-grade dysplasia in extensive, long-standing inflammatory bowel disease: a follow-up study. Dis Colon Rectum. 2002;45:615–620.
Kiran RP, Ali UA, Nisar PJ, et al. Risk and location of cancer in patients with preoperative colitis-associated dysplasia undergoing proctocolectomy. Ann Surg. 2014;259:302–309.
Chen YX, Qiao L. Adenoma-like and non-adenoma-like dysplasia-associated lesion or mass in ulcerative colitis. J Dig Dis. 2013;14:157–159.
Svrcek M, Cosnes J, Beaugerie L, et al. Colorectal neoplasia in Crohn’s colitis: a retrospective comparative study with ulcerative colitis. Histopathology. 2007;50:574–583.
Blackstone MO, Riddell RH, Rogers BH, Levin B. Dysplasia-associated lesion or mass (DALM) detected by colonoscopy in long-standing ulcerative colitis: an indication for colectomy. Gastroenterology. 1981;80:366–374.
Gerrits MM, Chen M, Theeuwes M, et al. Biomarker-based prediction of inflammatory bowel disease-related colorectal cancer: a case-control study. Cell Oncol (Dordr). 2011;34:107–117.
Odze RD, Brown CA, Hartmann CJ, Noffsinger AE, Fogt F. Genetic alterations in chronic ulcerative colitis-associated adenoma-like DALMs are similar to non-colitic sporadic adenomas. Am J Surg Pathol. 2000;24:1209–1216.
Bossard C, Denis MG, Bezieau S, et al. Involvement of the serrated neoplasia pathway in inflammatory bowel disease-related colorectal oncogenesis. Oncol Rep. 2007;18:1093–1097.
Odze RD, Brien T, Brown CA, Hartman CJ, Wellman A, Fogt F. Molecular alterations in chronic ulcerative colitis-associated and sporadic hyperplastic polyps: a comparative analysis. Am J Gastroenterol. 2002;97:1235–1242.
Boland CR. Chronic inflammation, colorectal cancer and gene polymorphisms. Dig Dis. 2010;28:590–595.
Knox SS. From ‘omics’ to complex disease: a systems biology approach to gene-environment interactions in cancer. Cancer cell international. 2010;10:11.
Molodecky NA, Kaplan GG. Environmental risk factors for inflammatory bowel disease. Gastroenterol Hepatol (NY). 2010;6:339–346.
Kaser A, Zeissig S, Blumberg RS. Genes and environment: how will our concepts on the pathophysiology of IBD develop in the future? Dig Dis. 2010;28:395–405.
Wang MH, Fiocchi C, Zhu X, et al. Gene-gene and gene-environment interactions in ulcerative colitis. Hum Genet. 2014;133:547–558.
Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014;20:91–99.
Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517.
Westbrook AM, Wei B, Braun J, Schiestl RH. Intestinal mucosal inflammation leads to systemic genotoxicity in mice. Cancer Res. 2009;69:4827–4834.
Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285.
Hofseth LJ, Saito S, Hussain SP, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci U S A. 2003;100:143–148.
Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–1816.
Ferguson LR. Chronic inflammation and mutagenesis. Mutat Res. 2010;690:3–11.
Jena G, Trivedi PP, Sandala B. Oxidative stress in ulcerative colitis: an old concept but a new concern. Free Radic Res. 2012;46:1339–1345.
Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274.
Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261.
Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370.
Cottliar A, Fundia A, Boerr L et al. High frequencies of telomeric associations, chromosome aberrations, and sister chromatid exchanges in ulcerative colitis. Am J Gastroenterol. 2000;95:2301–2307.
Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res. 1998;400:99–115.
Kinouchi Y, Hiwatashi N, Chida M, et al. Telomere shortening in the colonic mucosa of patients with ulcerative colitis. J Gastroenterol. 1998;33:343–348.
O’Sullivan J, Risques RA, Mandelson MT, et al. Telomere length in the colon declines with age: a relation to colorectal cancer? Cancer Epidemiol Biomark Prev. 2006;15:573–577.
Nair J, Barbin A, Velic I, Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat Res. 1999;424:59–69.
