
A new flavolignan acylglycoside, namely phillygenin 4-O-(2′′-trans-cinnamoyl)-α-L-rhamnopyranoside (1), along with a known phenolic acid (ellagic acid, 2) and three known flavonol glycosides (kaempferol 3-O-β-D-glucopyranoside (3), kaempferol 3-O-α-L-rhamnopyranoside (4), and quercetin 3-O-β-D-glucopyranoside (5), was isolated and purified from the fallen needles of Pinus banksiana by repeated column chromatography. The structures of these secondary metabolites were mainly elucidated by the extensive use of 1D and 2D NMR experiments, together with IR, UV, and FAB MS spectra. Among the four known compounds, 2 and 4 have never been found in this conifer species previously.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pinus banksiana Lam., a conifer species belonging to the family of Pinaceae, is widespread in Xiaoxing_an Ling, Daxing′an Ling, and Taibai Mountains of north China, northwest regions of America, and many forests of Canada [1,2,3]. Needles and barks of P. banksiana have long been used in Chinese folk medicine to treat and prevent cancer, cardiovascular and aging disorders, as well as to enhance immunity [4, 5]. Phytochemical investigations of P. banksiana have led to the isolation of several classes of secondary metabolites such as sesquiterpenes and flavonol glycosides [6,7,8]. In a previous study, we had reported the extraction and purification of trans-isoconiferin, caffeic acid, cedrusin, phillyrin, and phillyrin-6′′-α-rhamnopyranoside from the branch wood of P. banksiana [2]. The current work describes the isolation and structural elucidation of a novel flavolignan acylglycoside, phillygenin 4-O-(2′′-trans-cinnamoyl)-α-L-rhamnopyranoside (1) from the 70% acetone extract of the fallen needles of P. banksiana, together with four known compounds, ellagic acid (2), kaempferol 3-O-β-D-glucopyranoside (3), kaempferol 3-O-α-L-rhamnopyranoside (4), and quercetin 3-O-β-D-glucopyranoside (5). Compound 1 is a new flavolignan acylglycoside, and 2 and 4 are reported in this conifer species for the first time here.
Compound 1 was isolated as a white amorphous powder, with optical rotation \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –47.9° (c 0.5; MeOH) and mp 199–201°C. The molecular formula of compound 1 was assigned to be C36H40O11 on the basis of its positive FAB-MS spectrometric analysis (found [M + H]+, [M + K]+, and [M + Na]+ ion peaks at m/z 649, 687, and 671, respectively). In the TLC chromogenic reaction, R f values of compound 1 were 0.57 and 0.19 in solvents A and B, respectively), when spraying with 10% sulfuric acid (in EtOH) solution as reagent followed by heating [9, 10]. Its IR spectrum displayed the presence of phenyl (1525 and 1635 cm–1) and hydroxyl (3390 cm–1) moieties. The UV spectrum of compound 1 showed absorptions at 285 nm and 235 nm, which suggested its bis-perhydrofuran unit [11, 12].
In the 1H NMR spectrum (Table 1), compound 1 presented two pairs of ABX style aromatic proton signals at δ 6.99 (1H, d, J = 2.1 Hz, H-2), δ 7.04 (1H, d, J = 8.0 Hz, H-5), δ 6.90 (1H, dd, J = 2.1, 8.0 Hz, H-6) and δ 6.96 (1H, d, J = 2.2 Hz, H-2′), δ 6.79 (1H, d, J = 7.8 Hz, H-5′), δ 6.87 (1H, dd, J = 2.2, 7.8 Hz, H-6′). Proton peaks for benzylic oxymethine H-7 and H-7′ were found as two doublets at δ 4.34 (1H, J = 6.3 Hz) and 4.76 (1H, J = 6.1 Hz). Coupling signals at δ 2.80 (1H, m) and 3.06 (1H, m) were assignable to H-8 and H-8′ of the cis-diequatorial substituted 2,6-diaryl-3,7-dioxabicyclo[3,3,0]octane [13], whereas the oxygen-containing methylene protons H-9 and H-9′ were confirmed at δ 4.04, 3.68 (each 1H, m) and δ 3.71, 3.18 (each 1H, m) [14]. The three aromatic methoxyls were evidenced by their signals (δH 3.72 (3H, s) and δC 55.46 for 10-OMe), (δH 3.70 (3H, s) and δC 55.44 for 10′-OMe), and (δH 3.73 (3H, s) and δC 55.45 for 11′-OMe) in the 1H and 13C NMR spectra, respectively. These clues together with its 13C NMR data (Table 1) indicated that the aglycone moiety of compound 1 could be a flavolignan named phillygenin [15, 16]. The downfield shifts Δδ of approximately 1.11 and 3.14 ppm for C-3 and C-1, respectively, compared to phillygenin correponded well with the linkage of one substituent at C-4 of its equatorial ring in phillygenin [15].
In the 1H NMR spectrum of compound 1, an anomeric proton observed at δ 5.30 (1H, d, J = 1.6 Hz, H-1′′) and one secondary methyl moiety at δ 0.89 (3H, d, J = 6.1 Hz, H-6′′), along with the remaining sugar peaks ranging between δ 5.29 and 3.10 revealed the presence of one α-L-rhamnosyl residue [9]. This conclusion was also supported by the corresponding 13C NMR signals appearing at δ 98.41 (C-1′′) and 17.58 (C-6′′). Relative to the spectral data of an authentic rhamnosyl unit, the sharp downfield of H-2′′ and C-2′′ to δ 5.29 and 71.03, respectively, indicated that the hydroxyl of the rhamnosyl 2′′ position was replaced [17, 18].
