
Abstract
Colorectal cancer (CRC) is the third most common cancer and the fourth most common cause of cancer mortality worldwide. Colitis-associated colorectal cancer (CAC) is a subtype of CRC associated with inflammatory bowel disease (IBD). It is well known that individuals with IBD have a 2–3 times higher risk of developing CRC than those who do not, rendering CAC a major cause of death in this group. Although the etiology and pathogenesis of CAC are incompletely understood, animal models of chronic inflammation and human cohort data indicate that changes in the intestinal environment, including host response dysregulation and gut microbiota perturbations, may contribute to the development of CAC. Genomic alterations are a hallmark of CAC, with patterns that are distinct from those in sporadic CRC. The discovery of the biological changes that underlie the development of CAC is ongoing; however, current data suggest that chronic inflammation in IBD increases the risk of developing CAC. Therefore, a deeper understanding of the precise mechanisms by which inflammation triggers genetic alterations and disrupts intestinal homeostasis may provide insight into novel therapeutic strategies for the prevention of CAC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic, idiopathic, relapsing inflammatory disease of the gastrointestinal (GI) tract [1]. A subset of individuals with IBD (10–15%) develops colorectal cancer (CRC), referred to as colitis-associated CRC (CAC), which appears to carry a worse prognosis than CRC in patients with no history of IBD [2,3,4]. The knowledge that CAC is a major cause of death in IBD [5, 6] necessitates the development of novel therapy. The inflammation–dysplasia–carcinoma sequence of CAC is distinct from the normal–adenoma–adenocarcinoma sequence in sporadic CRC, with inflammation playing a central role in the development of CAC. A growing number of studies show that intestinal inflammation drives the initiation of cancer and conducts its development. With the recent advances in “omics” analyses and the detailed characterization of animal models of chronic GI inflammation, it is evident that intestinal inflammation can induce genetic alternation, oxidative stress-driven DNA damage, host immune response dysregulation, and gut microbiota/metabolite community disruption, which are strongly associated with CAC development [7]. In addition, clinical studies suggest that preventing the burden of inflammation may be essential to minimize the incidence and pathogenesis of CAC [8,9,10]. However, the development of inflammation-induced CAC follows a complicated trajectory. Many fundamental questions remain, including elucidation of disease immunopathogenesis. Hence, we will review the recent advances in the field, focusing on the mechanisms involved in the inflammation, immune responses, and gut dysbiosis in CAC.
2 Molecular pathogenesis of CAC and sporadic CRC
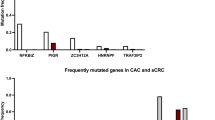
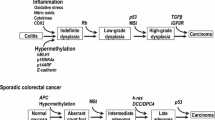
Studies using next-generation sequencing have reported several genomic alterations in patients with CAC and in patients with sporadic CRC [11, 12]. Both CAC and sporadic CRC arise in the precancerous dysplastic intestinal mucosa. However, CAC typically arises from flat dysplastic mucosa, whereas most sporadic CRC arises from polyps [13]. CAC develops in a chronically inflamed mucosa and follows the sequence of indefinite dysplasia to low-grade dysplasia to high-grade dysplasia to carcinoma. In comparison, most sporadic CRC develops from the dysplastic precursor, early adenoma, and progresses to invasive carcinoma [12,13,14]. Animal models show that inflammation induces both the initiation and progression of colonic neoplasia [15, 16], suggesting that chronic inflammation leads to genomic changes that increase CAC risk. Although multiple gene mutations are common between CAC and sporadic CRC, their timing varies [12, 13, 17, 18]. For example, the loss-of-function mutation of adenomatous polyposis coli (APC), the tumor suppressor gene, occurs early in the progression of sporadic CRC, whereas it usually occurs in the late phase of CAC development [19]. On the other hand, it is reported that the frequency and timing of microsatellite instability (MSI) is similar in CAC and sporadic CRC; i.e., the early stage of carcinoma progression [20]. The loss-of-function mutation of p53, an important step in the progression of carcinoma, occurs early in CAC, whereas it is a late phenomenon in sporadic CRC [21]. The loss of heterozygosity at p53 correlates with the malignant progression of CAC. The deletion of p53 was detected in 85% of biopsy specimens with carcinoma, whereas it was detected in 6% without dysplasia in CAC [22]. More than 50% of UC patients who do not have cancer have p53 mutations in the inflamed mucosa, indicating that chronic inflammation may cause these mutations [23]. DNA hypermethylation also is a critical feature of CAC. The methylation of CpG islands in several genes has been detected in chronically inflamed mucosal tissue, in the early phase of CAC development, without visible dysplasia. The hypermethylation of tumor suppressor genes, including hMLH1, p16INK4a, and p14ARF, has been observed in CAC patients [20, 24, 25]. Besides APC mutations, Kirsten rat sarcoma virus (KRAS) activation, the most common type of mutation reported in CRC, differs between CAC and sporadic CRC. KRAS mutation frequently occurs in the early stage of sporadic CRC, and in the late stage of CAC. In UC patients, the mutation of KRAS occurs in the advanced stages of CAC, such as high-grade dysplasia. Furthermore, KRAS mutations correlate with a disease duration longer than 10 years [26]. Previous studies indicate that alterations of the deleted in colorectal cancer (DCC) gene and the SMAD4 gene located on chromosome 18q are increased in CAC [27,28,29]. Of note, alteration in chromosome 18q genes occurs earlier in CAC (i.e., the indefinite dysplasia to low-grade dysplasia pathway) compared with sporadic CRC (i.e., the late adenoma to carcinoma pathway) [12, 30]. As with APC mutation, the alteration of the glycogen synthase kinase–3β (GSK3β) gene, which is involved in multiple inflammatory signaling pathways, is also required for the stage progression of CAC, from high-grade dysplasia to carcinoma [31, 32]. Moreover, the expression patterns of genes related to cancer stem cells (CSCs), which are involved in tumor development, progression, metastasis, and drug resistance [33], differ between sporadic CRCs and CACs. Leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), a crypt stem cell marker, is recognized as a CSC maker in the colon [34]. The expression of LGR5 is reported to be less frequent in CAC than in sporadic CRC [35]. Furthermore, the expression of CD133, a cell surface marker of colon CSCs, is also lower in CAC than in sporadic CRC [36]. A further understanding of the genomic events underlying the development of CAC may lead to the development of novel therapeutic approaches for the treatment of CAC and its early detection (Fig. 1).

Tumor development in sporadic CRC and CAC. Differences in molecular pathogenesis of sporadic CRC (upper) and CAC (lower). Intestinal inflammation is a hallmark of CAC, and the inflammation–dysplasia–carcinoma sequence of CAC is distinct from the sporadic sequence of CRC—normal to adenoma to adenocarcinoma. Loss of APC and mutations of KRAS occur in the early phase of the development of sporadic CRC, whereas these mutageneses occur late in the development of CAC. On the other hand, the mutation and loss of p53, SMAD4, and DCC are a late occurrence in sporadic CRC, and a characteristic early phenomenon in CAC. The occurrence and timing of MSI mutations is similar in sporadic CRC and CAC (i.e., the early stage of carcinoma progression). The mutations of GSK3β are required in the late stage of CAC development
3 The role of inflammation in CAC
In contrast to sporadic CRC, the pathogenesis of CAC involves the continuous stimulation of epithelial proliferation in an inflammatory environment [37]. Mounting evidence shows that chronic inflammation is a risk factor for cancer [7, 13,14,15]. Chronic inflammation enhances CAC development through various mechanisms, including oxidative stress, DNA damage, abnormal immune response, and perturbation of the gut microbiota.
3.1 Oxidative stress
IBD has been considered an oxyradical overload disease because chronic intestinal inflammation is related to the overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS), leading to oxidative stress. Oxidative stress, which is mainly produced by innate immune cells such as macrophages and neutrophils, promotes DNA damage in the intestinal epithelial cells (IECs), resulting in an increased tumorigenic alteration and accelerated growth of cancer cells [38, 39]. Inflammation-mediated oxidative stress is known to contribute to the pathogenesis of CAC in humans. Accordingly, elevated oxidative DNA damage induced by etheno-DNA adducts has been observed in colonic biopsy specimens from patients with CAC [40]. Furthermore, accumulated ROS and RNS are detected in the inflamed tissues of patients with active UC and CD [38, 41, 42]. ROS reacts with DNA leading to chromosomal breaks and increased chromosomal instability. Interestingly, chromosomal aberrations, represented by telomere shortening, are increased in biopsy specimens from UC patients who have progressed dysplasia or cancer compared to specimens from UC non-progressors or control individuals [43]. An animal study showed that mice deficient in nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2), a master regulator of antioxidant genes, develop enhanced CAC [44]. These findings indicate that inflammation-driven oxidative stress contributes to the development of CAC.
3.2 Immune-mediated pathways and signaling
3.2.1 STAT3 signaling
The phosphorylation activation of signal transducer and activator of transcription 3 (STAT3), a major oncogenic transcription factor in colonic epithelial cells, regulates antiapoptotic genes (e.g., Bcl-2 and Bcl-xl), cell cycle regulators (e.g., cyclin D1 and c-Myc), tissue-resistance factors (e.g., Hsp70, Reg3, and S100A9), and angiogenesis genes (e.g., bFGF and VEGF), which are all related to the tumor invasion, metastasis, and worsening progression of CAC [45,46,47,48,49,50]. STAT3 signaling is considered a specific pathway-targeted approach for CAC therapy because of the accumulating evidence of its pathogenic role in IBD and CRC [51] and the observation of activated STAT3 in patients with CAC [52]. Furthermore, in vivo loss-of-function and gain-of-function studies show that STAT3 regulates the impact of IL-6 and IL-11 on the initiation and development of CAC. In addition, inhibiting STAT3 in IECs results in the suppression of CAC induction and growth, demonstrating the critical oncogenic function of STAT3 in CAC [53]. Notably, STAT3 plays an essential role in CAC through crosstalk with other critical pathways and signaling related to inflammation and carcinogenesis (e.g., Wnt/β-catenin, NF-κB, PI3K/Akt/mTOR pathway, Notch pathway) [54, 55].
3.2.2 NF-κB pathway
The transcriptional factor nuclear factor kappa B (NF-κB) appears to be involved in cancer development. Deficiency in NF-κB results in increased IEC apoptosis, disturbed expression of antimicrobial peptides, and interruption of bacterial translocation to the mucosa, suggesting that NF-κB regulates epithelial integrity and interactions with the mucosal immune system [56]. IEC-specific inhibition of NF-κB in mice induces the spontaneous development of severe chronic inflammation [56]. A CAC mouse model involving azoxymethane (AOM) plus DSS treatment revealed that NF-κB activation in IECs is essential for the development of colonic adenomas [57, 58]. NF-κB also regulates the expression of various proinflammatory cytokines and chemokines, including TNF-α and IL-6, which contributes to the inflammatory processes of IBD [59]. Consistently, a high level of activated NF-κB is detected in IECs and macrophages in patients with IBD [60].
