
A 2,3-secotriterpenoid with a methylketone was prepared from 1-hydroxyiminodihydrobetulonic acid methyl ester via a Grignard reaction followed by a Beckmann rearrangement. Functionalization of it led to the formation of 3-hydroxy- and 31-bromo-substituted derivatives; intramolecular cyclization, to the formation of a five-membered ring A with an alkene-nitrile. The synthetic products included compounds (5a and 9) with moderate cytotoxicity (IC50 25.22–46.66 μM) against MCF-7, HCT116, A549, and PC-3 cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The significance of betulinic acid (3β-hydroxylup-20(29)-en-28-oic acid, BA) and semi-synthetic derivatives of BA for applications in medicinal chemistry derives mainly from their highly specific action against viruses and cancer cells with minimal harmful effects on healthy human cells [1,2,3,4]. The BA content in the extract of birch bark (Betula pendula) is usually less than 3–4% as compared to the elevated content (up to 80%) in the same extract of betulin, the biosynthetic precursor of BA [5]. BA can be produced in 86–92% yield by effective methods for chemical oxidation of betulin [6, 7]. Nevertheless, betulonic acid is most often used as starting material in laboratory syntheses. An effective method for preparative synthesis of it from betulin-containing birch-bark extract was recently published [8].
Previously, the ability to increase the antiviral and antitumor activity of pentacyclic triterpenoids by skeletal transformation of their C-3 alkylated derivatives was demonstrated by us [9,10,11,12,13]. In particular, a 2,3-secolupane α-bromo-substituted methylketone with pronounced cytotoxic activity (IC50 0.8–25.4 μM) against 11 tumor cell lines, including cancer cells with multi-drug resistance, was synthesized from betulonic acid methyl ester [14]. Also, introduction of a C-30 aldehyde into the isopropylidene fragment of lupane derivatives [10, 12, 14] most often increased the nonspecific cytotoxicity [15]. The present article involved the synthesis and screening of the cytotoxic properties of novel 3-methyl-substituted derivatives of dihydrobetulonic acid methyl ester (1) to assess the contribution of an oxidized isopropylidene moiety to the manifestation of cytotoxicity by the previously reported compounds [10, 12, 14].
The starting material for the work was α-hydroxyiminoketone 2, the synthesis of which from 1 was recently reported by us [16]. A Grignard reaction [10,11,12,13, 17] of 2 with the alkylating agent CH3MgI produced 3β-hydroxy-3α-methyl derivative 3 (Scheme 1). The IR spectrum of 3 exhibited bands for C=N−OH (1666 cm−1) and OH stretching vibrations (3326, 3505 cm−1). The 1H NMR spectrum of 3 showed doublets for the enantiomeric 2H-1 methylene protons with centers at 2.22 and 3.37 ppm (AB-system, SSCC 12.0 Hz); a broad singlet at 5.18 ppm for the hydroxyl; and a characteristic singlet at 1.37 ppm for the methyl protons. The 13C NMR spectrum of 3 exhibited resonances for C atoms bonded to hydroxyl (76.84 ppm) and hydroxyimine (163.98 ppm). In the next step, alkylated derivative 3 underwent a Beckmann rearrangement using SOCl2−CH2Cl2, which formed the corresponding methylketone 4. The IR spectrum of 4 contained characteristic absorption bands for C=O (1703 cm−1) and CN (2238 cm−1). The PMR spectrum of 4 showed two doublets for the methylene 2H-1 protons with centers at 2.43 and 2.55 ppm (AB-system, SSCC 16.0 Hz) and a singlet for the C-3 methyl at 2.27 ppm. The 13C NMR spectrum of 4 included characteristic resonances for the CN and C=O C atoms at 118.65 and 214.28 ppm, respectively.

Scheme 1.
