
A new oleanane-type triterpene glycoside, 3-O-α-L-arabinopyranosyl-(1→3)-α-L-rhamnopyranosyl-(1→2)-α-L-arabinopyranosyloleanolic acid 28-O-β-D-xylopyranosyl-(1→3)-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester, was isolated from the aqueous-ethanolic extract of the seeds of Nephelium lappaceum L. The structure elucidation of this compound was based on analyses of spectroscopic data, including 1D and 2D NMR and HR-ESI-MS techniques, and by comparing their NMR data with those reported in the literature.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Nephelium lappaceum L., belonging to the genus Nephelium and thus the Sapindaceae family, is widely distributed in Southeast Asia’s tropical regions, including Vietnam [1]. The fruit of this species, commonly known as “Rambutan”, is recommended for severe dysentery, as an astringent, an antifebrile, and a warm carminative in dyspepsia [2]. When rambutan fruit is processed, the fruits are deseeded first, and the seeds become a waste by-product [3, 4]. Interestingly, rambutan seeds have been reported to have narcotic effects, and roasted rambutan seeds are consumed in the Philippines [5]. Previous studies on the seeds of N. lappaceum reported the presence of alkaloids, phenolic compounds, glycosides, carbohydrates, and saponins that are responsible for these antioxidant and antibacterial activities [1, 6, 7]. As part of our ongoing search for new glycosides from natural sources, we report the isolation of a new oleanane-type triterpene glycoside from the seeds of N. lappaceum, and the spectral evidence leading to elucidating the structure was carried out.
Compound 1 was isolated as an amorphous powder, exhibiting a molecular ion (positive-ion HR-ESI-MS) at m/z 1315.6298 [M + Na]+ corresponding to the molecular formula C62H100O28. The 1H NMR spectrum of 1 displayed signals of seven tertiary methyl proton signals at δ 1.26 (s, H3-23), 1.13 (s, H3-24), 0.86 (s, H3-25), 0.97 (s, H3-26), 1.30 (s, H3-27), 0.95 (s, H3-29), 1.01 (s, H3-30), an olefinic proton at δ 5.46 (br.t, J = 3.5 Hz, H-12), corresponding to 13C NMR data of seven tertiary methyl carbon signals at δ 28.1 (C-23), 17.2 (C-24), 15.6 (C-25), 17.5 (C-26), 26.1 (C-27), 33.4 (C-29) and 23.6 (C-30), two olefinic carbon signals at δ 122.2 (C-12) and 144.6 (C-13), which were typical signals of the oleanolic acid skeleton [8]. The downfield chemical shift at δ 88.6 (C-3) and the upfield chemical shift at δ 178.1 (C-28) in the 13C NMR spectrum of 1 (Table 1) indicated that 1 was a 3,28-bidesmosidic glycoside [9].
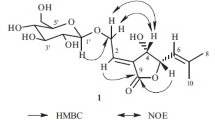
The 1H NMR spectrum of 1 showed six anomeric protons at δ 4.84 (d, J = 7.6 Hz), 4.86 (d, J = 6.0 Hz), 4.88 (d, J = 7.0 Hz), 5.01 (d, J = 7.6 Hz), 6.02 (d, J = 8.2 Hz), and 6.13 (br.s) which showed HSQC correlations with six anomeric carbon signals at δ 103.7, 104.9, 103.8, 105.0, 96.0, and 101.6, respectively, indicating the presence of six sugar units. These sugars were determined to be two L-arabinoses (Ara I, Ara II), two D-xyloses (Xyl I, Xyl II), one D-glucose (Glc), and one L-rhamnose (Rha) by acid hydrolysis and TLC comparison with authentic samples. The β-anomeric configurations for the Xyl and Glc units were deduced from their JH-1, 2 coupling constants ranging from 7.6 to 8.0 Hz, and the Ara units were determined to be α anomeric configurations