3.2.3 Wnt/β-catenin signaling
β-catenin signaling regulates vital cellular functions, including cell proliferation, polarity, migration, genetic stability, apoptosis, and tissue homeostasis [61]. The pivotal component of Wnt signaling is the cytoplasmic protein β-catenin, which is critical in CAC progression [61]. The inhibition of the Wnt/β-catenin pathway by 5-ASA, the most common anti-inflammatory therapy for UC patients, reduces colitis and is effective for therapy for CAC [62, 63]. Wnt signaling is detected only in polypoid lesions (91.3%), not in flat lesions (0%), in colitic mice [64, 65]. Moreover, the accumulation of nuclear β-catenin is observed in nondysplastic lesions in half of CAC patients [66], suggesting that dysregulation of the Wnt/β-catenin pathway plays a vital role in CAC progression, especially in the early phase.
3.2.4 TNF-α
Tumor necrosis factor (TNF)-α is a well-known proinflammatory cytokine that relates to a variety of cellular processes such as cell proliferation, differentiation, and cell death [67]. Considering that TNF neutralization is effective in the treatment of acute and chronic inflammatory conditions, the presence of TNF during the inflammatory response is important for the maintenance of chronic inflammation [68, 69]. TNF-α, which is produced by activated macrophages and T cells, acts as an initiator of carcinogenesis through DNA damage, stimulating tumor angiogenesis. It is striking that TNF-α activates NF-κB, which contributes to CAC development, as mentioned. Of note, TNF-α production is upregulated in UC patients, and it seems to contribute to the pathogenesis of IBD, an underlying condition for CAC development [70]. Administration of anti-TNF monoclonal antibodies has been shown to effectively treat CD and UC patients [71]. Moreover, elevated TNF expression occurs in the process of CAC carcinogenesis, and blockade of TNF-α signaling suppresses tumorigenicity and tumor growth in a CAC mouse model [72].
3.2.5 IL-1β
IL-1β is a proinflammatory cytokine that is upregulated in patients with IBD and in patients with colon cancer [73]. IL-1β, which is produced by macrophage and neutrophils, can induce the expression of IL-6, which is another important component in mediating CAC through the activation of STAT3 signaling pathway [74]. Consistent with this notion, blocking IL-1β activity reduces tumorigenesis in CAC mice by disordering macrophage-dependent IL-6 secretion [74]. Furthermore, the ablation of the TIR8 gene, a negative regulator of IL-1 receptor signaling, increased intestinal inflammation and CAC in AOM/DSS-treated mice [75]. IL-1β secretion in innate immune cells is regulated by the NOD-like receptor protein 3 (NLRP3) inflammasome [76]. A study demonstrated that inhibiting NLRP3 activation by small molecule andrographolide induces a decline in IL-1β production in vitro and in vivo, reducing the risk of CAC [77]. These findings imply that IL-1β may be a potential therapeutic target for CAC.
3.2.6 IL-6
Animal and human studies suggest that IL-6 plays a crucial role in IBD pathogenesis. It is conceivable that IL-6 can control the survival of colitogenic T cells, the differentiation of T helper 17 (Th17) cells, and the suppression of regulatory T cells (Tregs); all mechanisms that are associated with CAC progression [71, 78, 79]. The most crucial role of IL-6 is to activate STAT3 and NF-κB signaling, the keystone of CAC development, as mentioned. A correlation between IL-6-mediated STAT3 activation and CAC progression has been reported [80]. In the AOM/DSS-treated CAC mouse model, IL-6−/− mice have reduced tumor formation and growth compared to wild-type (WT) mice [53]. In addition, the inhibition of IL-6 signaling has been proven to ameliorate symptoms in patients with active CD [53, 81,82,83].
3.2.7 IL-17/IL-23
Accumulating evidence highlights the clinical significance of IL-17 to IBD and CAC. IL-17 family members, such as IL-17A, IL-17C, and IL-17F, are related to a proinflammatory signature, and they influence different aspects of the progression of IBD and colon cancer [84, 85]. Indeed, several studies have proven that IL-17A is a protumorigenic cytokine in various CAC mouse models, such as the APCMin model [86] and the AOM/DSS model [86, 87]. A remarkable feature of IL-17A is its ability to regulate intestinal inflammation and tumor growth, and further, IL-17A-deficient CAC mice have been shown to develop fewer tumors in the AOM/DSS CAC model [87, 88]. Also, the deletion of IL-17A reduces spontaneous small intestinal tumors in APCMin mice [89]. Similarly, epithelial IL-17C, driven by the microbiota, is associated with IEC survival and the development of colon cancer [90]. Contrary to IL-17A and IL-17C, colonic tumor development was enhanced in the absence of IL-17F in the AOM/DSS CAC model [91]. IL-23, produced by antigen-presenting cells, plays a role in tumor progression by inducing inflammation in the tumor microenvironment. IL-23 is upregulated in CAC patients and CAC model mice compared with the healthy colonic mucosa [92]. IL-23- or IL-23 receptor-deficient mice exhibit reduced secretion of proinflammatory cytokines, including IL-17A, in colon tissue, and decreased colon cancer development [92], whereas the overexpression of IL-23 promotes colitis and colon cancer by inducing IL-17 production [93]. These findings suggest that IL-23 regulates IL-17 production during inflammation and the IL-17/IL-23 axis plays an essential role in the development of chronic inflammation and CAC.
3.2.8 IL-22
Given that IL-22 plays an essential role in mucosal healing, epithelial barrier, and chronic inflammation, it likely acts as a tumor-promoting cytokine in various cancers [94,95,96]. For example, a study of Helicobacter hepaticus–infected Rag2−/− CAC mice showed that IL-22-driven nitric oxide causes DNA damage in crypt epithelial cells and leads to the development of dysplasia. Furthermore, depletion of IL-22 influences the severity of H. hepaticus-induced inflammation and tumorigenesis [97]. Notably, the loss of IL-22 binding protein (IL-22BP), a soluble IL-22 receptor that neutralizes the abilities of IL-22, in APCMin CAC mice, which have continuous activation of IL-22 signaling, promotes colonic tumorigenesis as compared to IL-22–deficient APCMin mice and WT mice, suggesting that the IL-22 and IL-22BP pathway regulates mucosal healing and tumor development [98]. Further, it has been reported that a dysregulated IL-22–STAT3 axis is related to UC-induced carcinogenesis, and a positive correlation is observed between IL-22 expression level and CAC progression [99, 100]. Thus, IL-22 can be used as a biomarker to detect the severity of CAC.
3.2.9 IL-33
The functions of IL-33 in the GI tract have also received much attention in recent years. IL-33 is a member of the IL-1 cytokine family, and it acts as an epithelial alarmin. Damaged or stressed tissue and necrosis cells release IL-33 to alert the immune system [101]. Several studies have shown that the expression of IL-33 and ST2, an IL-33 receptor, is increased in IBD patients and CAC patients [102, 103]. Activation of the IL-33–ST2 axis was observed in adenomatous polyps of APCMin mice, and deactivation of this signal decreased intestinal polyp development [104]. Similarly, in the AOM/DSS model, it was shown that the IL-33–ST2 pathway is closely associated with the development of colonic tumorigenesis by the induction of epithelial leakiness and IL-6 production. In addition, AOM/DSS mice with ST2 deletion, displayed delayed CAC formation compared to AOM/DSS mice with intact ST2 [105], suggesting that IL-33 may be a potential target of CAC therapy. On the other hand, IL-33 serves a protective function in CAC. IL-33 maintains gut homeostasis by regulating IgA production in the intestine. IL-33 deficient mice are highly susceptible to AOM/DSS-induced CAC. Of note, reduced intestinal IgA production in IL-33−/− mice treated with AOM and DSS induced gut dysbiosis characterized by an enhanced level of mucolytic and colitogenic bacteria, including Akkermansia muciniphila and segmented filamentous bacteria (SFB) [106]. Hence, the role and mechanism of IL-33 in CAC are important to elucidate the pathogenesis of CAC and to assess potential therapies for CAC.
4 The role of the gut microbiota in CAC
4.1 Pathobionts in CAC
Accumulating evidence suggests that the intestinal microbiota plays an essential role in maintaining intestinal homeostasis [107,108,109]. The gut microbiota takes on a role in beneficial functions for the host cells, including protection from pathogen colonization and shaping the immune cell network, and thus, its disruption, termed dysbiosis, is associated with the risk of intestinal inflammation and CAC [110, 111]. Interestingly, studies have reported that chronic inflammation induced by AOM/DSS treatment leads to gut dysbiosis in SPF mice. In this CAC model, bacterial diversity in the gut is significantly decreased and subsequently, the mice develop tumors in the gut. However, antibiotic exposure can reverse this phenotype. Consistently, APCMin CAC mice develop fewer tumors when housed in germ-free conditions [112]. Furthermore, gnotobiotic mice colonized with microbiota derived from CAC mice have an increased tumor burden and incidence compared to those colonized with microbiota derived from healthy mice [113]. Vancomycin administration in AOM/DSS mice reduces tumor development by the loss of neutrophils that induced DNA damage in the IECs [114]. These findings strongly suggest that inflammation-driven tumorigenesis is dependent on the gut microbiota. Another CAC mouse model, such as H. hepaticus infection, also proves that bacteria are required to initiate tumor development [115]. In clinical studies, it has been demonstrated that gut dysbiosis contributes to the pathogenesis of CAC. As observed in IBD patients, the composition of the mucosa-associated microbe population in CAC patients is strikingly different compared to that in the control individuals, such as a significant reduction in bacterial α-diversity [110]. The mucosa-associated microbiota in CAC patients is enriched in the Enterobacteriaceae family and Sphingomonas genus and diminished in Fusobacterium and Ruminococcus genera compared to the microbiota in patients with sporadic cancer [110]. Escherichia coli is the most well-studied procarcinogenic species in the Enterobacteriaceae family. It is known that some E. coli strains can produce the polyketide synthase (pks)-derived genotoxin colibactin, which causes chronic DNA damage in IECs [116]. It was reported that deletion of the pks genotoxic island from colibactin-producing E. coli reduced CRC in AOM–IL-10−/− and APCMin–IL-10−/− CAC mice [117, 118]. In addition to its genotoxic capacity, it was reported that mice colonized with pks+ E. coli had increased TNF expression compared with mice colonized with commensal E. coli. Given that the blockade of TNF signaling decreased colitis and colonic tumor development in pks+ E. coli colonized mice, proinflammatory cytokines induced by the colonization of pks+ E. coli also contribute to the tumorigenesis [119]. Interestingly, administration of sodium tungstate has a significant effect on the reduced intestinal inflammation and decreased tumorigenesis in AOM/DSS and AOM-treated IL-10−/− CAC models by inhibiting the gut colonization of colibactin-producing E. coli [120].