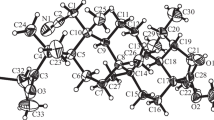
The nitrile of methylketone 4 remained inert upon reduction by LiAlH4 in the presence of AlCl3. The reaction products were a mixture of diastereomeric alcohols 3(R)-5a and 3(S)-5b (Scheme 1), which were isolated pure in a 1.3:1.0 ratio by column chromatography over SiO2. 13C NMR spectra of 5a and 5b had characteristic resonances for C-3 bonded to a hydroxyl at 73.11 or 72.92. Their IR spectra exhibited absorption bands for the OH at 3523 and 3521 cm−1, respectively. The relative (S)-configuration of C-3 bonded to the hydroxyl in 5b was confirmed by an X-ray crystal structure analysis (XSA) (Fig. 1). The resonances of the H-3 protons had practically the same shifts in 1H NMR spectra of (3R)-5a and (3S)-5b. The quartets corresponding to them appeared at 3.825 and 3.83 ppm, respectively. The weak-field shift of the (3S)-isomer protons was more noticeable for the 2H-1 protons, the doublets of which (AB-system, SSCC 18.0 Hz) were observed at 2.60 and 2.75 ppm for alcohol (3R)-5a or at 2.61 and 2.77 ppm for (3S)-5b, and the doublets for the 3H-32 protons (AB-system, SSCC 8.0 Hz) with centers at 1.14 or 1.24 ppm, respectively. Milder reduction of methylketone 4 by NaBH4 occurred stereoselectively to form the 3(R)-hydroxy derivative in 82% yield. This was consistent with data for the selective reduction by NaBH4 of oleanane- and lupane-type methylketones [10].

Structure of 3(S)-5b from XSA (N and O atoms refined as disordered).
The reaction of methylketone 4 with pyridinium tribromide in AcOH produced α-bromo-substituted derivative 6. Its IR spectrum showed characteristic bands for stretching vibrations of carbonyl at 1722 cm–1 and cyano at 2238 cm–1. The 1H NMR spectrum of 6 contained doublets for the methylene 2H-1 protons with centers at 2.37 and 2.60 ppm (AB-system, SSCC 18.0 Hz) and resonances for the methylene protons of the bromomethyl moiety as doublets of an AB-system with centers at 4.29 and 4.35 ppm (SSCC 12.0 Hz). The 13C NMR spectrum of 6 had characteristic resonances for the CN and C=O C atoms at 118.49 and 206.64 ppm, respectively.
Bromomethylketone 6 was O-alkylated by nicotinic acid with heating in an aprotic solvent in the presence of an excess of Et3N and K2CO3 to afford the corresponding β-ketoester 7 in 62% yield [12, 18]. The 13C NMR spectrum of 7 exhibited typical resonances for the CN (118.53 ppm) and C=O C atoms (206.76 ppm), for two esters (163.51 and 176.74 ppm), and for the heteroaromatic ring (124.42, 126.86, 139.92, 148.47, and 150.60 ppm). The 1H NMR spectrum included characteristic doublets of AB-systems for the two pairs of methylene protons 2H-1 and 2H-32 with centers at 2.49 and 2.66/5.27 and 5.35 ppm (SSCC 20.0/18.0 Hz) and resonances of the nicotinic acid aromatic protons in the range 7.57–9.30 ppm.
As expected [9,10,11, 13, 19], methylketone 4 underwent an intramolecular oxo-nitrile cyclization under basic conditions of t-BuOK−t-BuOH to form A-pentacyclic α,β-alkenenitrile 8 (Scheme 1). The IR spectrum of 8 exhibited characteristic bands for stretching vibrations at 1728 (COOCH3) and 2207 cm−1 (CN). The 1H NMR spectrum of 8 showed the resonance for the C-3 methyl at 1.85 ppm. Its 13C NMR spectrum gave characteristic resonances of the alkenenitrile C atoms C-1 (120.45), C-3 (118.36), and C-3 (167.10 ppm). Compound 9, the product of reduction of the C-28 carbomethoxyl group to hydroxyl, was obtained under reducing conditions (LiAlH4, AlCl3) from A-pentacyclic α,β-alkenenitrile 8. The 1H NMR spectrum of 9 exhibited a singlet at 1.86 ppm for the C-3 methyl protons and two doublets of an AB-system of the 2H-28 methylene protons at 3.32 and 3.75 ppm (SSCC 12.0 Hz). The 13C NMR spectrum of 9 gave characteristic resonances for the alkenenitrile moiety at 120.36 (C-1), 118.37 (C-2), and 167.19 ppm (C-3). The carbomethoxyl group of 4 remained inert under analogous conditions.