based on the JH-1, 2 values (6.0–7.0 Hz) observed in the 4C1 forms [10]. The α-configuration of the Rha unit was determined from the broad singlet observed for the anomeric proton and the JC-1, H-1 value of 168 Hz [8]. Assignment for all 1H and 13C NMR signals and determination of the structure were achieved by 2D NMR analyses, mainly in HMBC and ROESY. In the HMBC spectrum, a correlation between δH 4.86 (d, J = 6.0 Hz, Ara I H-1) and δC 88.6 (C-3 aglycone) indicated that Ara I was linked to the C-3 of the aglycone. The linkage of the Rha unit at the C-2 of Ara I was determined by the correlation between δH 6.13 (br.s, Rha H-1) and δC 75.5 (Ara I C-2). Similarly, the linkage of Ara II at C-3 of the Rha was indicated by the correlation between δH 4.88 (d, J = 7.0 Hz, Ara II H-1) and δC 83.1 (Rha C-3). This conclusion was further confirmed by the observation of three cross-peaks in the ROESY spectrum between δH 4.86 (d, J = 6.0 Hz, Ara I H-1) and 3.28 (H-3 aglycone), δH 6.13 (br.s, Rha H-1) and 4.53 (Ara I H-2), δH 4.88 (d, J = 7.0 Hz, Ara II H-1) and δH 4.67 (dd, J = 9.6, 2.1 Hz, Rha H-3). Thus, the glycosidic sequence linked to the C-3 of the aglycone was established as α-L-arabinopyranosyl-(1→3)-α-L-rhamnopyranosyl-(1→2)-α-L-arabinopyranosyl. The three remaining Xyl I, Xyl II, and Glc were identified as β-D-xylopyranosyl-(1→3)-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl sequence linked to C-28 of the aglycone. This assumption was verified according to the chemical shift at δC 96.0 (Glc C-1), a typical value suggesting an ester linkage with C-28 of the aglycone. The linkage was confirmed by the observation of three cross-peaks in the HMBC spectrum between δH 6.02 (d, J = 8.0 Hz, Glc H-1) and δC 178.1 (C-28 aglycone), δH 5.01 (d, J = 7.6 Hz, Xyl I H-1) and δC 75.8 (Glc C-2), δH 4.84 (d, J = 7.6 Hz) and δC 75.7 (Xyl I C-3), and two cross-peaks in the ROESY spectrum between δH 5.01 (d, J = 7.6 Hz, Xyl I H-1) and δC 4.28 (Glc H-2), δH 4.84 (d, J = 7.6 Hz) and δC 4.07 (Xyl I H-3) (Fig. 1). The structure of 1 was thus characterised as 3-O-α-L-arabinopyranosyl-(1→3)-α-L-rhamnopyranosyl-(1→2)-α-Larabinopyranosyloleanolic acid 28-O-β-D-xylopyranosyl-(1→3)-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester.

Key HMBC and ROESY correlations of compound 1.
Experimental
General Experimental Procedures. Optical rotation was measured with an AA-10R automatic polarimeter (Optical Activity LTD, Ramsey, UK). NMR spectra were measured on a Varian VNMRS 600 MHz spectrometer (Agilent Technologies, Santa Clara, California, USA). HR-ESI-MS spectrum was recorded on Bruker micrOTOF II mass spectrometer (Bruker, Mannheim, Germany). Separation and purification were performed by vacuum liquid chromatography (VLC) on RP-18 silica gel (75–200 μm, Silicycle, Quebec, Canada) and NP-60 silica gel 60 (60–200 μm, Merck, Germany), column chromatography (CC) on silica gel 60 (15–40 μm, Merck, Germany) and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Merck, Germany). Chemical shifts are given on the δ-scale with pyridine-d5 as the internal standard.
Plant Material. The seeds of N. lappaceum were collected in a fruit boutique in Thai Nguyen City, Vietnam, in 2021 (21°34′01′′ N, 105°48′35′′ E) and identified by one of the authors (Dr. Hung Duc Nguyen). A voucher specimen (No. NELASE1121) was deposited in our lab.