Bacteroides fragilis is known to harbor metalloprotease enterotoxin, which induces the cleavage of E-cadherin, disrupting the intestinal barrier. The colonization of enterotoxigenic B. fragilis (ETBF) promotes Th17 responses, resulting in the activation of NF-κB–STAT3 signaling in IECs following CAC development in APCMin mice. The neutralization of Th17 cell-derived IL-17, alone or in combination with IL-23 receptor blocking, suppressed ETBF-induced colonic tumorigenesis [86, 121]. ETBF was also shown to elicit polyp development in an AOM/DSS CAC model through elevated protumorigenic inflammatory cytokine production (e.g., IFN-γ, IL-17A, IL-6, TNF-α) [122].
More recently, the formation of a bacterial biofilm has been recognized as a driving factor in the early phase of CRC development [123, 124]. Interestingly, a study using mucosal tissue samples from patients with familial adenomatous polyposis (FAP) revealed that pks+ E. coli and ETBF dominated the bacterial population of the colonic biofilm in FAP polyps. In line with this finding, co-colonization of pks+ E. coli and ETBF enhanced the development of CRC. In vitro experiments further suggested that mucus degradation by ETBF enhances the colonization of pks+ E. coli in the gut, and induces epithelial damage as mentioned [125]. This indicates that bacterial biofilms and toxins promote the development of CRC.
Although Fusobacterium nucleatum is a major pathogen involved in chronic periodontitis, recent studies have shown that this bacterium plays a crucial role in CAC progression. For instance, in APCMin mice, colonization by F. nucleatum promotes the development of colonic tumors by recruiting tumor-infiltrating myeloid cells [126]. Moreover, F. nucleatum is known to promote epithelial–mesenchymal transition (EMT), an important process in colonic tumorigenesis [127].
Porphyromonas gingivalis, which often coexists with F. nucleatum in oral biofilms, is recognized as an oral pathobiont [128], and was recently reported to be enriched in feces and tissue samples from CRC patients compared with the samples from healthy subjects. Cohort research showed a positive correlation between P. gingivalis abundance in carcinoma tissue and a poor prognosis [129]. In addition, a study of APCMin mice revealed that P. gingivalis facilitates protumorigenic IL-1β production via NLRP3 inflammasome activation in recruited tumor-infiltrating myeloid cells, resulting in the promotion of CAC formation [130]. Like P. gingivalis, Streptococcus gallolyticus, previously known as S. bovis, an oral pathobiont that forms an oral biofilm community, can promote cancer progression [131]. Given that S. gallolyticus can degrade tannic acids, inhibiting the proliferation of tumor cells such as CRC, this bacterium appears to be a tumorigenic player. Cohort analyses indicate that S. gallolyticus is enriched in CAC tissue compared with sporadic CRC tissue [110]. Consist with human studies, the accumulation of S. gallolyticus was observed in the colonic mucosa of carcinoma tissue from AOM/DSS mice (3 cycles DSS) compared to the adenoma tissues from AOM/DSS mice (2 cycles DSS) and healthy tissues from normal mice. Colonization by S. gallolyticus aggravates the development of CAC in AOM/DSS mice. In this model, the recruitment of CD11b+ myeloid cells was induced by S. gallolyticus to promote CAC progression [132].
Infection with Helicobacter species is endemic in many animal facilities and may influence the penetrance of CAC phenotypes. Numerous studies report the relationship between Helicobacter spp. and IBD pathogenesis, and the apparent association between Helicobacter spp. and CAC pathogenesis. In IL-10−/− mice, infection with H. rodentium or H. typhlonius causes CAC development, which can be prevented by antibiotic treatment [133]. Furthermore, it has been reported that H. hepaticus can cause CAC in Rag2−/− mice through the induction of TNF-α and IL-6 [115]. Another group found that infection with H. hepaticus in Rag2−/− mice led to an accumulation of nitric oxide (NO) and H. hepaticus-driven inflammation and carcinogenesis, which was suppressed by TNF-α neutralization, acting upstream of NO production [134]. In contrast, H. pylori infection in AOM/DSS CAC mice can attenuate the development of CAC by reducing the infiltration of tumor-associated macrophages [135].
Akkermansia muciniphila is a mucin-degrading bacterium that has important regulatory effects on gut homeostasis [136, 137]. A. muciniphila is thought to be inversely correlated with inflammation status; however, the relationship between A. muciniphila and CAC remains controversial. It has been reported that A. muciniphila abundance was decreased in IBD patients and AOM/DSS mice, and the incidence of CAC in mice colonized by A. muciniphila was ameliorated by reducing the infiltration of macrophages [138]. However, several studies of the effects of A. muciniphila in CAC have drawn opposite conclusions. The abundance of A. muciniphila is significantly increased in IL-33-deficient AOM/DSS CAC mice, which are highly susceptible to colitis and CAC [106]. In addition, a recent study has revealed that A. muciniphila acts as a promoter in CAC development. In AOM/DSS mice, the administration of A. muciniphila enhanced the susceptibility of mice to CAC by enhancing the proliferation of IECs [139]. Clearly, further large-scale animal studies and human studies are needed to clarify the role of A. muciniphila in CAC pathogenesis.
Lipocalin-2 (Lcn2) is a host defense protein that is upregulated during inflammation. The antimicrobial peptide Lcn2 was induced in IL-10−/− colitis mice and Lcn2 deficiency aggravated IL-10−/− colitis and CAC. This study also demonstrated that Lcn2−/−IL-10−/− mice harbored increased Alistipes spp., and A. finegoldii challenge to IL-10−/− mice yielded increased susceptibility to CAC via activation of IL-6–STAT3 signaling [140]. Hydrogen sulfide (H2S) is a mediator of inflammation, homeostasis, and repair in the GI tract [141]. Although the function of endogenously and exogenously produced H2S in IBD remains controversial, it has been shown that the abundance of H2S-producing Atopobium parvulum positively correlates with the severity of pediatric IBD. A. parvulum induced colitis in IL-10−/− mice, and was mitigated by administrating the H2S scavenger bismuth [142] (Table 1).
4.2 Beneficial microbiota in CAC
As mentioned, gut dysbiosis may closely contribute to CAC pathogenesis. Various publications have reported that the normalization of gut dysbiosis by fecal microbiota transplantation (FMT) ameliorates the incidence and symptoms of GI disease, including Clostridioides difficile infection [150, 151]. Notably, revision of a perturbed gut microbiota by FMT of the gut microbiota derived from healthy control mice markedly suppressed CAC tumorigenesis in an AOM/DSS model. In addition, FMT-treated CAC mice had increased anti-inflammatory responses through suppression of NF-κB activation. Moreover, FMT triggered the accumulation of butyrate-producing regulatory T cells (Tregs), but not colitogenic Th17 cells in CAC mice [149]. These findings imply that FMT has potential as a therapeutic approach to CAC.
Not only the bulk population of microbiota transplantation but also transplantation of a single bacterium may be effective in cancer management. For example, a study using a CAC model revealed that Bifidobacterium lactis, a commensal-derived probiotic species, inhibits NF-κB activation in IECs and suppresses acute colitis and CAC development [143].
B. bifidum is also a widely used probiotic bacterium. Administration of B. bifidum has been shown to attenuate intestinal tumorigenesis by modulating the gut microbiome and metabolome compositions. Enrichment of Ruminococcaceae and Lachnospiraceae, two major families known as anti-inflammatory factors, was observed in B. bifidum-colonized AOM/DSS mice compared with the control group. In addition, metabolome analysis demonstrated that fatty acid metabolism pathways related to CRC formation were also regulated by the administration of B. bifidum in CAC mice [144].
Like Bifidobacterium spp., Lactobacillus genera are broadly acknowledged to have health-promoting and immunomodulatory properties. Lactobacillus bulgaricus treatment reduces tumor progression and intestinal inflammation in AOM/DSS mice by regulating the inflammatory response, and reducing proinflammatory cytokines in tumors [145]. In addition, a pilot study revealed that supplementation of the L. casei strain Shirota in fermented milk ameliorated disease activity in patients with active UC [152]. A component of the polysaccharide peptidoglycan complex in L. casei Shirota inhibits IL-6–STAT3 axis activation, thereby attenuating colonic tumor development in CAC-induced BALB/c mice. Consistent with this finding, a mutant strain of the L. casei Shirota strain lacking the polysaccharide peptidoglycan complex had no effect on the suppression of CAC development [146]. L. reuteri has the unique capacity to convert histidine to histamine. Given that the lack of histidine decarboxylase (HDC) has been shown to promote CAC by accumulating CD11b+ Gr-1+ immature myeloid cells, histamine appears to be a potential antitumorigenic factor [153]. Note, the administration of histamine-producing L. reuteri reduced tumor number and size in AOM/DSS-induced Hdc-deficient CAC mice, whereas the Hdc-deficient L. reuteri mutant, which cannot generate histamine, failed to suppress colitis and CAC. Furthermore, colonization of L. reuteri decreased the expression of inflammation- and cancer-related genes, including IL-6 and IL-22, and it reduced the infiltration of CD11b+ Gr-1+ immature myeloid cells in the spleen [147]. These findings indicate that the regulation of luminal histamine may be considered as one of the therapeutic approaches to intestinal inflammation and CAC. Furthermore, a combination of L. acidophilus and L. fermentum reduces intestinal tumorigenesis by decreasing cell proliferation and suppressing β-catenin signal activation in tumor cells [148, 154].
Besides the microbiota per se, the microbiota’s metabolic by-products are also an important factor in CAC development. Indeed, administration of the microbial metabolites short-chain fatty acids (SCFAs), such as acetate, butyrate, and propionate, results in the decreased expression of IL-6, IL-17A, and TNF-α, thereby reducing colitis and tumor incidence in the AOM/DSS-induced CAC mouse model [155] (Table 1).
4.3 The role of pattern recognition receptors in CAC
Innate immunity acts as a primary host response to microbial invasion. The pivotal elements of this process are pattern recognition receptors (PRRs), particularly toll-like receptors (TLRs). A study has shown that TLR2−/− mice display higher IL-6, IL-17A, and STAT3 activation following enhanced tumor development in the AOM/DSS CAC model, indicating the protective role of TLR2 in CAC [156]. The protumorigenic role of TLR4 in CAC is well established. TLR4 expression in IECs is upregulated in patients with dysplasia and CAC. In support of this human data, TLR4 deletions significantly decreased gut inflammation and tumorigenesis in AOM/DSS-induced CAC mice compared with control mice, and antibody blockade of TLR4 ameliorated the incidence of CAC [157]. Further, the constitutive activation of TLR4 signaling in IECs augmented colitis-associated tumorigenesis in a transgenic mouse model [158]. In addition, in the absence of myeloid differentiation factor 88 (MyD88), a downstream mediator of TLR4, colonic tumors failed to develop in AOM-treated IL-10−/− CAC mice by decreasing protumorigenic cytokines (e.g., IL-12 and TNF-α) [159]. These findings indicate that the bidirectional roles of the bacterial recognition sensors—PRRs—are related to the CAC protection or development process. Further detailed functional analysis of TLRs in CAC biology is needed to establish novel therapeutic strategies for CAC patients.