Compounds 2, 4, and 6–8 were nontoxic (IC50 > 100 μM) according to a study of the cytotoxic activity of synthetic products 2–9 against seven tumor cell lines. The lack of cytotoxic properties for bromo-substituted methylketone 6 indicated that the specific activity of the previously reported lupane methylketone [10] was due largely to the presence in the triterpenoid of the C-30 aldehyde of the isopropylidene moiety or a combination in the triterpene structure of the aldehyde and a modified ring A. Compounds 5a and 9 in the series of synthesized 2–9 were moderately active (IC50 25.22–46.66 μM) against cancer cell lines MCF-7, HCT116, A549, and PC-3. Alcohol 5a appeared more selective, considering its low toxicity against normal HEK293 cells (Table 1).
Experimental
1H NMR, 13C NMR, and DEPT spectra of the synthesized compounds in CDCl3 with TMS internal standard were taken on a Bruker Avance II NMR spectrometer (400 and 100 MHz, respectively). IR spectra (ν, cm−1) were recorded from thin films obtained by evaporation of CHCl3 solutions of the compounds on a Bruker IFS 66/S FT-IR spectrometer (Germany). Optical rotation in CHCl3 solution was measured at 589 nm on a PerkinElmer 341 polarimeter (USA). The threshold melting point at heating rate 1°C/min was determined on an OptiMelt MPA100 apparatus (USA). GC-MS spectra were analyzed by an Agilent Technologies 6890N instrument with a DB-35ms capillary column (30 m × 0.25 mm) at vaporizer temperature 240°C with programmed temperature of 20–40°C/min and He carrier gas. Elemental analyses (C, H, N) used a Vario EL cube analyzer (Germany). The course of reactions was monitored by TLC on Sorbfil plates (Russia) after treatment with H2SO4 (5%) followed by heating at 95–100°C for 2–3 min. Column chromatography used Macherey-Nagel silica gel (60–200 μm) and petroleum ether (40–70°)–EtOAc or petroleum ether (40–70°)−EtOAc−CHCl3 eluent that was selected individually for each reaction product.
An X-ray crystal structure analysis (XSA) of 5b used an Xcalibur Ruby single-crystal diffractometer (Agilent Technologies) and the standard method [Mo Kα-radiation, 295(2) K, ω-scanning in 1° steps]. Absorption corrections were applied empirically using the SCALE3 ABSPACK algorithm [CrysAlisPro, Agilent Technologies, version 1.171.37.33 (release 27-03-2014 CrysAlis171.NET)]. The crystal (C32H53NO3, MM 499.75) was monoclinic, space group P21, a = 7.0686(16), b = 17.595(4), c = 11.924(3) Å, β = 92.48(2) Å, V = 1481.6(6) Å 3, Z = 2, dcalcd = 1.12 g/cm3, μ = 0.070 mm−1. The final refinement parameters were R1 = 0.0598 [for 1935 reflections with I > 2σ(I)], wR2 = 0.1837 (for all 5470 independent reflections), S = 0.781, ratio of twinning components 0.7473(15):0.2527 (15). The structure was solved using the SHELXS program [20] and refined by anisotropic full-matrix least squares methods over F2 for all nonhydrogen atoms using the SHELXL program [21] with an OLEX2 graphics interface [22]. H atoms were refined using a rider model. The crystal was refined using a dataset with reflection intensities in HKLF 5 format as a twin with two components. The XSA results for 5b were deposited in the Cambridge Crystallographic Data Centre under No. CCDC 2174771 and can be requested at www.ccdc.cam.ac.uk/data_request/cif.
Anhydrous solvents were prepared by standard methods [23]. Dihydrobetulin was obtained in 70% yield by catalytic (Pd−C) hydrogenation of betulin [24]. Oxidation of dihydrobetulin by Jones reagent gave dihydrobetulonic acid, which was methylated by CH3I in Me2CO to the corresponding methyl ester [25].
