
A 1-cyano-2,3-secolupane derivative with an ethylketone fragment was synthesized from 2-hydroxyoximodihydrobetulonic acid methyl ester via a Grignard reaction followed by a Beckmann rearrangement and was used further to prepare α-bromo-substituted diastereomers. Nitrile-anionic cyclization of the ethylketone and α-bromo isomers produced cyclic derivatives with an alkenenitrile and an α,β-unsaturated ketone in ring A, respectively. The obtained compounds did not exhibit cytotoxic activity according to biological screening data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Reductive allyl(alkyl)ation of triterpenoids has been achieved a few times, indicating that this approach could be used successfully to synthesize new biologically active compounds. For example, C-3 allylation under Grignard reaction conditions was reported and helped to improve the antidiabetic, cytotoxic, and anti-inflammatory activity of triterpenoids [1,2,3,4,5], including betulinic acid. Previously, 3-alkyl-substituted triterpenoids were shown by us to be useful as intermediates for preparing bioactive triterpene derivatives with a fragmented or five(six)-membered ring A [6,7,8,9,10], including lupane derivatives with pronounced antitumor properties such as methyl 3-(1-bromoethyl)-3-oxo-1-cyano-2,3-seco-2-norlup-20(29)-en-30-al-28-oate and methyl 1-cyano-3-ethyl-2-norlup-1(3),20(29)-dien-30-al-28-oate [10]. New fragmented and cyclic 3-ethyl-substituted lupane-type derivatives were synthesized and their cytotoxic activity was evaluated in continuation of research on the structure-activity (biological) relationship of C-3 alkylated triterpenoids.
The starting material for preparing the C-3 ethyl-substituted triterpenoids was hydroxyiminoketone 1, which was prepared by sequential transformations from 20,29-dihydrogenated betulin [11]. The corresponding 3β-hydroxy-3α-ethyl derivative 2 was prepared using a Grignard reaction with C2H5MgBr as the alkylating agent [9, 10]. The IR spectrum of 2 exhibited bands for C=N (1644 cm-1) and OH stretching vibrations (3313, 3437 cm-1). The 1H NMR spectrum of 2 contained characteristic doublets for the methylene H-1 protons at δ 1.52 and 3.38 ppm (AB-system, J = 12.6 Hz); a broad singlet for the OH at δ 5.89 ppm; and a resonance for the methylene H-32 protons as a doublet of quartets with centers at 1.76 and 1.85 ppm (J = 13.8, 7.2 Hz). The 13C NMR spectrum of 2 displayed resonances for C atoms bonded to OH (79.59 ppm) and hydroxyimine groups (162.63 ppm).
The ethyl derivative 2 underwent a Beckmann rearrangement using SOCl2-CH2Cl2 to form the corresponding 2,3-seco-derivative 3. The IR spectrum of 3 had characteristic absorption bands for C=O (1695 cm-1) and C≡N (2234). The 1H NMR spectrum of seco-derivative 3 exhibited two doublets for the methylene H-1 protons with centers at 2.34 and 2.54 ppm (AB-system, J = 18.0 Hz) and two types of resonances for the ethyl moiety, i.e., a triplet for the methyl at δ 1.08 ppm (J = 7.2 Hz) and a doublet of quartets at δ 2.55 and 2.74 ppm (J = 19.6 and 7.2 Hz). The 13C NMR spectrum of 3 included characteristic resonances of cyano and keto C atoms at δ 118.55 and 216.82 ppm, respectively.
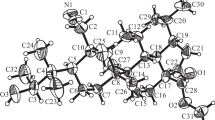
The ethylketone of 2,3-seco-derivative 3 was activated by halogenation (Scheme 1) using the brominating agent pyridinium bromide perbromide [7, 9, 10]. The reaction proceeded with heating and stirring. Pure reaction products were isolated by column chromatography and included isomeric bromo-substituted derivatives 4 and 5, the NMR spectra of which differed slightly. For example, 1H NMR spectra of 4 and 5 showed the protons of the bromo-substituted ethyl group as a doublet for the methyl at 1.74 ppm (J = 6.6 Hz). The methine H-32 proton resonated as a quartet with SSCC 6.6 Hz at 5.04 (4) or 5.03 (5). Also, doublets for the methylene H-1 protons of 5 (2.58 and 2.69 ppm, J = 18.0 Hz) as compared to those of 4 (2.60 and 2.71 ppm, J = 17.9 Hz) appeared at stronger field. The 13C NMR spectrum contained resonances at stronger field for the nitrile and ester of bromo derivative 4 at 118.58 (C-2) and 176.74 (C-28) as compared to the analogous resonances of 5 at 118.72 (C-2) and 176.76 ppm (C-28). The absolute configuration of asymmetric C-32 in diastereomers 4 and 5 was confirmed based on an X-ray crystal structure analysis (XSA) of 4 (Fig. 1).