5 Antitumorigenic colitis therapy
The development of CAC is thought to be multifactorial—a combination of genetic, host immunity, and microbial influences. Consequently, the control of these factors may be a strategy for the prevention of CAC.
5.1 5-ASA
5–Aminosalicylic acid (5-ASA), the most used medication for UC treatment, attenuates tumor development by reducing oxidative stress, inhibiting cell proliferation, β-catenin accumulation, and promoting apoptosis [160]. Accordingly, 5-ASA was shown to decrease tumor incidence and growth of CAC in a mouse model [161]. Consistent with the animal study, the risk of CAC in humans was decreased by 5-ASA treatment [162, 163]. However, a meta-analysis yielded an inconsistent result, suggesting that 5-ASA treatment is not protective against CAC [164]; hence, additional studies are needed to analyze the effect of 5-ASA on the risk of CAC.
5.2 Anti–TNF-α
In CAC, chronic inflammation induces TNF-α production, which induces DNA damage and IEC disproportion, resulting in the generation of dysplastic lesions. CAC animal studies have shown that TNF-α blockade decreases colitis and the risk of CAC development [165]. TNF-α antagonists are one of the most effective medications for the treatment of patients with IBD. Notably, a clinical study has shown that 734 IBD patients treated long-term with infliximab, a chimeric monoclonal antibody against TNF-α, did not develop CRC, while 8 of 666 IBD patients without exposure to infliximab developed CRC [166]. The protective effect of anti-TNF agents was also validated by another group [167]. As clinical studies of the use of anti–TNF-α agents for the treatment of patients with CAC are limited, we cannot draw a concrete conclusion regarding the protective effect of this medication. However, long-term treatment of infliximab may be highly protective against CAC development.
5.3 Thiopurines
Thiopurines are antimetabolites and immunomodulators, and their efficacy against CAC remains controversial. In a cohort study including the data from 144 IBD patients with CRC, the treatment of thiopurines did not protect against CAC. In contrast, a meta-analysis of 24 observational studies involving 76,999 patients with IBD revealed that thiopurine exposure significantly decreased the risk of high-grade dysplasia and CRC, particularly in those patients with UC [168]. Further studies are needed to explore the protective effects of thiopurines on CRC development.
5.4 Antioxidants
Antioxidants can also prevent cancer cells from oxidation-induced death [169]. Long-term administration of antioxidants such as N-acetylcysteine decreases oxidative damage in colonic tissue and protects against CAC development. In addition, the synthetic compound growth factor 9 (GF-9), a flavonoid derivative with antioxidant properties, protects against CAC by deactivating NLRP3 inflammasome via autophagy [170]. Among natural antioxidants, vitamin C attenuates oxidative DNA damage by neutralizing ROS and protecting against inflammation-associated tumorigenesis in different animal models of CAC [171]. Although epidemiological studies have shown the protective effects of antioxidants against CAC, further studies are required to validate their association with disease pathophysiology.
6 Conclusion and perspective
In patients with IBD, chronic inflammation is a major risk factor for CAC development. The pathogenesis of CAC is distinct from that of sporadic CRC, and the critical molecular mechanisms underlying this process have yet to be elucidated. Accumulating evidence shows that chronic inflammation enhances the elements related to CAC development by various mechanisms, including oxidative stress-driven DNA damage, abnormal immune responses, and gut dysbiosis. This suggests that inflammation plays a pivotal role in creating a favorable environment for tumor initiation and development. Remarkably, the intertwined relationship between inflammation and the microbiota is complex. As mentioned, intestinal inflammation is a critical factor that drives gut dysbiosis. On the other hand, gut dysbiosis can, in turn, induce inflammation. Therefore, it will be necessary to consider an overview of scientific findings of the interaction of gut dysbiosis and inflammation to use the gut microbiota as a target for CAC treatment. Biological knowledge of the intestinal mucosal system has advanced markedly in the past decade with powerful technologies such as improved anaerobic bacterial culture methods, next-generation bacterial sequencing, high-throughput analysis of luminal metabolites, and the widespread use of gnotobiotic mouse models. In the future, primary epithelial cells derived from CAC patients can be cultured in vitro in an organoid system to test the effects of genetic mutations in humans. The tissue organoid system can also be applied to the study of epithelial-microbial interactions in the context of tumorigenesis. In addition, single-cell RNA sequencing technologies will enable a more sophisticated understanding of the functional impact of intestinal epithelial and immune cells networks on host physiology, inflammation, and tumorigenesis.
Taken together, a better understanding of the genomic events and the intestinal environment underlying the development of CAC could have implications for the early detection of CAC and the development of new therapeutic strategies for more advanced CAC.
Abbreviations
- APC:
-
Adenomatous polyposis coli
- CD:
-
Crohn’s disease
- CAC:
-
Colitis-associated colorectal cancer
- CRC:
-
Colorectal cancer
- CSCs:
-
Cancer stem cells
- DCC:
-
Deleted in colon cancer
- DSS:
-
Dextran sulfate sodium
- EMT:
-
Epithelial–mesenchymal transition
- ETBF:
-
Enterotoxigenic Bacteroides fragilis
- FAP:
-
Familial adenomatous polyposis
- FMT:
-
Fecal microbiota transplantation
- GI:
-
Gastrointestinal
- GSK3β:
-
Glycogen synthase kinase-3β
- HDC:
-
Histidine decarboxylase
- H2S:
-
Hydrogen sulfide
- IL-22BP:
-
IL-22 binding protein
- IBD:
-
Inflammatory bowel disease
- IEC:
-
Intestinal epithelial cell
- KRAS:
-
Kirsten rat sarcoma virus
- Lcn2:
-
Lipocalin-2
- LGR5:
-
Leucine-rich repeat-containing G-protein-coupled receptor 5
- MSI:
-
Microsatellite instability
- Myd88:
-
Myeloid differentiation factor 88
- NF-κB:
-
Nuclear factor kappa-light-chain enhancer
- NLRP3:
-
NOD-like receptor protein 3
- NOS:
-
Nitric oxide synthase
- pks:
-
Polyketide synthase
- PRR:
-
Pattern recognition receptor
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- STAT3:
-
Signal transducer and activator of transcription 3
- Th:
-
T helper
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- Tregs:
-
Regulatory T cells
- UC:
-
Ulcerative colitis
- 5-ASA:
-
5-Aminosalicylic acid
References
Kaplan, G. G. (2015). The global burden of IBD: From 2015 to 2025. Nature reviews Gastroenterology & hepatology, 12(12), 720–727.
Eaden, J. A., Abrams, K. R., & Mayberry, J. F. (2001). The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut, 48(4), 526–535. https://doi.org/10.1136/gut.48.4.526
Jess, T., Loftus, E. V., Jr., Velayos, F. S., Harmsen, W. S., Zinsmeister, A. R., Smyrk, T. C., et al. (2006). Incidence and prognosis of colorectal dysplasia in inflammatory bowel disease: A population-based study from Olmsted County. Minnesota. Inflamm Bowel Dis, 12(8), 669–676. https://doi.org/10.1097/00054725-200608000-00001
Andersen, N. N., & Jess, T. (2013). Has the risk of colorectal cancer in inflammatory bowel disease decreased? World Journal of Gastroenterology, 19(43), 7561–7568. https://doi.org/10.3748/wjg.v19.i43.7561
Lewis, J. D., Deren, J. J., & Lichtenstein, G. R. (1999). Cancer risk in patients with inflammatory bowel disease. Gastroenterology Clinics of North America, 28(2), 459–477, x, https://doi.org/10.1016/s0889-8553(05)70065-0.
Wanders, L. K., Dekker, E., Pullens, B., Bassett, P., Travis, S. P., & East, J. E. (2014). Cancer risk after resection of polypoid dysplasia in patients with longstanding ulcerative colitis: A meta-analysis. Clinical Gastroenterology and Hepatology, 12(5), 756–764. https://doi.org/10.1016/j.cgh.2013.07.024
Fantini, M. C., & Guadagni, I. (2021). From inflammation to colitis-associated colorectal cancer in inflammatory bowel disease: Pathogenesis and impact of current therapies. Digestive and Liver Disease, 53(5), 558–565. https://doi.org/10.1016/j.dld.2021.01.012
Waldner, M. J., & Neurath, M. F. (2008). Cytokines in colitis associated cancer: Potential drug targets? Inflammation & Allergy: Drug Targets, 7(3), 187–194. https://doi.org/10.2174/187152808785748137
Bezzio, C., Festa, S., Saibeni, S., & Papi, C. (2017). Chemoprevention of colorectal cancer in ulcerative colitis: Digging deep in current evidence. Expert Review of Gastroenterology & Hepatology, 11(4), 339–347. https://doi.org/10.1080/17474124.2017.1292129
Lopez, A., Pouillon, L., Beaugerie, L., Danese, S., & Peyrin-Biroulet, L. (2018). Colorectal cancer prevention in patients with ulcerative colitis. Best Practice & Research Clinical Gastroenterology, 32–33, 103–109. https://doi.org/10.1016/j.bpg.2018.05.010
Yaeger, R., Shah, M. A., Miller, V. A., Kelsen, J. R., Wang, K., Heins, Z. J., et al. (2016). Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology, 151(2), 278-287.e276. https://doi.org/10.1053/j.gastro.2016.04.001
Kameyama, H., Nagahashi, M., Shimada, Y., Tajima, Y., Ichikawa, H., Nakano, M., et al. (2018). Genomic characterization of colitis-associated colorectal cancer. World Journal of Surgical Oncology, 16(1), 121. https://doi.org/10.1186/s12957-018-1428-0
RJ Porter MJ Arends AMD Churchhouse S Din 2021 Inflammatory bowel disease-associated colorectal cancer: Translational risks from mechanisms to medicines Journal of Crohn's and Colitis https://doi.org/10.1093/ecco-jcc/jjab102
Ullman, T. A., & Itzkowitz, S. H. (2011). Intestinal inflammation and cancer. Gastroenterology, 140(6), 1807–1816. https://doi.org/10.1053/j.gastro.2011.01.057
Itzkowitz, S. H., & Yio, X. (2004). Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. American Journal of Physiology-Gastrointestinal and Liver Physiology, 287(1), G7–17, https://doi.org/10.1152/ajpgi.00079.2004.