Preparation of 3β-Hydroxy-2-hydroxyimino-3α-methyllup-28-oic Acid (3). A freshly prepared solution of CH3MgI (5.0 mmol) in Et2O (5 mL) was treated dropwise with a solution (15 mL) of 2 (4.0 mmol) in a mixture (2:1) of Et2O and THF. The reaction mixture was stirred for 2 h, chilled, treated dropwise with ice water (25 mL) and HCl solution (17–20%, 20 mL), and stirred until the precipitate dissolved completely. The reaction products were extracted with EtOAc (2 × 50 mL). The organic layer was separated; washed with saturated Na2S2O3 solution, NaHCO3 solution (5%), and a small amount of H2O; and dried over anhydrous MgSO4. The solvent was distilled off. The solid was purified by column chromatography (CC) with elution by petroleum ether−EtOAc (15:1). Colorless crystals, yield 86%, Rf 0.46 (hexane−EtOAc, 7:3), mp 230.8°C (hexane−EtOAc), [α]20D –32.6° (c 0.5, CHCl3). IR (ν, cm–1): 3505 (OH), 3326 (OH), 1726 (COOCH3), 1666 (C=N–OH). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.86 (3H each, d, J = 8.0, CH3), 0.76 (6H, s, CH3), 0.91, 0.97, 0.98 (3H each, s, CH3), 1.37 (3H, s, H-32), 2.22, 3.37 (1H each, d, J = 12.0, H-1, AB system), 2.21–2.28 (2H, m, H-19, 20), 3.64 (3Í, s, Í-31), 5.18 (1H, br.s, OH). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.67, 14.76, 15.79, 16.82, 18.61, 18.93, 21.36, 22.83, 22.89, 23.46, 24.01, 26.80, 29.77, 29.79, 32.16, 34.43, 36.38, 37.33, 38.23, 41.28, 41.55, 42.75, 44.33, 44.54, 49.10, 50.39, 51.05, 53.45, 57.09, 76.84 (C-3), 163.98 (C-2), 176.79 (C-28). Found, %: C, 74.52; H, 10.36; N, 2.72. C32H53NO4. Calcd, %: C, 74.81; H, 10.39; N, 2.62. Mass spectrum m/z 515.4 [M]+. MM 515 g/mol.
Preparation of 3-Methyl-3-oxo-1-cyano-2,3-seco-2-norlup-28-oic Acid Methyl Ester (4). A solution of 3 (3.8 mmol) in CH2Cl2 (40 mL) was treated with SOCl2 (7.1 mmol). The reaction mixture was stirred at room temperature for 30 min. Formation of products was monitored by TLC. The solvent was evaporated. The solid was rinsed with CH2Cl2 (2 × 20 mL) and purified by CC with elution by petroleum ether−EtOAc (15:1). Colorless crystals, 82%, Rf 0.55 (hexane−EtOAc, 7:3), mp 211.7°C (hexane−EtOAc), [α]20D +1.2° (c 0.5, CHCl3). IR (ν, cm–1): 2238 (C≡N), 1725 (COOCH3), 1703 (C=O). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.85 (3H each, d, J = 8.0, CH3), 0.91, 0.95, 1.01, 1.14, 1.21 (3H each, s, CH3), 1.95–2.02 (2H, m, H-19, 20), 2.27 (3H, s, H-32), 2.43, 2.55 (1H each, d, J = 16.0, H-1, AB system), 3.63 (3H, s, H-31). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.62, 14.67, 15.74, 18.83, 21.38, 21.82, 22.75, 22.92, 23.70, 24.04, 25.74, 26.89, 29.46, 29.70, 29.71, 31.83, 33.45, 37.19, 38.14, 40.59, 42.49, 43.01, 44.12, 45.06, 48.76, 48.78, 51.14, 53.07, 57.01, 118.65 (C-2), 176.76 (C-28), 214.28 (C-3). Found, %: C, 77.22; H, 10.33; N, 2.81. C32H51NO3. Calcd, %: C, 77.92; H, 10.23; N, 2.76. Mass spectrum m/z 497.4 [M]+. MM 497 g/mol.
Preparation of 3(R)-Hydroxy-3-methyl-1-cyano-2,3-seco-2-norlup-28-oic Acid Methyl Ester (5a). A solution of 4 (4.0 mmol) in MeOH (40 mL) was stirred and treated in portions with NaBH4 (42 mmol). The reaction mixture was stirred at room temperature for 40 min and refluxed for 5 min. The formation of products was monitored by TLC. The solvent was evaporated. The resulting solid was dissolved in HCl solution (10%, 100 mL). The products were extracted with EtOAc (2 × 50 mL). The organic layer was separated, washed with H2O, and dried over anhydrous MgSO4. The solvent was evaporated. The solid was purified by CC with elution by petroleum ether−EtOAc−CHCl3 (20:1:1). Yield, 82%.