Scheme 1

General view of methyl (3R)-3-(1-bromoethyl)-3-oxo-1-cyano-2,3-seco-2-norlup-28-oate (4) from an XSA with 30% probability thermal ellipsoids.
Ethylketone 3 and bromo derivative 4 underwent intramolecular nitrile-anion cyclization [12] in the basic system t-BuOK-t-BuOH [13]. The cyclization product of 3 was α,β-alkenenitrile 6. Its IR spectrum showed characteristic absorptions at 1594 (C=C), 1729 (C=O), and 2206 cm-1 (C≡N). The 1H NMR spectrum of α,β-alkenenitrile 6 lacked doublets for the methylene H-1 protons of the starting compound and retained the characteristic resonances of the ethyl moiety as a triplet for the Me-32 protons with a center at 1.13 ppm (J = 7.6 Hz) and a quartet for the methylene H-31 protons with a center at 2.28 ppm (J = 7.6 Hz). The 13C NMR spectrum of 6 exhibited resonances for the nitrile (118.33 ppm) and double-bond C atoms (120.52 and 172.41).
Cyclization of bromo derivative 4 occurred with loss of the nitrile, which led to formation of an α,β-unsaturated ketone in six-membered ring A of 7. The IR spectrum of 7 lacked bands for the nitrile and exhibited a carbonyl absorption band at 1667 cm-1. The 1H NMR spectrum included two characteristic singlets corresponding to the C-3 methyl protons (1.73 ppm) and olefinic H-1 proton (6.84 ppm). The 13C NMR spectrum exhibited resonances for C-2 and C-1 of the double bond at 130.95 and 154.85 ppm, respectively, and for the carbonyl C atom at 205.62 ppm.
A study of the cytotoxic activity against eight tumor cell lines found that compounds 3–6 were nontoxic (IC50 > 100 μM). Thus, the C-30 aldehyde group played a decisive role in the manifestation of cytotoxic activity by the previously reported ethyl-substituted lupane derivatives [10].
Experimental
1H NMR, 13C NMR, and DEPT spectra of the synthesized compounds were recorded in CDCl3 solutions (DMSO-d6 was also used for 2) with TMS internal standard on a Bruker Avance II NMR spectrometer (400 and 100 MHz, respectively). IR spectra were recorded from thin films prepared by evaporating CHCl3 solutions of the compounds on a Bruker IFS 66/S FT-IR spectrometer (Germany). The threshold melting point at heating rate 1°C/min was determined on an OptiMelt MPA100 apparatus (USA). Specific optical rotation was measured in CHCl3 solutions of the compounds on a PerkinElmer model 341 polarimeter at 589 nm. Elemental analyses (C, H, N) were performed in a Vario EL cube analyzer (Germany). The course of reactions was monitored by TLC on Sorbfil plates (Russia) after treatment with H2SO4 (5%) followed by heating at 95–100°C for 2–3 min. Column chromatography used Macherey-Nagel silica gel (60–200 μm) with elution by petroleum ether–EtOAc mixtures, the ratio of which was selected individually for each reaction product.