Cooks, T., Pateras, I. S., Tarcic, O., Solomon, H., Schetter, A. J., Wilder, S., et al. (2013). Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell, 23(5), 634–646. https://doi.org/10.1016/j.ccr.2013.03.022
Khor, B., Gardet, A., & Xavier, R. J. (2011). Genetics and pathogenesis of inflammatory bowel disease. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Review]. Nature, 474(7351), 307–317, https://doi.org/10.1038/nature10209.
Chandrasinghe, P., Cereser, B., Moorghen, M., Al Bakir, I., Tabassum, N., Hart, A., et al. (2018). Role of SMAD proteins in colitis-associated cancer: From known to the unknown. Oncogene, 37(1), 1–7. https://doi.org/10.1038/onc.2017.300
Cooper, H. S., Everley, L., Chang, W. C., Pfeiffer, G., Lee, B., Murthy, S., et al. (2001). The role of mutant Apc in the development of dysplasia and cancer in the mouse model of dextran sulfate sodium-induced colitis. Gastroenterology, 121(6), 1407–1416. https://doi.org/10.1053/gast.2001.29609
Flaeisher, A. S., Esteller, M., Harpaz, N., Leytin, A., Rashid, A., Xu, Y., et al. (2000). Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Research, 60(17), 4864–4868.
Caooks, T., Pateras, I. S., Tarcic, O., Solomon, H., Schetter, A. J., Wilder, S., et al. (2013). Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell, 23(5), 634–646. https://doi.org/10.1016/j.ccr.2013.03.022
Burmer, G. C., Rabinovitch, P. S., Haggitt, R. C., Crispin, D. A., Brentnall, T. A., Kolli, V. R., et al. (1992). Neoplastic progression in ulcerative colitis: Histology, DNA content, and loss of a p53 allele. Gastroenterology, 103(5), 1602–1610. https://doi.org/10.1016/0016-5085(92)91184-6
Hussain, S. P., Amstad, P., Raja, K., Ambs, S., Nagashima, M., Bennett, W. P., et al. (2000). Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Research, 60(13), 3333–3337.
Hsieh, C. J., Klump, B., Holzmann, K., Borchard, F., Gregor, M., & Porschen, R. (1998). Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Research, 58(17), 3942–3945.
Moriyama, T., Matsumoto, T., Nakamura, S., Jo, Y., Mibu, R., Yao, T., et al. (2007). Hypermethylation of p14 (ARF) may be predictive of colitic cancer in patients with ulcerative colitis. Diseases of the Colon and Rectum, 50(9), 1384–1392. https://doi.org/10.1007/10350-007-0302-x
Lang, S. M., Stratakis, D. F., Heinzlmann, M., Heldwein, W., Wiebecke, B., & Loeschke, K. (1999). Molecular screening of patients with long standing extensive ulcerative colitis: Detection of p53 and Ki-ras mutations by single strand conformation polymorphism analysis and differential hybridisation in colonic lavage fluid. Gut, 44(6), 822–825. https://doi.org/10.1136/gut.44.6.822
Bläker, H., von Herbay, A., Penzel, R., Groß, S., & Otto, H. F. (2002). Genetics of adenocarcinomas of the small intestine: Frequent deletions at chromosome 18q and mutations of the SMAD4 gene. Oncogene, 21(1), 158–164. https://doi.org/10.1038/sj.onc.1205041
Maru, D., Wu, T. T., Canada, A., Houlihan, P. S., Hamilton, S. R., & Rashid, A. (2004). Loss of chromosome 18q and DPC4 (Smad4) mutations in appendiceal adenocarcinomas. Oncogene, 23(3), 859–864. https://doi.org/10.1038/sj.onc.1207194
Perreault, N. (2018). Ulcerative colitis-associated carcinoma: Epithelial SMAD4-mediated signaling is a key guardian. Cellular and molecular gastroenterology and hepatology, 6(3), 350–351. https://doi.org/10.1016/j.jcmgh.2018.06.004
Tarafa, G., Villanueva, A., Farré, L., Rodríguez, J., Musulén, E., Reyes, G., et al. (2000). DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene, 19(4), 546–555. https://doi.org/10.1038/sj.onc.1203353
Itzkowitz, S. H. (2006). Molecular biology of dysplasia and cancer in inflammatory bowel disease. Gastroenterology Clinics of North America, 35(3), 553–571. https://doi.org/10.1016/j.gtc.2006.07.002
Foersch, S., & Neurath, M. F. (2014). Colitis-associated neoplasia: Molecular basis and clinical translation. Cellular and Molecular Life Sciences, 71(18), 3523–3535. https://doi.org/10.1007/s00018-014-1636-x
Bayik, D., & Lathia, J. D. (2021). Cancer stem cell–immune cell crosstalk in tumour progression. Nature Reviews Cancer, 21(8), 526–536. https://doi.org/10.1038/s41568-021-00366-w
Barker, N., Ridgway, R. A., van Es, J. H., van de Wetering, M., Begthel, H., van den Born, M., et al. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature, 457(7229), 608–611. https://doi.org/10.1038/nature07602
Iwaya, M., Ota, H., Nakajima, T., Uehara, T., Riddell, R., & Conner, J. (2021). Most colitis associated carcinomas lack expression of LGR5: A preliminary study with implications for unique pathways of carcinogenesis compared to sporadic colorectal carcinoma. BMC Cancer, 21(1), 119. https://doi.org/10.1186/s12885-021-07835-3
Yasuda, H., Tanaka, K., Okita, Y., Araki, T., Saigusa, S., Toiyama, Y., et al. (2011). CD133, OCT4, and NANOG in ulcerative colitis-associated colorectal cancer. Oncology Letters, 2(6), 1065–1071. https://doi.org/10.3892/ol.2011.415
Grivennikov, S. I. (2013). Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin Immunopathol, 35(2), 229–244. https://doi.org/10.1007/s00281-012-0352-6
Hofseth, L. J., Saito, S., Hussain, S. P., Espey, M. G., Miranda, K. M., Araki, Y., et al. (2003). Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci U S A, 100(1), 143–148. https://doi.org/10.1073/pnas.0237083100
Hussain, S. P., Hofseth, L. J., & Harris, C. C. (2003). Radical causes of cancer. Nature Reviews Cancer, 3(4), 276–285. https://doi.org/10.1038/nrc1046
Bartsch, H., & Nair, J. (2005). Accumulation of lipid peroxidation-derived DNA lesions: Potential lead markers for chemoprevention of inflammation-driven malignancies. Mutation Research, 591(1–2), 34–44. https://doi.org/10.1016/j.mrfmmm.2005.04.013
Rachmilewitz, D., Stamler, J. S., Bachwich, D., Karmeli, F., Ackerman, Z., & Podolsky, D. K. (1995). Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Gut, 36(5), 718–723. https://doi.org/10.1136/gut.36.5.718
Kimura, H., Hokari, R., Miura, S., Shigematsu, T., Hirokawa, M., Akiba, Y., et al. (1998). Increased expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in colonic mucosa of patients with active ulcerative colitis. Gut, 42(2), 180–187. https://doi.org/10.1136/gut.42.2.180
O’Sullivan, J. N., Bronner, M. P., Brentnall, T. A., Finley, J. C., Shen, W. T., Emerson, S., et al. (2002). Chromosomal instability in ulcerative colitis is related to telomere shortening. Nature Genetics, 32(2), 280–284. https://doi.org/10.1038/ng989
Osburn, W. O., Karim, B., Dolan, P. M., Liu, G., Yamamoto, M., Huso, D. L., et al. (2007). Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. International Journal of Cancer, 121(9), 1883–1891. https://doi.org/10.1002/ijc.22943
Kusaba, T., Nakayama, T., Yamazumi, K., Yakata, Y., Yoshizaki, A., Inoue, K., et al. (2006). Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncology Reports, 15(6), 1445–1451.
Grivennikov, S. I., & Karin, M. (2010). Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine & Growth Factor Reviews, 21(1), 11–19. https://doi.org/10.1016/j.cytogfr.2009.11.005
Kusaba, T., Nakayama, T., Yamazumi, K., Yakata, Y., Yoshizaki, A., Nagayasu, T., et al. (2005). Expression of p-STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. Journal of Clinical Pathology, 58(8), 833–838. https://doi.org/10.1136/jcp.2004.023416
Corvinus, F. M., Orth, C., Moriggl, R., Tsareva, S. A., Wagner, S., Pfitzner, E. B., et al. (2005). Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia, 7(6), 545–555. https://doi.org/10.1593/neo.04571
Lassmann, S., Schuster, I., Walch, A., Göbel, H., Jütting, U., Makowiec, F., et al. (2007). STAT3 mRNA and protein expression in colorectal cancer: Effects on STAT3-inducible targets linked to cell survival and proliferation. Journal of Clinical Pathology, 60(2), 173–179. https://doi.org/10.1136/jcp.2005.035113
Yu, H., Pardoll, D., & Jove, R. (2009). STATs in cancer inflammation and immunity: A leading role for STAT3. Nature Reviews Cancer, 9(11), 798–809. https://doi.org/10.1038/nrc2734
Atreya, R., & Neurath, M. F. (2008). Signaling molecules: The pathogenic role of the IL-6/STAT-3 trans signaling pathway in intestinal inflammation and in colonic cancer. Current Drug Targets, 9(5), 369–374. https://doi.org/10.2174/138945008784221116
Han, J., & Theiss, A. L. (2014). Stat3: Friend or foe in colitis and colitis-associated cancer? Inflammatory bowel diseases, 20(12), 2405–2411. https://doi.org/10.1097/MIB.0000000000000180
Grivennikov, S., Karin, E., Terzic, J., Mucida, D., Yu, G. Y., Vallabhapurapu, S., et al. (2009). IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell, 15(2), 103–113. https://doi.org/10.1016/j.ccr.2009.01.001
Yu, H., Kortylewski, M., & Pardoll, D. (2007). Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nature Reviews Immunology, 7(1), 41–51. https://doi.org/10.1038/nri1995
Jin, W. (2020). Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells, 9(1), 217. https://doi.org/10.3390/cells9010217
Nenci, A., Becker, C., Wullaert, A., Gareus, R., van Loo, G., Danese, S., et al. (2007). Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature, 446(7135), 557–561. https://doi.org/10.1038/nature05698
Greten, F. R., Eckmann, L., Greten, T. F., Park, J. M., Li, Z. W., Egan, L. J., et al. (2004). IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell, 118(3), 285–296. https://doi.org/10.1016/j.cell.2004.07.013
Greten, F. R., & Karin, M. (2004). The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Letters, 206(2), 193–199. https://doi.org/10.1016/j.canlet.2003.08.029
Schottelius, A. J., & Dinter, H. (2006). Cytokines, NF-kappaB, microenvironment, intestinal inflammation and cancer. Cancer Treatment and Research, 130, 67–87. https://doi.org/10.1007/0-387-26283-0_3
Rogler, G., Brand, K., Vogl, D., Page, S., Hofmeister, R., Andus, T., et al. (1998). Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology, 115(2), 357–369. https://doi.org/10.1016/s0016-5085(98)70202-1
Lee, G., Goretsky, T., Managlia, E., Dirisina, R., Singh, A. P., Brown, J. B., et al. (2010). Phosphoinositide 3-kinase signaling mediates beta-catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology, 139(3), 869–881, 881.e861–869, https://doi.org/10.1053/j.gastro.2010.05.037.