General Method for Preparing 5a,b and 9. A mixture of LiAlH4 (12.5 eq) and AlCl3 (4 eq) in anhydrous Et2O was chilled in an ice bath, stirred for 15–20 min, treated with 4 (4.0 mmol, for 5a,b) or 8 (4.2 mmol, for 9), and stirred and heated for 2 h. The formation of products was monitored by TLC. The reaction mixture was diluted with aqueous NaOH solution (1%, 20 mL). The products were extracted by EtOAc (2 × 50 mL). The organic layer was separated and dried over anhydrous MgSO4. The solvent was evaporated. The solid was purified by CC with elution by petroleum ether− EtOAc−CHCl3 (20:1:1).
3(R)-Hydroxy-3-methyl-1-cyano-2,3-seco-2-norlup-28-oic acid methyl ester (5a), colorless crystals, yield 46%, Rf 0.41 (hexane−EtOAc, 7:3), mp 176.1°C (hexane−EtOAc), [α]20D –3.0° (c 0.5, CHCl3). IR (ν, cm–1): 3523 (OH), 2239 (C≡N), 1726 (COOCH3). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.74, 0.84 (3H each, d, J = 8.0, CH3), 0.83, 0.93, 0.98, 1.00, 1.08 (3H each, s, CH3), 1.14 (3H, d, J = 8.0, Í-32), 2.19–2.30 (2H, m, H-19, 20), 2.60, 2.75 (1H each, d, J = 18.0, H-1, AB system), 3.62 (3H, s, HÍ-31), 3.825 (1H, q, J = 8.0, H-3). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.54, 14.66, 16.08, 17.63, 17.84, 21.18, 21.29, 22.19, 22.75, 22.92, 23.01, 27.17, 29.52, 29.68 (2C), 31.87, 33.61, 37.21, 38.31, 40.83, 42.39, 42.66, 43.05, 44.11, 45.50, 47.32, 48.74, 51.12, 57.06, 73.11 (C-3), 119.16 (C-2), 176.84 (C-28). Found, %: C, 76.90; H, 10.69; N, 2.80. C32H53NO3. Calcd, %: C, 76.58; H, 10.89; N, 2.73. Mass spectrum m/z 499.4 [M]+. MM 499 g/mol.
3(S)-Hydroxy-3-methyl-1-cyano-2,3-seco-2-norlup-28-oic acid methyl ester (5b), colorless crystals, yield 35%, Rf 0.33 (hexane−EtOAc, 7:3), mp 180.7°C (hexane−EtOAc), [α]20D +3.0° (c 0.5, CHCl3). IR (ν, cm–1): 3521 (OH), 2239 (C≡N), 1726 (COOCH3). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.74, 0.85 (3H each, d, J = 8.0, CH3), 0.93, 0.98 (6Í, s, CO3), 0.99, 1.05 (3H, s, CH3), 1.24 (3H, d, J = 8.0, Í-32), 2.19–2.30 (2H, m, H-19, 20), 2.61, 2.77 (1H each, d, J = 18.0, H-1, AB system), 3.63 (3H, s, H-31), 3.83 (1Í, q, J = 8.0, H-3). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.56, 14.67, 16.06, 18.36, 18.79, 20.99, 21.23, 22.03, 22.17, 22.76, 22.93, 27.16, 29.70 (3Ñ), 31.87, 33.58, 37.22, 38.31, 40.74, 42.71, 42.76, 43.04, 44.12, 45.34, 47.49, 48.75, 51.14, 57.06, 72.92 (C-3), 119.20 (C-2), 176.84 (C-28). Found, %: C, 76.90; H, 10.69; N, 2.80. C32H53NO3. Calcd, %: C, 76.62; H, 10.81; N, 2.63. Mass spectrum m/z 499.5 [M]+. MM 499 g/mol.
3-Methyl-1-cyano-2-norlup-1,3-en-28-ol (9), colorless crystals, yield 48%, Rf 0.41 (hexane−EtOAc, 7:3), mp 123.8°C (hexane−EtOAc), [α]20D –4.8° (c 0.5, CHCl3). IR (ν, cm–1): 3319 (OH), 2208 (C≡N). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.76, 0.84 (3H each, d, J = 4.0, CH3), 0.92, 0.97, 1.02, 1.06, 1.10 (3H each, s, CH3), 1.86 (3H, s, H-31), 3.32, 3.75 (1H each, d, J = 12.0, H-28, AB system). 13C NMR (100 MHz, CDCl3, δ, ppm): 12.72, 14.80, 14.84, 17.41, 18.68, 19.60, 21.71, 22.24, 22.86, 26.42, 27.05, 27.09, 27.12, 29.38, 29.46, 34.07, 35.11, 36.69, 42.63, 43.35, 44.57, 46.79, 46.84, 47.84, 48.10, 50.85, 60.66, 62.17, 118.37 (C-2), 120.36 (C-1), 167.19 (C-3). Found, %: C, 82.42; H, 10.93; N, 3.10. C31H49NO. Calcd, %: C, 82.12; H, 10.99; N, 3.01. Mass spectrum m/z 451.1 [M]+. MM 451 g/mol.