The XSA of 4 was performed on an Xcalibur Ruby single-crystal diffractometer (Agilent Technologies) by the standard method [Mo Kα-radiation, 295(2) K, ω-scanning in 1° steps]. Absorption corrections were applied empirically using the SCALE3 ABSPACK algorithm [14]. The structure was solved using the SHELXT program [15] and refined by anisotropic full-matrix least-squares methods over F2 for all nonhydrogen atoms using the SHELXL program [16] and the OLEX2 graphics interface [17]. H atoms were refined using a rider model. The crystal (C33H52BrNO3, MM 590.66) was monoclinic, space group P21, a = 7.705(2), b = 17.834(5), c = 11.818(3) Å , β = 92.74(2) °, V = 1621.9(7) Å3, Z = 2, dcalcd = 1.209 g/cm3, μ = 1.298 mm-1. The final refinement parameters were R1 0.0677 [for 2890 reflections with I > 2σ(I)], wR2 0.1694 (for all 5882 independent reflections, Rint 0.0297), S = 1.035, Flack parameter 0.018(8). Results for the XSA of 4 were deposited in the Cambridge Crystallographic Data Centre under No. CCDC 2190242 and can be requested at www.ccdc.cam.ac.uk/data_request.cif.
Anhydrous solvents were prepared by standard methods [18]. Ketoxime 1 was prepared according to the literature method [11].
Preparation of Methyl 3β-Hydroxy-2-hydroxyimino-3α-ethyllup-28-oate (2). A solution of 1 (2.5 mmol) in a mixture of anhydrous Et2O and THF (2:1, 14 mL) was added dropwise to a freshly prepared solution of C2H5MgBr (5.0 mmol) in anhydrous Et2O (5 mL). The mixture was stirred with heating (60°C) for 2 h, cooled, worked up dropwise with ice water (25 mL) and HCl solution (20 mL, 17–20%), and stirred until the precipitate was fully dissolved. The resulting solution was extracted with EtOAc (3 × 30 mL). The organic layer was separated; washed with saturated Na2S2O3 solution, NaHCO3 solution (5%), and a small amount of H2O; and dried over anhydrous MgSO4. The EtOAc was evaporated in a rotary evaporator. The dry solid was purified by column chromatography (CC) with elution by petroleum ether−EtOAc (15:1). Colorless crystals, mp 206.4°C (hexane−EtOAc), yield 40%, Rf 0.21 (hexane−EtOAc, 5:1), [α]25D +1.7° (c 0.4, CHCl3). IR (ν, cm–1): 1644 (C=N), 1722 (COOCH3), 3313 (OH), 3437 (NOH). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.73–0.78 (9H, m, H-29, 30, 33), 0.84, 0.86, 0.90 (3H each, s, CH3), 0.97 (6H, s, CH3), 1.52, 3.38 (1H each, d, J = 12.6, H-1, AB system), 1.76, 1.85 (1H each, dq, J = 14.3, 7.3, H-32), 2.19–2.27 (2H, m, H-19, 20), 3.64 (3H, s, H-31), 5.89 (1H, br.s, OH). 1H NMR (400 MHz, DMSO-d6, δ, ppm, J/Hz): 0.64 (3H, t, J = 7.4, H-33), 0.69, 0.76 (3H each, d, J = 7.2, H-29, 30), 0.84 (3H, s, CH3), 0.86 (6H, s, CH3), 0.88, 0.99 (3H each, s, CH3), 10.64 (1H, s, NOH). 13C NMR (100 MHz, CDCl3, δ, ppm): 7.38, 14.65, 14.74, 15.74, 16.86, 18.89, 19.02, 21.28, 22.76, 22.93, 23.98, 26.71, 26.78, 29.73, 32.08, 34.34, 36.62, 37.29, 38.07, 41.17, 41.23, 41.63, 42.67, 44.19, 45.13, 48.94, 50.28, 51.17, 53.10, 57.01, 79.59 (C-3), 162.63 (C-2), 176.88 (C-28). Found, %: C, 74.43; H, 10.52; N, 2.75. C33H55NO4. Calcd, %: C, 74.81; H, 10.46; N, 2.64.