Eaden, J., Abrams, K., Ekbom, A., Jackson, E., & Mayberry, J. (2000). Colorectal cancer prevention in ulcerative colitis: A case-control study. Alimentary Pharmacology & Therapeutics, 14(2), 145–153. https://doi.org/10.1046/j.1365-2036.2000.00698.x
Bernstein, C. N., Eaden, J., Steinhart, A. H., Munkholm, P., & Gordon, P. H. (2002). Cancer prevention in inflammatory bowel disease and the chemoprophylactic potential of 5-aminosalicylic acid. Inflammatory Bowel Diseases, 8(5), 356–361. https://doi.org/10.1097/00054725-200209000-00007
Cooper, H. S., Murthy, S., Kido, K., Yoshitake, H., & Flanigan, A. (2000). Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis, 21(4), 757–768, https://doi.org/10.1093/carcin/21.4.757.
Chang, W. C., Coudry, R. A., Clapper, M. L., Zhang, X., Williams, K. L., Spittle, C. S., et al. (2007). Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis, 28(11), 2375–2381. https://doi.org/10.1093/carcin/bgm134
Claessen, M. M., Schipper, M. E., Oldenburg, B., Siersema, P. D., Offerhaus, G. J., & Vleggaar, F. P. (2010). WNT-pathway activation in IBD-associated colorectal carcinogenesis: Potential biomarkers for colonic surveillance. Cellular Oncology, 32(4), 303–310. https://doi.org/10.3233/clo-2009-0503
Liu, Z. G. (2005). Molecular mechanism of TNF signaling and beyond. Cell Research, 15(1), 24–27. https://doi.org/10.1038/sj.cr.7290259
Kruglov, A. A., Kuchmiy, A., Grivennikov, S. I., Tumanov, A. V., Kuprash, D. V., & Nedospasov, S. A. (2008). Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: Mouse models. Cytokine & Growth Factor Reviews, 19(3–4), 231–244. https://doi.org/10.1016/j.cytogfr.2008.04.010
Feldmann, M. (2009). Translating molecular insights in autoimmunity into effective therapy. Annual Review of Immunology, 27, 1–27. https://doi.org/10.1146/annurev-immunol-082708-100732
Kollias, G. (2004). Modeling the function of tumor necrosis factor in immune pathophysiology. Autoimmunity Reviews, 3(Suppl 1), S24-25.
Ford, A. C., Sandborn, W. J., Khan, K. J., Hanauer, S. B., Talley, N. J., & Moayyedi, P. (2011). Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. American Journal of Gastroenterology, 106(4), 644–659, quiz 660, https://doi.org/10.1038/ajg.2011.73.
Popivanova, B. K., Kitamura, K., Wu, Y., Kondo, T., Kagaya, T., Kaneko, S., et al. (2008). Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. The Journal of Clinical Investigation, 118(2), 560–570. https://doi.org/10.1172/jci32453
Voronov, E., & Apte, R. N. (2015). IL-1 in colon inflammation, colon carcinogenesis and invasiveness of colon cancer. Cancer microenvironment : Official journal of the International Cancer Microenvironment Society, 8(3), 187–200. https://doi.org/10.1007/s12307-015-0177-7
Wang, Y., Wang, K., Han, G. C., Wang, R. X., Xiao, H., Hou, C. M., et al. (2014). Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunology, 7(5), 1106–1115. https://doi.org/10.1038/mi.2013.126
Garlanda, C., Riva, F., Veliz, T., Polentarutti, N., Pasqualini, F., Radaelli, E., et al. (2007). Increased susceptibility to colitis-associated cancer of mice lacking TIR8, an inhibitory member of the interleukin-1 receptor family. Cancer Research, 67(13), 6017–6021. https://doi.org/10.1158/0008-5472.can-07-0560
Mao, L., Kitani, A., Strober, W., & Fuss, I. J. (2018). The role of NLRP3 and IL-1β in the pathogenesis of inflammatory bowel disease. [Review]. Frontiers in Immunology, 9(2566), https://doi.org/10.3389/fimmu.2018.02566.
Guo, W., Sun, Y., Liu, W., Wu, X., Guo, L., Cai, P., et al. (2014). Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy, 10(6), 972–985. https://doi.org/10.4161/auto.28374
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T. B., Oukka, M., et al. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature, 441(7090), 235–238. https://doi.org/10.1038/nature04753
Dominitzki, S., Fantini, M. C., Neufert, C., Nikolaev, A., Galle, P. R., Scheller, J., et al. (2007). Cutting edge: Trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. The Journal of Immunology, 179(4), 2041–2045. https://doi.org/10.4049/jimmunol.179.4.2041
Li, Y., de Haar, C., Chen, M., Deuring, J., Gerrits, M. M., Smits, R., et al. (2010). Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut, 59(2), 227–235. https://doi.org/10.1136/gut.2009.184176
Reinisch, W., Gasché, C., Tillinger, W., Wyatt, J., Lichtenberger, C., Willheim, M., et al. (1999). Clinical relevance of serum interleukin-6 in Crohn’s disease: Single point measurements, therapy monitoring, and prediction of clinical relapse. American Journal of Gastroenterology, 94(8), 2156–2164. https://doi.org/10.1111/j.1572-0241.1999.01288.x
Atreya, R., Mudter, J., Finotto, S., Müllberg, J., Jostock, T., Wirtz, S., et al. (2000). Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in crohn disease and experimental colitis in vivo. Nature Medicine, 6(5), 583–588. https://doi.org/10.1038/75068
Ito, H., Takazoe, M., Fukuda, Y., Hibi, T., Kusugami, K., Andoh, A., et al. (2004). A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology, 126(4), 989–996; discussion 947, https://doi.org/10.1053/j.gastro.2004.01.012.
De Simone, V., Pallone, F., Monteleone, G., & Stolfi, C. (2013). Role of T(H)17 cytokines in the control of colorectal cancer. Oncoimmunology, 2(12), e26617. https://doi.org/10.4161/onci.26617
Ernst, M., & Putoczki, T. (2014). IL-17 cuts to the chase in colon cancer. Immunity, 41(6), 880–882. https://doi.org/10.1016/j.immuni.2014.12.004
Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature Medicine, 15(9), 1016–1022. https://doi.org/10.1038/nm.2015
Hyun, Y. S., Han, D. S., Lee, A. R., Eun, C. S., Youn, J., & Kim, H.-Y. (2012). Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis, 33(4), 931–936. https://doi.org/10.1093/carcin/bgs106
Leppkes, M., Becker, C., Ivanov, I. I., Hirth, S., Wirtz, S., Neufert, C., et al. (2009). RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology, 136(1), 257–267. https://doi.org/10.1053/j.gastro.2008.10.018
Chae, W. J., & Bothwell, A. L. (2011). IL-17F deficiency inhibits small intestinal tumorigenesis in ApcMin/+ mice. Biochemical and Biophysical Research Communications, 414(1), 31–36. https://doi.org/10.1016/j.bbrc.2011.09.016
Song, X., Gao, H., Lin, Y., Yao, Y., Zhu, S., Wang, J., et al. (2014). Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity, 40(1), 140–152. https://doi.org/10.1016/j.immuni.2013.11.018
Yang, X. O., Chang, S. H., Park, H., Nurieva, R., Shah, B., Acero, L., et al. (2008). Regulation of inflammatory responses by IL-17F. Journal of Experimental Medicine, 205(5), 1063–1075. https://doi.org/10.1084/jem.20071978
Grivennikov, S. I., Wang, K., Mucida, D., Stewart, C. A., Schnabl, B., Jauch, D., et al. (2012). Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature, 491(7423), 254–258. https://doi.org/10.1038/nature11465
Chan, I. H., Jain, R., Tessmer, M. S., Gorman, D., Mangadu, R., Sathe, M., et al. (2014). Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunology, 7(4), 842–856. https://doi.org/10.1038/mi.2013.101
Keir, M., Yi, Y., Lu, T., & Ghilardi, N. (2020). The role of IL-22 in intestinal health and disease. Journal of Experimental Medicine, 217(3), e20192195. https://doi.org/10.1084/jem.20192195
Sabihi, M., Böttcher, M., Pelczar, P., & Huber, S. (2020). Microbiota-dependent effects of IL-22. Cells, 9(10), https://doi.org/10.3390/cells9102205.
Doulabi, H., Masoumi, E., Rastin, M., Foolady Azarnaminy, A., Esmaeili, S. A., & Mahmoudi, M. (2022). The role of Th22 cells, from tissue repair to cancer progression. Cytokine, 149, 155749. https://doi.org/10.1016/j.cyto.2021.155749
Wang, C., Gong, G., Sheh, A., Muthupalani, S., Bryant, E. M., Puglisi, D. A., et al. (2017). Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunology, 10(6), 1504–1517. https://doi.org/10.1038/mi.2017.9
Huber, S., Gagliani, N., Zenewicz, L. A., Huber, F. J., Bosurgi, L., Hu, B., et al. (2012). IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature, 491(7423), 259–263. https://doi.org/10.1038/nature11535
Jiang, R., Wang, H., Deng, L., Hou, J., Shi, R., Yao, M., et al. (2013). IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer, 13, 59. https://doi.org/10.1186/1471-2407-13-59
Wei, H.-X., Wang, B., & Li, B. (2020). IL-10 and IL-22 in mucosal immunity: driving protection and pathology. [Review]. Frontiers in Immunology, 11(1315), https://doi.org/10.3389/fimmu.2020.01315.
Lüthi, A. U., Cullen, S. P., McNeela, E. A., Duriez, P. J., Afonina, I. S., Sheridan, C., et al. (2009). Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity, 31(1), 84–98. https://doi.org/10.1016/j.immuni.2009.05.007
O’Donnell, C., Mahmoud, A., Keane, J., Murphy, C., White, D., Carey, S., et al. (2016). An antitumorigenic role for the IL-33 receptor, ST2L, in colon cancer. British Journal of Cancer, 114(1), 37–43. https://doi.org/10.1038/bjc.2015.433
Chen, J., He, Y., Tu, L., & Duan, L. (2020). Dual immune functions of IL-33 in inflammatory bowel disease. Histology and Histopathology, 35(2), 137–146, https://doi.org/10.14670/hh-18-149.