Preparation of 3-Bromomethyl-3-oxo-1-cyano-2,3-seco-2-norlup-28-oic Acid Methyl Ester (6). A solution of 4 (4.0 mmol) in AcOH (40 mL) was treated with C5H6Br3N (4.0 mmol). The reaction mixture was stirred at room temperature for 2 h. The formation of product was monitored by TLC. When the reaction was finished, the reaction mixture was diluted with H2O. The product was extracted with EtOAc (2 × 50 mL). The organic layer was washed with aqueous NaHCO3 solution (5%) and a small amount of H2O and dried over anhydrous MgSO4. The solvent was evaporated. The solid was purified by CC with elution by petroleum ether−EtOAc (10:1). Colorless crystals, yield 92%, Rf 0.46 (hexane−EtOAc, 7:3), mp 209.4°C (hexane−EtOAc), [α]20D +2.4° (c 0.5, CHCl3). IR (ν, cm–1): 2238 (C≡N), 1722 (COOCH3, C=O). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.85 (3H each, d, J = 8.0, CH3), 0.92, 0.96, 1.01, 1.25, 1.31 (3H each, s, CH3), 1.93–2.00 (2H, m, H-19, 20), 2.37, 2.60 (1H each, d, J = 18.0, H-1, AB system), 3.63 (3H, s, H-31), 4.29, 4.35 (1H each, d, J = 12.0, H-32, AB system). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.56, 14.65, 15.80, 18.59, 21.72, 21.87, 22.73, 22.91, 23.95, 24.11, 26.89, 29.60, 29.65, 29.71, 31.78, 32.38, 33.27, 37.16, 38.12, 40.55, 42.50, 43.02, 44.09, 45.05, 48.72, 49.05, 51.14, 53.36, 56.99, 118.49 (C-2), 176.74 (C-28), 206.64 (C-3). Found, %: C, 66.65; H, 8.74; N, 2.43. C32H50BrNO3. Calcd, %: Ñ, 66.25; H, 8.63; N, 2.43. Mass spectrum m/z 576.1 [M]+. MM 576 g/mol.
Preparation of 31-O-Nicotinate-1-cyano-2,3-seco-2-norlup-28-oic Acid Methyl Ester (7). A solution of 6 (3.4 mmol) in anhydrous Me2CO (10 mL) was treated with nicotinic acid (10.4 mmol), a 10-fold excess of Et3N, and a four-fold excess of K2CO3. The reaction mixture was refluxed for 12 h. The course of the reaction was monitored by TLC. When the reaction was finished, the K2CO3 was filtered off. The reaction mixture was washed with HCl solution (10%) and with H2O until neutral. The product was extracted with EtOAc (2 × 50 mL). The organic layer was separated, dried over anhydrous MgSO4, and evaporated. The solid was purified by CC with elution by petroleum ether−EtOAc−CHCl3 (5:1:1). Colorless crystals, yield 62%, Rf 0.15 (hexane−EtOAc, 7:3), mp 86.7°C (hexane−EtOAc), [α]20D +0.4° (c 0.5, CHCl3). IR (ν, cm–1): 2238 (C≡N), 1721 (COOCH3, C=O). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.86 (3H each, d, J = 8.0, CH3), 0.94, 1.00, 1.02, 1.26, 1.39 (3H each, s, CH3), 2.49, 2.66 (1H each, d, J = 20.0, H-1, AB system), 3.64 (3H, s, H-31), 5.27, 5.35 (1H each, d, J = 18.0, H-32, AB system), 7.57–7.60 (1H, m, H-37), 8.52 (1H, d, J = 8.0, H-38), 8.84 (1H, d, J = 4.0, H-36), 9.30 (1H, br.s, H-35). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.56, 14.64, 15.83, 18.85, 21.86 (2C), 22.73 (2C), 22.89, 24.40, 26.90, 29.64, 29.70, 29.75, 31.78, 33.33, 37.15, 38.12, 40.57, 42.52, 43.02, 44.09, 45.12, 48.71, 48.77, 51.13, 52.02, 56.98, 65.91 (C-32), 118.53 (C-2), 124.42, 126.86, 139.92, 148.47, 150.60, 163.51, 176.74 (C-28), 206.76 (C-3). Found, %: C, 73.75; H, 8.80; N, 4.53. C38H54N2O5. Calcd, %: C, 73.95; H, 8.65; N, 4.30. Mass spectrum m/z 618.4 [M]+. MM 618 g/mol.