Hussain SP, Harris CC. Molecular epidemiology of human cancer: contribution of mutation spectra studies of tumor suppressor genes. Cancer Res. 1998;58:4023–4037.
Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721.
Nishikawa M, Oshitani N, Matsumoto T, Nishigami T, Arakawa T, Inoue M. Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br J Cancer. 2005;93:331–337.
Craven PA, Pfanstiel J, Saito R, DeRubertis FR. Actions of sulfasalazine and 5-aminosalicylic acid as reactive oxygen scavengers in the suppression of bile acid-induced increases in colonic epithelial cell loss and proliferative activity. Gastroenterology. 1987;92:1998–2008.
van Staa TP, Card T, Logan RF, Leufkens HG. 5-Aminosalicylate use and colorectal cancer risk in inflammatory bowel disease: a large epidemiological study. Gut. 2005;54:1573–1578.
Eaden J, Abrams K, Ekbom A, Jackson E, Mayberry J. Colorectal cancer prevention in ulcerative colitis: a case-control study. Aliment Pharmacol Ther. 2000;14:145–153.
Andersen NN, Jess T. Has the risk of colorectal cancer in inflammatory bowel disease decreased? World J Gastroenterol. 2013;19:7561–7568.
Lopez A, Peyrin-Biroulet L. 5-Aminosalicylic acid and chemoprevention: does it work? Dig Dis. 2013;31:248–253.
Kaiser GC, Yan F, Polk DB. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor kappaB activation in mouse colonocytes. Gastroenterology. 1999;116:602–609.
Oudkerk Pool M, Bouma G, Visser JJ, et al. Serum nitrate levels in ulcerative colitis and Crohn’s disease. Scand J Gastroenterol. 1995;30:784–788.
Rachmilewitz D, Stamler JS, Bachwich D, Karmeli F, Ackerman Z, Podolsky DK. Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Gut. 1995;36:718–723.
Kimura H, Miura S, Shigematsu T, et al. Increased nitric oxide production and inducible nitric oxide synthase activity in colonic mucosa of patients with active ulcerative colitis and Crohn’s disease. Dig Dis Sci. 1997;42:1047–1054.
Singer II, Kawka DW, Scott S, et al. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–885.
Ding X, Hiraku Y, Ma N, et al. Inducible nitric oxide synthase-dependent DNA damage in mouse model of inflammatory bowel disease. Cancer Sci. 2005;96:157–163.
Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89–95.
Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–893.
Ljung T, Lundberg S, Varsanyi M, et al. Rectal nitric oxide as biomarker in the treatment of inflammatory bowel disease: responders versus nonresponders. World J Gastroenterol. 2006;12:3386–3392.
Fantini MC, Pallone F. Cytokines: from gut inflammation to colorectal cancer. Curr Drug Targets. 2008;9:375–380.
Kishimoto T. Interleukin-6: from basic science to medicine–40 years in immunology. Annu Rev Immunol. 2005;23:1–21.
Schnur DM. Recent trends in library design: ‘rational design’ revisited. Curr Opin Drug Discov Dev. 2008;11:375–380.
Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113.
Atreya R, Neurath MF. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin Rev Allergy Immunol. 2005;28:187–196.
Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–1319.
Yan B, Wang H, Rabbani ZN, et al. Tumor necrosis factor-alpha is a potent endogenous mutagen that promotes cellular transformation. Cancer Res. 2006;66:11565–11570.
Fan W, Maoqing W, Wangyang C, et al. Relationship between the polymorphism of tumor necrosis factor-alpha-308 G>A and susceptibility to inflammatory bowel diseases and colorectal cancer: a meta-analysis. Eur J Hum Genet. 2011;19:432–437.
Schottelius AJ, Dinter H. Cytokines, NF-kappaB, microenvironment, intestinal inflammation and cancer. Cancer Treat Res. 2006;130:67–87.
Wang S, Liu Z, Wang L, Zhang X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6:327–334.
Tang W, Wang W, Zhang Y, Liu S, Liu Y, Zheng D. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced chemokine release in both TRAIL-resistant and TRAIL-sensitive cells via nuclear factor kappa B. FEBS J. 2009;276:581–593.