Maywald, R. L., Doerner, S. K., Pastorelli, L., De Salvo, C., Benton, S. M., Dawson, E. P., et al. (2015). IL-33 activates tumor stroma to promote intestinal polyposis. Proceedings of the National Academy of Sciences of the United States of America, 112(19), E2487-2496. https://doi.org/10.1073/pnas.1422445112
Mertz, K. D., Mager, L. F., Wasmer, M. H., Thiesler, T., Koelzer, V. H., Ruzzante, G., et al. (2016). The IL-33/ST2 pathway contributes to intestinal tumorigenesis in humans and mice. Oncoimmunology, 5(1), e1062966. https://doi.org/10.1080/2162402x.2015.1062966
Malik, A., Sharma, D., Zhu, Q., Karki, R., Guy, C. S., Vogel, P., et al. (2016). IL-33 regulates the IgA-microbiota axis to restrain IL-1α-dependent colitis and tumorigenesis. The Journal of Clinical Investigation, 126(12), 4469–4481. https://doi.org/10.1172/JCI88625
Kamada, N., & Núñez, G. (2013). Role of the gut microbiota in the development and function of lymphoid cells. The Journal of Immunology, 190(4), 1389–1395. https://doi.org/10.4049/jimmunol.1203100
Kamada, N., Seo, S. U., Chen, G. Y., & Núñez, G. (2013). Role of the gut microbiota in immunity and inflammatory disease. Nature Reviews Immunology, 13(5), 321–335. https://doi.org/10.1038/nri3430
Nagao-Kitamoto, H., & Kamada, N. (2017). Host-microbial Cross-talk in Inflammatory Bowel Disease. Immune network, 17(1), 1–12. https://doi.org/10.4110/in.2017.17.1.1
Richard, M. L., Liguori, G., Lamas, B., Brandi, G., da Costa, G., Hoffmann, T. W., et al. (2018). Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes, 9(2), 131–142. https://doi.org/10.1080/19490976.2017.1379637
Priyamvada, P. (2021). Dysbiosis in microbiome leading to colitis-associated cancer: gut microbiome correlation with CAC. In P. Ashok Kumar (Ed.), Diagnostic and Treatment Methods for Ulcerative Colitis and Colitis-Associated Cancer (pp. 142–169). Hershey, PA, USA: IGI Global.
Dove, W. F., Clipson, L., Gould, K. A., Luongo, C., Marshall, D. J., Moser, A. R., et al. (1997). Intestinal neoplasia in the ApcMin mouse: Independence from the microbial and natural killer (beige locus) status. Cancer Research, 57(5), 812–814.
Zackular, J. P., Baxter, N. T., Iverson, K. D., Sadler, W. D., Petrosino, J. F., Chen, G. Y., et al. (2013). The gut microbiome modulates colon tumorigenesis. mBio, 4(6), e00692–00613, https://doi.org/10.1128/mBio.00692-13.
Tanaka, Y., Ito, S., & Isobe, K.-I. (2016). Vancomycin-sensitive bacteria trigger development of colitis-associated colon cancer by attracting neutrophils. Scientific Reports, 6(1), 23920. https://doi.org/10.1038/srep23920
Poutahidis, T., Haigis, K. M., Rao, V. P., Nambiar, P. R., Taylor, C. L., Ge, Z., et al. (2007). Rapid reversal of interleukin-6-dependent epithelial invasion in a mouse model of microbially induced colon carcinoma. Carcinogenesis, 28(12), 2614–2623. https://doi.org/10.1093/carcin/bgm180
Arthur, J. C., & Jobin, C. (2013). The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes, 4(3), 253–258. https://doi.org/10.4161/gmic.24220
Arthur, J. C., Perez-Chanona, E., Muhlbauer, M., Tomkovich, S., Uronis, J. M., Fan, T. J., et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Science, 338(6103), 120–123, https://doi.org/10.1126/science.1224820.
Tomkovich, S., Yang, Y., Winglee, K., Gauthier, J., Mühlbauer, M., Sun, X., et al. (2017). Locoregional effects of microbiota in a preclinical model of colon carcinogenesis. Cancer Research, 77(10), 2620–2632. https://doi.org/10.1158/0008-5472.CAN-16-3472
Yang, Y., Gharaibeh, R. Z., Newsome, R. C., & Jobin, C. (2020). Amending microbiota by targeting intestinal inflammation with TNF blockade attenuates development of colorectal cancer. Nat Cancer, 1(7), 723–734. https://doi.org/10.1038/s43018-020-0078-7
Zhu, W., Miyata, N., Winter, M. G., Arenales, A., Hughes, E. R., Spiga, L., et al. (2019). Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. Journal of Experimental Medicine, 216(10), 2378–2393. https://doi.org/10.1084/jem.20181939
Chung, L., Thiele Orberg, E., Geis, A. L., Chan, J. L., Fu, K., DeStefano Shields, C. E., et al. (2018). Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host & Microbe, 23(2), 203-214.e205. https://doi.org/10.1016/j.chom.2018.01.007
Hwang, S., Lee, C. G., Jo, M., Park, C. O., Gwon, S. Y., Hwang, S., et al. (2020). Enterotoxigenic Bacteroides fragilis infection exacerbates tumorigenesis in AOM/DSS mouse model. International Journal of Medical Sciences, 17(2), 145–152. https://doi.org/10.7150/ijms.38371
Li, S., Peppelenbosch, M. P., & Smits, R. (2019). Bacterial biofilms as a potential contributor to mucinous colorectal cancer formation. Biochimica et Biophysica Acta - Reviews on Cancer, 1872(1), 74–79. https://doi.org/10.1016/j.bbcan.2019.05.009
Chew, S. S., Tan, L. T., Law, J. W., Pusparajah, P., Goh, B. H., Ab Mutalib, N. S., et al. (2020). Targeting gut microbial biofilms-a key to hinder colon carcinogenesis? Cancers (Basel), 12(8), https://doi.org/10.3390/cancers12082272.
Dejea, C. M., Fathi, P., Craig, J. M., Boleij, A., Taddese, R., Geis, A. L., et al. (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science, 359(6375), 592–597. https://doi.org/10.1126/science.aah3648
Kostic, A. D., Chun, E., Robertson, L., Glickman, J. N., Gallini, C. A., Michaud, M., et al. (2013). Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host & Microbe, 14(2), 207–215. https://doi.org/10.1016/j.chom.2013.07.007
Yu, M. R., Kim, H. J., & Park, H. R. (2020). Fusobacterium nucleatum accelerates the progression of colitis-associated colorectal cancer by promoting EMT. Cancers (Basel), 12(10), https://doi.org/10.3390/cancers12102728.
Flynn, K. J., Baxter, N. T., & Schloss, P. D. (2016). Metabolic and community synergy of oral bacteria in colorectal cancer. mSphere, 1(3), https://doi.org/10.1128/mSphere.00102-16.
Li, X., Zhu, S., Zhang, T., & Chen, X. (2021). Association between oral microflora and gastrointestinal tumors (Review). Oncology Reports, 46(2), https://doi.org/10.3892/or.2021.8111.
Wang, X., Jia, Y., Wen, L., Mu, W., Wu, X., Liu, T., et al. (2021). Porphyromonas gingivalis promotes colorectal carcinoma by activating the hematopoietic NLRP3 inflammasome. Cancer Research, 81(10), 2745–2759. https://doi.org/10.1158/0008-5472.can-20-3827
Kitamoto, S., Nagao-Kitamoto, H., Hein, R., Schmidt, T. M., & Kamada, N. (2020). The bacterial connection between the oral cavity and the gut diseases. Journal of Dental Research, 99(9), 1021–1029. https://doi.org/10.1177/0022034520924633
Zhang, Y., Weng, Y., Gan, H., Zhao, X., & Zhi, F. (2018). Streptococcus gallolyticus conspires myeloid cells to promote tumorigenesis of inflammatory bowel disease. Biochemical and Biophysical Research Communications, 506(4), 907–911. https://doi.org/10.1016/j.bbrc.2018.10.136
Chichlowski, M., Sharp, J. M., Vanderford, D. A., Myles, M. H., & Hale, L. P. (2008). Helicobacter typhlonius and Helicobacter rodentium differentially affect the severity of colon inflammation and inflammation-associated neoplasia in IL10-deficient mice. Comparative Medicine, 58(6), 534–541.
Erdman, S. E., Rao, V. P., Poutahidis, T., Rogers, A. B., Taylor, C. L., Jackson, E. A., et al. (2009). Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proceedings of the National Academy of Sciences of the United States of America, 106(4), 1027–1032. https://doi.org/10.1073/pnas.0812347106
Li, L. N., Liu, Y., Zhang, H. C., Wu, T., Dai, Y., & Wang, W. H. (2020). Helicobacter pylori infection reduces TAMs infiltration in a mouse model of AOM/DSS induced colitis-associated cancer. PLoS ONE, 15(11), e0241840. https://doi.org/10.1371/journal.pone.0241840
Karamzin, A. M., Ropot, A. V., Sergeyev, O. V., & Khalturina, E. O. (2021). Akkermansia muciniphila and host interaction within the intestinal tract. Anaerobe, 72, 102472. https://doi.org/10.1016/j.anaerobe.2021.102472
Zhang, T., Ji, X., Lu, G., & Zhang, F. (2021). The potential of Akkermansia muciniphila in inflammatory bowel disease. Applied Microbiology and Biotechnology, 105(14–15), 5785–5794. https://doi.org/10.1007/s00253-021-11453-1
Wang, L., Tang, L., Feng, Y., Zhao, S., Han, M., Zhang, C., et al. (2020). A purified membrane protein from Akkermansia muciniphila or the pasteurised bacterium blunts colitis associated tumourigenesis by modulation of CD8(+) T cells in mice. Gut, 69(11), 1988–1997. https://doi.org/10.1136/gutjnl-2019-320105
Wang, F., Cai, K., Xiao, Q., He, L., Xie, L., & Liu, Z. (2022). <i>Akkermansia muciniphila</i> administration exacerbated the development of colitis-associated colorectal cancer in mice. [Research Paper]. Journal of Cancer, 13(1), 124–133, https://doi.org/10.7150/jca.63578.