Preparation of 3-Methyl-1-cyano-2-norlup-1(3)-en-28-oic Acid Methyl Ester (8). A solution of 4 (4.0 mmol) in t-BuOH (15 mL) was treated with t-BuOK (12.0 mmol). The reaction mixture was refluxed for 2 h. The course of the reaction was monitored by TLC. When the reaction was finished, the mixture was diluted with HCl solution (5%) until neutral. The product was extracted by EtOAc (2 × 50 mL). The organic layer was separated, dried over anhydrous MgSO4, and evaporated. The solid was purified by CC with elution by petroleum ether−EtOAc (15:1). Colorless crystals, yield 98%, Rf 0.43 (hexane−EtOAc, 5:1), mp 89.3°C (hexane−EtOAc), [α]20D –5.2° (c 0.5, CHCl3). IR (ν, cm–1): 2207 (C≡N), 1728 (COOCH3). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.74, 0.85 (3H each, d, J = 4.0, CH3), 0.92, 0.94, 0.96, 1.01, 1.09 (3H each, s, CH3), 1.85 (3H, s, H-32), 2.18–2.25 (2H, m, H-19, 20), 3.64 (3H, s, H-31). 13C NMR (100 MHz, CDCl3, δ, ppm): 12.70, 14.60, 14.74, 17.40, 17.41, 18.69, 19.57, 22.32, 22.78, 22.88, 26.43, 27.04, 29.62, 29.75, 32.27, 35.14, 37.39, 38.08, 42.37, 43.03, 44.28, 46.81, 47.00, 49.01, 50.91, 51.11, 56.88, 62.32, 118.36 (C-2), 120.45 (C-1), 167.10 (C-3), 176.81 (C-28). Found, %: C, 80.12; H, 10.30; N, 2.92. C32H49NO2. Calcd, %: C, 80.92; H, 10.18; N, 2.82. Mass spectrum m/z 479.4 [M]+. MM 479 g/mol.
Screening for Cytotoxic Activity of 3–9. Cytotoxic activity of the synthesized compounds was determined by the classical MTT assay [26] using HEpG2, HCT116, MS, RD-TE32, MCF-7, A549, and PC-3 tumor cell lines and normal HEK293 cells. Cells were cultivated in DMEM (HEpG2, HCT116, MCF-7) and RPMI-1640 medium (RD TE32, MS, A549, PC-3) with added fetal bovine serum (10%), L-glutamine (2 mM), and penicillin-streptomycin solution (1%) at 37°C and 5% CO2 in a humid atmosphere for 24 h. Then, the cell cultures were treated with the tested compounds in DMSO at concentrations of 3.125–100 μM. The controls were wells with added DMSO, the final concentration of which was ≤ 1% and was nontoxic to the cells. Cell survival was assessed after incubation for 72 h with the tested compounds by adding to each well a solution of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 5 mg/mL] (20 μL) with subsequent determination of the optical density of the formed formazan at 544 nm on a FLUOstar Optima spectrophotometer (BMG Labtech, Germany). The 50% inhibitory concentrations (IC50) of the tested compounds were determined using dose-dependent curves and GraphPad Prism 6.0 software. Results were given as means of three independent tests ± SD (Table 1).
References
S. Xiao, Z. Tian, Y. Wang, L. Si, L. Zhang, and D. Zhou, Med. Res. Rev., 38, 951 (2018).
A. Hordyjewska, A. Ostapiuk, A. Horecka, and J. Kurzepa, Phytochem. Rev., 18, 929 (2019).
S. Amiri, S. Dastghaib, M. Ahmadi, P. Mehrbod, F. Khadem, H. Behrooj, and S. Ghavami, Biotechnol. Adv., 38, 107409 (2020).
A. Lombrea, A. D. Scurtu, S. Avram, I. Z. Pavel, M. Turks, J. Lugioina, and C. Danciu, Int. J. Mol. Sci., 22 (7), 3676 (2021).