Synthesis of Methyl 3-Oxo-1-cyano-3-ethyl-2,3-seco-2-norlup-28-oate (3). A solution of 2 (1 mmol) in anhydrous CH2Cl2 (50 mL) was stirred and treated with SOCl2 (3 mmol). The mixture was stirred for 30 min at room temperature. The solvent was evaporated in a rotary evaporator. Traces of SOCl2 were removed by rinsing the precipitate twice with CH2Cl2. The dry solid was purified by CC with elution by petroleum ether−EtOAc (15:1). Yield 75%, Rf 0.34 (hexane−EtOAc, 5:1), colorless crystals, mp 164.7°C (hexane−EtOAc), [α]25D –1.7° (c 0.4, CHCl3). IR (ν, cm–1): 1695 (C=O), 1725 (COOCH3), 2234 (C≡N). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.85 (3H each, d, J = 6.8, CH3), 0.91, 0.95, 1.01, 1.16, 1.22 (3H each, s, CH3), 1.08 (3H, t, J = 7.2, H-33), 2.19–2.30 (2H, m, H-19, 20), 2.34, 2.54 (1H, d, J = 18.0, H-1, AB system), 2.55, 2.74 (2H, dq, J = 19.6, 7.2, H-32), 3.63 (3H, s, H-31). 13C NMR (100 MHz, CDCl3, δ, ppm): 8.71, 14.60, 14.67, 15.74, 18.78, 21.57, 21.82, 22.75, 22.91, 24.00, 24.07, 26.90, 29.49, 29.69, 29.71, 30.37, 31.82, 33.41, 37.18, 38.14, 40.55, 42.40, 43.00, 44.12, 45.06, 48.75, 48.88, 51.13, 52.87, 57.01, 118.55 (C-2), 176.76 (C-28), 216.82 (C-3). Found, %: C, 77.03; H, 10.38; N, 2.80. C33H53NO3. Calcd, %: C, 77.45; H, 10.44; N, 2.74.
Method for Preparing 4 and 5. A solution of 2,3-seco-derivative 3 (0.4 mmol) in AcOH (30 mL) was stirred and treated with C5H6Br3N. The reaction mixture was left on a magnetic stirrer with heating (115°C) for 6 h, diluted with H2O, and extracted with EtOAc (3 × 20 mL). The organic layer was washed with NaHCO3 solution (5%) and H2O and dried over anhydrous MgSO4. The solvent was evaporated in a rotary evaporator. The dry solid was purified by CC with elution by petroleum ether−EtOAc (15:1).
Methyl (3R)-3-(1-Bromoethyl)-3-oxo-1-cyano-2,3-seco-2-norlup-28-oate (4). Yield 67%, Rf 0.43 (hexane−EtOAc, 5:1, double elution), colorless crystals, mp 172.3°C (hexane−EtOAc), [α]D22 +38.5° (c 0.5, CHCl3). IR (ν, cm–1): 1718 (COOCH3, C=H), 2239 (C≡N). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.85 (6H, d, J = 6.8, CH3), 0.91, 0.98, 1.00, 1.23, 1.50 (3H each, s, CH3), 1.74 (3H, d, J = 6.6, H-33), 2.19–2.32 (2H, m, H-19, 20), 2.60, 2.71 (1H, d, J = 17.9, H-1), 3.63 (3H, s, H-31), 5.04 (1H, q, J = 6.6, H-32). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.51, 14.67, 15.93, 18.95, 21.29, 21.92, 22.20, 22.41, 22.74, 22.92, 25.85, 26.99, 29.65, 29.73, 30.02, 31.78, 33.14, 37.16, 38.18, 40.34, 40.59, 42.64, 42.98, 44.10, 45.25, 46.01, 48.72, 51.16, 54.33, 57.00, 118.58 (C-2), 176.74 (C-28), 208.51 (C-3). Found, %: C, 66.83; H, 8.98; N, 2.50. C33H52BrNO3. Calcd, %: C, 67.10; H, 8.87; N, 2.37.