Moschen, A. R., Gerner, R. R., Wang, J., Klepsch, V., Adolph, T. E., Reider, S. J., et al. (2016). Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host & Microbe, 19(4), 455–469. https://doi.org/10.1016/j.chom.2016.03.007
Wallace, J. L. (2010). Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxidants & Redox Signaling, 12(9), 1125–1133. https://doi.org/10.1089/ars.2009.2900
Mottawea, W., Chiang, C. K., Mühlbauer, M., Starr, A. E., Butcher, J., Abujamel, T., et al. (2016). Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nature Communications, 7, 13419. https://doi.org/10.1038/ncomms13419
Kim, S. W., Kim, H. M., Yang, K. M., Kim, S. A., Kim, S. K., An, M. J., et al. (2010). Bifidobacterium lactis inhibits NF-kappaB in intestinal epithelial cells and prevents acute colitis and colitis-associated colon cancer in mice. Inflammatory Bowel Diseases, 16(9), 1514–1525. https://doi.org/10.1002/ibd.21262
Wang, Q., Wang, K., Wu, W., Lv, L., Bian, X., Yang, L., et al. (2020). Administration of Bifidobacterium bifidum CGMCC 15068 modulates gut microbiota and metabolome in azoxymethane (AOM)/dextran sulphate sodium (DSS)-induced colitis-associated colon cancer (CAC) in mice. Applied Microbiology and Biotechnology, 104(13), 5915–5928. https://doi.org/10.1007/s00253-020-10621-z
Silveira, D. S. C., Veronez, L. C., Lopes-Júnior, L. C., Anatriello, E., Brunaldi, M. O., & Pereira-da-Silva, G. (2020). Lactobacillus bulgaricus inhibits colitis-associated cancer via a negative regulation of intestinal inflammation in azoxymethane/dextran sodium sulfate model. World Journal of Gastroenterology, 26(43), 6782–6794. https://doi.org/10.3748/wjg.v26.i43.6782
Matsumoto, S., Hara, T., Nagaoka, M., Mike, A., Mitsuyama, K., Sako, T., et al. (2009). A component of polysaccharide peptidoglycan complex on Lactobacillus induced an improvement of murine model of inflammatory bowel disease and colitis-associated cancer. Immunology, 128(1 Suppl), e170–e180. https://doi.org/10.1111/j.1365-2567.2008.02942.x
Gao, C., Ganesh, B. P., Shi, Z., Shah, R. R., Fultz, R., Major, A., et al. (2017). Gut microbe–mediated suppression of inflammation-associated colon carcinogenesis by luminal histamine production. The American Journal of Pathology, 187(10), 2323–2336. https://doi.org/10.1016/j.ajpath.2017.06.011
Kahouli, I., Malhotra, M., Westfall, S., Alaoui-Jamali, M. A., & Prakash, S. (2017). Design and validation of an orally administrated active L. fermentum-L. acidophilus probiotic formulation using colorectal cancer Apc (Min/+) mouse model. Applied Microbiology and Biotechnology, 101(5), 1999–2019, https://doi.org/10.1007/s00253-016-7885-x.
Wang, Z., Hua, W., Li, C., Chang, H., Liu, R., Ni, Y., et al. (2019). Protective role of fecal microbiota transplantation on colitis and colitis-associated colon cancer in mice is associated with Treg cells. [Original Research]. Frontiers in Microbiology, 10(2498), https://doi.org/10.3389/fmicb.2019.02498.
Borody, T. J., & Khoruts, A. (2011). Fecal microbiota transplantation and emerging applications. [Research Support, Non-U.S. Gov't Review]. Nature Reviews Gastroenterology & Hepatology, 9(2), 88–96, https://doi.org/10.1038/nrgastro.2011.244.
van Nood, E., Vrieze, A., Nieuwdorp, M., Fuentes, S., Zoetendal, E. G., de Vos, W. M., et al. (2013). Duodenal infusion of donor feces for recurrent Clostridium difficile. New England Journal of Medicine, 368(5), 407–415. https://doi.org/10.1056/NEJMoa1205037
Mann, E. R., You, J., Horneffer-van der Sluis, V., Bernardo, D., Omar Al-Hassi, H., Landy, J., et al. (2013). Dysregulated circulating dendritic cell function in ulcerative colitis is partially restored by probiotic strain <i>Lactobacillus casei</i> Shirota. Mediators of Inflammation, 2013, 573576. https://doi.org/10.1155/2013/573576
Yang, X. D., Ai, W., Asfaha, S., Bhagat, G., Friedman, R. A., Jin, G., et al. (2011). Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of CD11b+Ly6G+ immature myeloid cells. Nature Medicine, 17(1), 87–95. https://doi.org/10.1038/nm.2278
Ghanavati, R., Asadollahi, P., Shapourabadi, M. B., Razavi, S., Talebi, M., & Rohani, M. (2020). Inhibitory effects of Lactobacilli cocktail on HT-29 colon carcinoma cells growth and modulation of the Notch and Wnt/β-catenin signaling pathways. Microbial Pathogenesis, 139, 103829. https://doi.org/10.1016/j.micpath.2019.103829
Tian, Y., Xu, Q., Sun, L., Ye, Y., & Ji, G. (2018). Short-chain fatty acids administration is protective in colitis-associated colorectal cancer development. Journal of Nutritional Biochemistry, 57, 103–109. https://doi.org/10.1016/j.jnutbio.2018.03.007
Lowe, E. L., Crother, T. R., Rabizadeh, S., Hu, B., Wang, H., Chen, S., et al. (2010). Toll-like receptor 2 signaling protects mice from tumor development in a mouse model of colitis-induced cancer. PLoS ONE, 5(9), e13027. https://doi.org/10.1371/journal.pone.0013027
Pastille, E., Faßnacht, T., Adamczyk, A., Ngo Thi Phuong, N., Buer, J., & Westendorf, A. M. (2021). Inhibition of TLR4 signaling impedes tumor growth in colitis-associated colon cancer. [Original Research]. Frontiers in Immunology, 12(1608), https://doi.org/10.3389/fimmu.2021.669747.
Fukata, M., Shang, L., Santaolalla, R., Sotolongo, J., Pastorini, C., España, C., et al. (2011). Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflammatory Bowel Diseases, 17(7), 1464–1473. https://doi.org/10.1002/ibd.21527
Uronis, J. M., Mühlbauer, M., Herfarth, H. H., Rubinas, T. C., Jones, G. S., & Jobin, C. (2009). Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE, 4(6), e6026. https://doi.org/10.1371/journal.pone.0006026
Lopez, A., & Peyrin-Biroulet, L. (2013). 5-Aminosalicylic acid and chemoprevention: Does it work? Digestive Diseases, 31(2), 248–253. https://doi.org/10.1159/000353806
Koelink, P. J., Robanus-Maandag, E. C., Devilee, P., Hommes, D. W., Lamers, C. B. H. W., & Verspaget, H. W. (2009). 5-Aminosalicylic acid inhibits colitis-associated but not sporadic colorectal neoplasia in a novel conditional Apc mouse model. Carcinogenesis, 30(7), 1217–1224. https://doi.org/10.1093/carcin/bgp113
Velayos, F. S., Terdiman, J. P., & Walsh, J. M. (2005). Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: A systematic review and metaanalysis of observational studies. American Journal of Gastroenterology, 100(6), 1345–1353. https://doi.org/10.1111/j.1572-0241.2005.41442.x
Zhao, L. N., Li, J. Y., Yu, T., Chen, G. C., Yuan, Y. H., & Chen, Q. K. (2014). 5-Aminosalicylates reduce the risk of colorectal neoplasia in patients with ulcerative colitis: An updated meta-analysis. PLoS ONE, 9(4), e94208. https://doi.org/10.1371/journal.pone.0094208
Nguyen, G. C., Gulamhusein, A., & Bernstein, C. N. (2012). 5-Aminosalicylic acid is not protective against colorectal cancer in inflammatory bowel disease: A meta-analysis of non-referral populations. Official journal of the American College of Gastroenterology | ACG, 107(9).
Lopetuso, L. R., Petito, V., Zinicola, T., Graziani, C., Gerardi, V., Arena, V., et al. (2016). Infliximab does not increase colonic cancer risk associated to murine chronic colitis. World Journal of Gastroenterology, 22(44), 9727–9733. https://doi.org/10.3748/wjg.v22.i44.9727
Biancone, L., Petruzziello, C., Calabrese, E., Zorzi, F., Naccarato, P., Onali, S., et al. (2009). Long-term safety of Infliximab for the treatment of inflammatory bowel disease: Does blocking TNFalpha reduce colitis-associated colorectal carcinogenesis? Gut, 58(12), 1703. https://doi.org/10.1136/gut.2008.176461
Baars, J. E., Looman, C. W., Steyerberg, E. W., Beukers, R., Tan, A. C., Weusten, B. L., et al. (2011). The risk of inflammatory bowel disease-related colorectal carcinoma is limited: Results from a nationwide nested case-control study. American Journal of Gastroenterology, 106(2), 319–328. https://doi.org/10.1038/ajg.2010.428
Lu, M. J., Qiu, X. Y., Mao, X. Q., Li, X. T., & Zhang, H. J. (2018). Systematic review with meta-analysis: Thiopurines decrease the risk of colorectal neoplasia in patients with inflammatory bowel disease. Alimentary Pharmacology & Therapeutics, 47(3), 318–331. https://doi.org/10.1111/apt.14436
Zhang, Q., Pi, J., Woods, C. G., & Andersen, M. E. (2010). A systems biology perspective on Nrf2-mediated antioxidant response. Toxicology and Applied Pharmacology, 244(1), 84–97. https://doi.org/10.1016/j.taap.2009.08.018
Zhao, Y., Guo, Q., Zhao, K., Zhou, Y., Li, W., Pan, C., et al. (2017). Small molecule GL-V9 protects against colitis-associated colorectal cancer by limiting NLRP3 inflammasome through autophagy. Oncoimmunology, 7(1), e1375640–e1375640. https://doi.org/10.1080/2162402X.2017.1375640
Irrazabal, T., Thakur, B. K., Kang, M., Malaise, Y., Streutker, C., Wong, E. O. Y., et al. (2020). Limiting oxidative DNA damage reduces microbe-induced colitis-associated colorectal cancer. Nature Communications, 11(1), 1802. https://doi.org/10.1038/s41467-020-15549-6
Acknowledgements
This work was supported by the Crohn’s and Colitis Foundation grant 632826 (to H.N.-K.), the Office of the Assistant Secretary of Defense for Health Affairs endorsed by the Department of Defense through the Peer-Reviewed Cancer Research Program under Award No. W81XWH2010547 and the Prevent Cancer Foundation (to S.K.), and the National Institutes of Health grants DK108901, DK119219, AI142047, and DK125087 (to N.K.).
Author information
Authors and Affiliations
Contributions
H.N.-K., S.K., and N.K. wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nagao-Kitamoto, H., Kitamoto, S. & Kamada, N. Inflammatory bowel disease and carcinogenesis. Cancer Metastasis Rev 41, 301–316 (2022). https://doi.org/10.1007/s10555-022-10028-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-022-10028-4