Extraction and Isolation. Dried seeds (156.8 g) of N. lappaceum were successively extracted three times with 500 mL of EtOH–H2O (75%–35%) each time. The aqueous-ethanolic extract was evaporated under reduced pressure to give a thick syrup, then dissolved in a minimum volume of H2O and freeze-dried to yield 8.9 g of crude extract. This was dissolved in a minimum volume of H2O and submitted to a VLC over silica gel RP-18 eluting with each 500 mL of solvents, including EtOH–H2O (0:1, 1:1, 0:1), yielding 3 fractions (NLS.1, NLS.2 and NLS.3). Fraction NLS.2 (617.1 mg) was dissolved in a minimum volume of CHCl3–MeOH–H2O, 75:25:3 and subjected to a VLC over silica gel NP-60 eluted with CHCl3–MeOH– H2O, 75:25:3, 70:30:5, 60:32:7 (300 mL each) to collect three subfractions (NLS.2.1–NLS.2.3). Subfraction NLS.2.2 (89.2 mg) was subjected again on a CC over silica gel NP-60 eluted with CHCl3–MeOH–H2O, 70:30:5 affording five subfractions (NLS.2.2.1–NLS.2.2.5). Subfraction NLS.2.2.2 (6.3 mg) rich in saponin was applied to a CC over Sephadex LH-20 with EtOH 96% to remove pigments to afford 1 (2.9 mg) as a pure compound.
3-O-α-L-Arabinopyranosyl-(1→3)-α-L-rhamnopyranosyl-(1→2)-α-L-arabinopyranosyloleanolic acid 28-O-β-D-xylopyranosyl-(1→3)-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester (1), white amorphous powder, [α]25D –22° (c 0.75, MeOH). 1H (600 MHz, Py-d5) and 13C (150 MHz, Py-d5) NMR spectral data are shown in Table 1. HR-ESI-MS m/z 1315.6298 [M + Na]+ (calcd for C62H100NaO28, 1315.6293).
Acid Hydrolysis and GC Analysis. The protocol of identification of sugar moieties and those absolute configurations of isolated compound was detailed as previously referenced [11,12,13].
References
Y. X. Zhao, W. J. Liang, H. J. Fan, Q. Y. Ma, W. X. Tian, H. F. Dai, H. Z. Jiang, N. Li, and X. F. Ma, Carbohydr. Res., 346 (11), 1302 (2011).
U. Palanisamy, H. M. Cheng, T. Masilamani, T. Subramaniam, L. T. Ling, and A. K. Radhakrishnan, Food Chem., 109 (1), 54 (2008).
J. A. Solis-Fuentes, G. Camey-Ortiz, M. del R. Hernandez-Medel, F. Perez-Mendoza, and C. Duran-de-Bazua, Bioresour. Technol., 101 (2), 799 (2010).
N. Lourith, M. Kanlayavattanakul, K. Mongkonpaibool, T. Butsaratrakool, and T. Chinmuang, Ind. Crops Prod., 83, 149 (2016).
K. F. Chai, N. Mohd Adzahan, R. Karim, Y. Rukayadi, and H. M. Ghazali, Int. J. Food Prop., 21 (1), 1091 (2018).
A. Rajasekaran, S. Ganesan, N. Kamini, C. Lavanya, L. Lee Yoon, and H. Shian Oh, Orient Pharm. Exp. Med., 13 (2), 149 (2013).
N. Thitilertdecha, A. Teerawutgulrag, J. D. Kilburn, and N. Rakariyatham, Molecules, 15 (3), 1453 (2010).
H. D. Nguyen, J. Asian Nat. Prod. Res., 25 (3), 218 (2023).
T. Q. Tu, H. D. Nguyen, and M. H. Chu, Phytochem. Lett., 55, 142 (2023).
A. E. Moffi Biang, L. M. Messi, E. Le Doux Kamto, L. M. Simo, P. Lavedan, M. Vedrenne, J. N. Mbing, D. E. Pegnyemb, M. Haddad, and O. P. Note, Carbohydr. Res., 508, 108393 (2021).
T. T. Quang, M. C. Hoang, and H. D. Nguyen, Chem. Nat. Compd., 59, 520 (2023).
Thuy Thi Xuan Vi, Hung Duc Nguyen, Tan Quang Tu, Yen Thi Hai Nguyen, Quan Huu Nguyen, and Mau Hoang Chu, Chem. Nat. Compd., 60, 283 (2024).
T. H. Y. Nguyen, H. M. Chu, and D. H. Nguyen, Nat. Prod. Res., 38 (7), 1191 (2024).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 4, July–August, 2024, pp. 566–568.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vi, T.T.X., Nguyen, H.D., Tu, T.Q. et al. A New Oleanane-Type Triterpene Glycoside from Nephelium lappaceum. Chem Nat Compd 60, 653–656 (2024). https://doi.org/10.1007/s10600-024-04406-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-024-04406-3