P. A. Krasutsky, Nat. Prod. Rep., 23, 919 (2006).
L. A. Baltina, O. B. Flekhter, L. R. Nigmatullina, E. I. Boreko, N. I. Pavlova, S. N. Nikolaeva, O. V. Savinova, and G. A. Tolstikov, Bioorg. Med. Chem. Lett., 13, 3549 (2003).
R. Csuk, K. Schmuck, and R. Schafer, Tetrahedron Lett., 47, 8769 (2006).
S. A. Popov, L. P. Kozlova, L. M. Kornaukhova, and A. V. Shpatov, Ind. Crops. Prod., 92, 197 (2016).
A. V. Pereslavtseva, I. A. Tolmacheva, P. A. Slepukhin, O. S. El′tsov, I. I. Kucherov, V. F. Eremin, and V. V. Grishko, Chem. Nat. Compd., 49, 1059 (2014).
A. V. Konysheva, V. O. Nebogatikov, I. A. Tolmacheva, M. V. Dmitriev, and V. V. Grishko, Eur. J. Med. Chem., 140, 74 (2017).
A. V. Konysheva, A. E. Zhukova, M. V. Dmitriev, and V. V. Grishko, Chem. Nat. Compd., 54, 1094 (2018).
D. V. Eroshenko, G. F. Krainova, A. V. Konysheva, M. V. Dmitriev, and V. V. Grishko, Bioorg. Med. Chem. Lett., 28, 3752 (2018).
A. V. Konysheva, D. V. Eroshenko, and V. V. Grishko, Nat. Prod. Commun., 14 (10), 1 (2019).
E. Y. Rybalkina, N. I. Moiseeva, A. F. Karamysheva, D. V. Eroshenko, A. V. Konysheva, A. V. Nazarov,
and V. V. Grishko, Chem. Biol. Interact., 348, 109645 (2021).
15. J. Pokorny, S. Krajcovicova, M. Hajduch, M. Holoubek, S. Gurska, P. Dzubak, T. Volna, I. Popa, and M. Urban, Future Med. Chem., 10 (5), 483 (2018).
16. G. F. Krainova, O. N. Gagarskikh, and V. V. Grishko, Chem. Nat. Compd., 58, 693 (2022).
R. Csuk, C. Nitsche, R. Sczepek, S. Schwarz, and B. Siewer, Arch. Pharm., Chem. Life Sci., 346, 232 (2013).
18. G. C. Souza, Jr., G. C. Franchi, A. E. Nowill, L. S. Santos, C. N. Alves, L. E. S. Barata, and C. K. Z. Andrade, J. Braz. Chem. Soc., 28 (11), 2229 (2017).
19. A. V. Konysheva, I. A. Tolmacheva, D. V. Eroshenko, and V. V. Grishko, Chem. Nat. Compd., 53, 497 (2017).
G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 64, 112 (2008).
G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 71, 3 (2015).
O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann, J. Appl. Crystallogr., 42, 339 (2009).
B. Keil, Laboratoriumstechnik der organische Chemie, Akademie-Verlag, Berlin (1961) [Russian translation, Mir, Moscow, 1966, 591 pp].
I. Y. Strobykina, B. F. Garifullin, R. R. Sharipova, A. D. Voloshina, A. S. Strobykina, A. B. Dobrynin, and V. E. Kataev, Chem. Nat. Compd., 53, 1101 (2017).
I. A. Tolmacheva, A. V. Nazarov, O. A. Maiorova, and V. V. Grishko, Chem. Nat. Compd., 44, 606 (2008).
T. Mosmann, J. Immunol. Methods, 65, 55 (1983).
Acknowledgment
The work was financially supported by Russian Science Foundation grant No. 21-13-00161. We thank the Center for Collective Use of PFRC, UrB, RAS, Research on Materials and Compounds, for facilitating the spectral and analytical studies and Candidate M. V. Dmitriev, Department of Organic Chemistry, Perm State Natl. Res. Univ., for performing the XSA of 5b.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2023, pp. 71–76.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Krainova, G.F., Beloglazova, Y.A. & Grishko, V.V. Synthesis of 3-Methyl Derivatives from Dihydrobetulonic Acid Methyl Ester. Chem Nat Compd 59, 80–86 (2023). https://doi.org/10.1007/s10600-023-03923-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-023-03923-x