Amit S, Ben-Neriah Y. NF-kappaB activation in cancer: a challenge for ubiquitination- and proteasome-based therapeutic approach. Semin Cancer Biol. 2003;13:15–28.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241.
Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–877.
Kamba A, Lee IA, Mizoguchi E. Potential association between TLR4 and chitinase 3-like 1 (CHI3L1/YKL-40) signaling on colonic epithelial cells in inflammatory bowel disease and colitis-associated cancer. Curr Mol Med. 2013;13:1110–1121.
Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20.
Marnett LJ, DuBois RN. COX-2: a target for colon cancer prevention. Annu Rev Pharmacol Toxicol. 2002;42:55–80.
Azer SA. Overview of molecular pathways in inflammatory bowel disease associated with colorectal cancer development. Eur J Gastroenterol Hepatol. 2013;25:271–281.
Wang D, Wang H, Brown J, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203:941–951.
Serafino A, Moroni N, Zonfrillo M, et al. WNT-pathway components as predictive markers useful for diagnosis, prevention and therapy in inflammatory bowel disease and sporadic colorectal cancer. Oncotarget. 2014;5:978–992.
Kohno H, Suzuki R, Sugie S, Tanaka T. Suppression of colitis-related mouse colon carcinogenesis by a COX-2 inhibitor and PPAR ligands. BMC Cancer. 2005;5:46.
Makita H, Mutoh M, Maruyama T, et al. A prostaglandin E2 receptor subtype EP1-selective antagonist, ONO-8711, suppresses 4-nitroquinoline 1-oxide-induced rat tongue carcinogenesis. Carcinogenesis. 2007;28:677–684.
Yang T, Owen JL, Lightfoot YL, Kladde MP, Mohamadzadeh M. Microbiota impact on the epigenetic regulation of colorectal cancer. Trends Mol Med. 2013;19:714–725.
Azcarate-Peril MA, Sikes M, Bruno-Barcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301:G401–G424.
Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904.
Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022.
Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients. 2012;4:1095–1119.
Tomasello G, Tralongo P, Damiani P, et al. Dismicrobism in inflammatory bowel disease and colorectal cancer: Changes in response of colocytes. World J Gastroenterol. 2014;20:18121–18130.
Ghadimi D, Helwig U, Schrezenmeir J, Heller KJ, de Vrese M. Epigenetic imprinting by commensal probiotics inhibits the IL-23/IL-17 axis in an in vitro model of the intestinal mucosal immune system. J Leukoc Biol. 2012;92:895–911.
Tomasello G, Sciume C, Rappa F, et al. Hsp10, Hsp70, and Hsp90 immunohistochemical levels change in ulcerative colitis after therapy. Eur J Histochem. 2011;55:e38.
Knox SS, Ochs MF. Implications of systemic dysfunction for the etiology of malignancy. Gene Regul Syst Biol. 2013;7:11–22.
Poullis A, Foster R, Shetty A, Fagerhol MK, Mendall MA. Bowel inflammation as measured by fecal calprotectin: a link between lifestyle factors and colorectal cancer risk. Cancer Epidemiol Biomark Prev. 2004;13:279–284.
Amre D. Gene-environment interactions in the etiopathogenesis of inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2005;40:S39.
Slattery ML, Curtin K, Ma K, et al. Diet activity, and lifestyle associations with p53 mutations in colon tumors. Cancer Epidemiol Biomark Prev. 2002;11:541–548.
Slattery ML, Curtin K, Anderson K, et al. Associations between dietary intake and Ki-ras mutations in colon tumors: a population-based study. Cancer Res. 2000;60:6935–6941.
Slattery ML, Anderson K, Curtin K, Ma KN, Schaffer D, Samowitz W. Dietary intake and microsatellite instability in colon tumors. Int J Cancer. 2001;93:601–607.
Garg M, Rosella O, Lubel JS, Gibson PR. Association of circulating vitamin D concentrations with intestinal but not systemic inflammation in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2634–2643.
Leslie WD, Miller N, Rogala L, Bernstein CN. Vitamin D status and bone density in recently diagnosed inflammatory bowel disease: the Manitoba IBD Cohort Study. Am J Gastroenterol. 2008;103:1451–1459.