Methyl (3S)-3-(1-Bromoethyl)-3-oxo-1-cyano-2,3-seco-2-norlup-28-oate (5). Yield 22%, Rf 0.34 (hexane−EtOAc, 5:1, double elution), colorless crystals, mp 201.8°C (hexane−EtOAc), [α]25D +5.7° (c 0.4, CHCl3). IR (ν, cm–1): 1718 (COOCH3, C=O), 2237 (C≡N). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75, 0.85 (6H, d, J = 6.8, CH3), 0.92, 1.00, 1.15, 1.24, 1.37 (3H each, s, CH3), 1.74 (3H, d, J = 6.6, H-33), 2.15–2.31 (2H, m, H-19, 20), 2.58, 2.69 (1H each, d, J = 18.0, H-1, AB system), 3.62 (3H, s, H-31), 5.03 (1H, q, J = 6.6, H-32). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.42, 14.66, 16.03, 19.13, 21.41, 21.83, 22.56, 22.68, 22.74, 22.92, 24.75, 26.99, 29.59, 29.71, 29.96, 31.80, 32.70, 37.17, 38.17, 40.51, 40.55, 42.68, 43.02, 44.11, 45.18, 46.36, 48.74, 51.13, 54.40, 57.01, 118.72 (C-2), 176.76 (C-28), 208.07 (C-3). Found, %: C, 66.88; H, 8.95; N, 2.47. C33H52BrNO3. Calcd, %: C, 67.10; H, 8.87; N, 2.37.
General Method for Preparing 6 and 7. A solution of 3 or 4 (1 mmol) in t-BuOH (15 mL) was treated with t-BuOK (3 mmol). The reaction mixture was refluxed for 2 h, diluted with HCl solution (5%), and extracted with EtOAc (3 × 15 mL). The organic layer was washed with NaHCO3 solution and dried over anhydrous MgSO4. The solvent was evaporated in a rotary evaporator. The solid was purified by CC with elution by petroleum ether−EtOAc (30:1).
Methyl 1-Cyano-3-ethyl-2-norlup-1(3)-en-28-oate (6). Yield 73%, Rf 0.52 (hexane−EtOAc, 10:1), colorless crystals, mp 223.9°C (hexane−EtOAc), [α]25D –6.7° (c 0.3, CHCl3). IR (ν, cm–1): 1594 (C=C), 1729 (COOCH3), 2206 (C≡N). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.73, 0.84 (6H, d, J = 6.8, CH3), 0.94, 0.95, 0.97, 1.04, 1.10 (3H each, s, CH3), 1.13 (3H, t, J = 7.7, H-33), 2.15–2.24 (2H, m, H-19, 20), 2.28 (2H, q, J = 7.7, H-32), 3.64 (3H, s, H-31). 13C NMR (100 MHz, CDCl3, δ, ppm): 13.72, 14.59, 14.78, 17.34, 17.37, 18.81, 20.49, 20.96, 22.25, 22.77, 22.87, 26.42, 27.21, 29.61, 29.74, 32.27, 35.08, 37.38, 38.09, 42.40, 43.04, 44.27, 46.98, 47.31, 48.99, 50.68, 51.09, 56.87, 62.52, 118.33 (C-2), 120.52 (C-1), 172.41 (C-3), 176.80 (C-28). Found, %: C, 80.34; H, 10.61; N, 2.75. C33H51NO2. Calcd, %: C, 80.27; H, 10.41; N, 2.84.
2-Methyl-3-oxolup-1(2)-en-28-oic Acid Methyl Ester (7). Yield 43%, Rf 0.57 (hexane−EtOAc, 5:1), colorless crystals, mp 175.3°C (hexane−EtOAc), [α]D22 +9.3° (c 0.5, CHCl3). IR (ν, cm–1): 1594 (C=C), 1667 (C=O), 1729 (COOCH3). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.74, 0.86 (3H each, d, J = 6.8, CH3), 0.95, 0.97, 1.00, 1.07, 1.08 (3H each, s, CH3), 1.73 (3H, s, H-32), 2.15–2.31 (2H, m, H-19, 20), 3.64 (3H, s, H-31), 6.84 (1H, s, H-1). 13C NMR (100 MHz, CDCl3, δ, ppm): 14.54, 14.65, 16.41, 16.92, 19.17, 19.37, 21.31, 21.54, 22.76, 22.93, 26.94, 28.35, 29.61, 29.73, 32.06, 33.94, 37.30, 38.39, 38.77, 41.56, 42.84, 44.19, 44.45, 44.70, 48.89, 51.14, 53.43, 57.00, 130.95 (C-2), 154.85 (C-1), 176.77 (C-28), 205.62 (C-3). Found, %: C, 79.44; H, 10.56. C32H50O3. Calcd, %: Ñ, 79.62; H, 10.44.
Screening for Cytotoxic Activity of 3–6. The cytotoxic activity of the synthesized compounds was determined by the classical MTT assay [19] using HEpG2, HCT116, MS, RD TE32, MCF-7, A549, and PC-3 cancer cell lines and HEK293 normal cells. A detailed description of the method has been published [11].
References
C. Genet, A. Strehle, C. Schmidt, G. Boudjelal, A. Lobstein, K. Schoonjans, M. Souchet, J. Auwerx, R. Saladin, and A. Wagner, J. Med. Chem., 53, 178 (2010).
C. Genet, C. Schmidt, A. Strehle, K. Schoonjans, J. Auwerx, R. Saladin, and A. Wagner, ChemMedChem, 5, 1983 (2010).
R. Csuk, C. Nitsche, R. Sczepek, S. Schwarz, and B. Siewert, Arch. Pharm. (Weinheim, Ger.), 346, 232 (2013).
A. I. Govdi, N. V. Sokolova, I. V. Sorokina, D. S. Baev, T. G. Tolstikova, V. I. Mamatyuk, D. S. Fadeev, S. F. Vasilevsky, and V. G. Nenajdenko, MedChemComm, 6, 230 (2015).
G. Fontana, M. Bruno, M. Notarbartolo, M. Labbozzetta, P. Poma, A. Spinella, and S. Rosselli, Bioorg. Chem., 90, 103054 (2019).
A. V. Pereslavtseva, I. A. Tolmacheva, P. A. Slepukhin, O. S. El′ tsov, I. I. Kucherov, V. F. Eremin, and V. V. Grishko, Chem. Nat. Compd., 49, 1059 (2014).
A. V. Konysheva, V. O. Nebogatikov, I. A. Tolmacheva, M. V. Dmitriev, and V. V. Grishko, Eur. J. Med. Chem., 140, 74 (2017).
A. V. Konysheva, I. A. Tolmacheva, D. V. Eroshenko, and V. V. Grishko, Chem. Nat. Compd., 53, 497 (2017).
A. V. Konysheva, A. E. Zhukova, M. V. Dmitriev, and V. V. Grishko, Chem. Nat. Compd., 54, 1094 (2018).
A. V. Konysheva, D. V. Eroshenko, and V. V. Grishko, Nat. Prod. Commun., 14, 1 (2019).
G. F. Krainova, O. N. Gagarskikh, and V. V. Grishko, Chem. Nat. Compd., 58, 693 (2022).
F. F. Fleming, L. A. Funk, R. Altundas, and V. Sharief, J. Org. Chem., 67, 9414 (2002).
V. V. Grishko, N. V. Galaiko, I. A. Tolmacheva, I. I. Kucherov, V. F. Eremin, E. I. Boreko, O. V. Savinova, and P. A. Slepukhin, Eur. J. Med. Chem., 83, 601 (2014).
CrysAlisPro, Agilent Technologies, Version 1.171.37.33 (release 27-03-2014 CrysAlis171.NET).
G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 71, 3 (2015).
G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 71, 3 (2015).
O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann, J. Appl. Crystallogr., 42, 339 (2009).
B. Keil, Laboratoriumstechnik der organische Chemie, Akademie-Verlag, Berlin (1961) [Russian translation, Mir, Moscow, 1966, 591 pp].
T. Mosmann, J. Immunol. Methods, 65, 55 (1983).
Acknowledgment
The work was financially supported by a grant from the Russian Science Foundation No. 21-13-00161. We thank the Center for Collective Use, PFRC, UrB, RAS, Research on Materials and Compounds, for facilitating the spectral and analytical studies and Candidate M. V. Dmitriev, Department of Organic Chemistry, Perm State Natl. Res. Univ., for performing the XSA of 4.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ttranslated from Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2023, pp. 83–86.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Galaiko, N.V., Beloglazova, Y.A. & Grishko, V.V. Synthesis and Intramolecular Cyclization of 2,3-Seco-Lupane Triterpenoids with an Ethylketone Fragment. Chem Nat Compd 59, 94–98 (2023). https://doi.org/10.1007/s10600-023-03925-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-023-03925-9