Hutter CM, Chang-Claude J, Slattery ML, et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res. 2012;72:2036–2044.
Aune D, Lau R, Chan DS, et al. Nonlinear reduction in risk for colorectal cancer by fruit and vegetable intake based on meta-analysis of prospective studies. Gastroenterology. 2011;141:106–118.
Chan DS, Lau R, Aune D, et al. Red and processed meat and colorectal cancer incidence: meta-analysis of prospective studies. PLoS ONE. 2011;6:e20456.
Baron S, Turck D, Leplat C, et al. Environmental risk factors in paediatric inflammatory bowel diseases: a population based case control study. Gut. 2005;54:357–363.
Sakamoto N, Kono S, Wakai K, et al. Dietary risk factors for inflammatory bowel disease: a multicenter case-control study in Japan. Inflamm Bowel Dis. 2005;11:154–163.
Amre DK, D’Souza S, Morgan K, et al. Imbalances in dietary consumption of fatty acids, vegetables, and fruits are associated with risk for Crohn’s disease in children. Am J Gastroenterol. 2007;102:2016–2025.
Slattery ML, Wolff RK, Herrick JS, Curtin K, Caan BJ, Samowitz W. Alcohol consumption and rectal tumor mutations and epigenetic changes. Dis Colon Rectum. 2010;53:1182–1189.
Mahid SS, Minor KS, Soto RE, Hornung CA, Galandiuk S. Smoking and inflammatory bowel disease: a meta-analysis. Mayo Clin Proc. 2006;81:1462–1471.
Aldhous MC, Drummond HE, Anderson N, et al. Smoking habit and load influence age at diagnosis and disease extent in ulcerative colitis. Am J Gastroenterol. 2007;102:589–597.
Slattery ML, Curtin K, Anderson K, et al. Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J Natl Cancer Inst. 2000;92:1831–1836.
Campbell PT, Curtin K, Ulrich CM, et al. Mismatch repair polymorphisms and risk of colon cancer, tumour microsatellite instability and interactions with lifestyle factors. Gut. 2009;58:661–667.
Mawdsley JE, Rampton DS. Psychological stress in IBD: new insights into pathogenic and therapeutic implications. Gut. 2005;54:1481–1491.
Peters S, Grunwald N, Rummele P, et al. Chronic psychosocial stress increases the risk for inflammation-related colon carcinogenesis in male mice. Stress. 2012;15:403–415.
Mawdsley JE, Macey MG, Feakins RM, Langmead L, Rampton DS. The effect of acute psychologic stress on systemic and rectal mucosal measures of inflammation in ulcerative colitis. Gastroenterology. 2006;131:410–419.
Soderholm JD, Yang PC, Ceponis P, et al. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology. 2002;123:1099–1108.
Timmer A, Preiss JC, Motschall E, Rucker G, Jantschek G, Moser G. Psychological interventions for treatment of inflammatory bowel disease. Cochrane Database Syst Rev. 2011. doi:10.1002/14651858.CD006913.pub2.
Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy and protection against ulcerative colitis. N Engl J Med. 2001;344:808–814.
Frisch M, Pedersen BV, Andersson RE. Appendicitis, mesenteric lymphadenitis, and subsequent risk of ulcerative colitis: cohort studies in Sweden and Denmark. BMJ. 2009;338:b716.
Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of appendicitis and appendectomy in the pathogenesis of ulcerative colitis: a critical review. Inflamm Bowel Dis. 2002;8:277–286.
Gardenbroek TJ, Eshuis EJ, Ponsioen CI, Ubbink DT, D’Haens GR, Bemelman WA. The effect of appendectomy on the course of ulcerative colitis: a systematic review. Colorectal Dis. 2012;14:545–553.
Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy is followed by increased risk of Crohn’s disease. Gastroenterology. 2003;124:40–46.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research (KAKENHI, 23390329) from the Ministry of Education, Science, Sports, Culture, and Technology of Japan, and by Priority Research Fund of Osaka City University.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yashiro, M. Molecular Alterations of Colorectal Cancer with Inflammatory Bowel Disease. Dig Dis Sci 60, 2251–2263 (2015). https://doi.org/10.1007/s10620-015-3646-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-015-3646-4