The presence of a trans-cinnamoyl unit in compound 1 was revealed by its 1H and 13C NMR spectra: a monosubstituted benzene ring was confirmed by the symmetrical 1H NMR signals at δ 7.59 (1H each, dd, J = 2.0, 6.9 Hz, H-2′′′/6′′′), 7.26 (1H each, m, H-3′′′/5′′′), and 7.28 (1H, m, H-4′′′) and 13C NMR peaks at δ 128.56 (C-2′′′/6′′′), 128.94 (C-3′′′/5′′′), 133.82 (C-1′′′) and 130.65 (1H, m, H-4′′′) [18, 19]. In addition, the olefinic proton signals characteristically resonated at δ 7.49 (1H, d, H-7′′′) and 6.55 (1H, d, H-8′′′) with a low coupling constant of J = 16.2 Hz, suggesting the trans-configuration for the cinnamoyl unit [17,18,19].
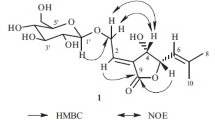
In the H→C couplings of the HMBC spectrum of compound 1, correlations were observed between anomeric H-1′′ δ 5.30 (1H, d, J = 1.6 Hz) of the rhamnose and C-4 (δ 145.93) of the aglycone, and between H-2′′ δ 5.29 (1H, dd, J = 1.6, 3.2 Hz) of the sugar and C-9′′′ (δ 165.56) of the cinnamoyl residue, as demonstrated in Fig. 1, verifying that the rhamnosyl moiety is attached to C-4 of phillygenin, and the trans-cinnamoyl unit is linked to C-2′′ of the rhamnosyl sugar. The DEPT spectrum data, as shown in Table 1, classified 36 carbon signals of compound 1 into two methylenes (CH2), 22 methines (CH), four methyls (CH3) and eight quaternary carbons (qC), with each signal agreeing well with the corresponding carbons in phillygenin 4-O-(2′′-trans-cinnamoyl)-α-L-rhamnopyranoside.

Key HMBC correlations observed in compound 1.
Thus, compound 1 was established as phillygenin 4-O-(2′′-trans-cinnamoyl)-α-L-rhamnopyranoside, which is a new flavolignan acylglycoside isolated and elucidated for the first time here.
The chemical structures of the four known compounds, ellagic acid (2), kaempferol 3-O-β-D-glucopyranoside (3), kaempferol 3-O-α-L-rhamnopyranoside (4), and quercetin 3-O-β-D-glucopyranoside (5), were determined mainly by comparing their MS and NMR data with those in previously published literature [20,21,22]. To the best of our knowledge, compounds 2 and 4 have never been reported from P. banksiana previously.
Experimental
General Experimental Procedures. UV spectra were measured in MeOH on a Jenway 6405 spectrophotometer. IR experiments were done in KBr on a PerkinElmer BX FT-IR spectrometer. NMR spectra were recorded in DMSO-d6 with TMS as an internal standard on a Bruker Avance DPX 400 spectrometer at frequencies of 400 and 100 MHz for 1H and 13C NMR, respectively. Optical rotation data were determined by a Jasco DIP-1000 digital polarimeter. An Electro Thermal 9100 apparatus was used to obtain the compounds′ melting points (mp). Sephadex LH-20 (Sigma, USA) and silica gel (Shanhai Anyan Biological Company, China) were used for column chromatography. BS-100A fraction collectors were used to collect the fraction eluent. In TLC, DC-Plastikfolien Cellulose F (Merck Co.) plates were used with t-BuOH–AcOH–H2O (3:1:1, solvent A) and AcOH–H2O (3:47, solvent B) as developing solvents. Spots were detected on TLC under UV light (365 and 254 nm) or by heating after spraying with 1% FeCl3 in EtOH.
Plant Material. Fallen needles of P. banksiana were collected in Jiageda Qi, Daxin_an District, Heilongjiang Province, P. R. China, in October 2012. The plant materials were identified by Prof. Dan Wang at the Institute of the Chemical Industry of Forest Products, P.R. China. A voucher specimen (No. 201210008) has been deposited in the Herbarium of the Tianjin Key Laboratory of Pulp and Paper, College of Papermaking Science and Technology, Tianjin University of Science and Technology, P. R. China.
Extraction and Isolation. A 5.62 g portion of the fallen needles of P. banksiana was dried and finely powdered, then extracted in 70% acetone for more than 3 days. After evaporation of the aqueous acetone under vacuum, the dark brown residue was suspended in water and sequentially fractionated in separation funnels with n-hexane, CHCl3, and EtOAc, then freeze dried to afford EtOAc soluble faction (ESF) powders. A portion of the resulting ESF (32.91 g) was separated by column chromatography over silica gel using a gradient of CHCl3–MeOH (99:1→49:1→1:1) as washing eluent to give five fractions (ESF1–ESF5) according to their TLC profiles. Fraction ESF3 was further subjected to silica gel column chromatography with CHCl3–H2O–MeOH (40:9:1→20:4:1→1:1:1) to yield six fractions (ESF31–ESF36). Fraction ESF35 was then loaded on a chromatographic column packed with Sephadex LH-20 using MeOH–H2O (3:1) as eluting solvent to give four fractions (ESF351–ESF354). Fraction ESF353 was further subjected to Sephadex LH-20 column chromatography using n-hexane–EtOH (2:1, 1:2) and MeOH–H2O (1:1, 1:4) as eluting solvents to obtain 39 mg of compound 1 and 62 mg of compound 2 as amorphous powders. Fraction ESF33 was also subjected to Sephadex LH-20 column chromatography with MeOH–H2O (2:1, 1:2, 1:5) and n-hexane–EtOH (3:1, 1:1, 1:4) as mobile solvents to yield yellowish amorphous compounds 3 (71 mg), 4 (28 mg), and 5 (46 mg).
Phillygenin 4- O -(2′′- trans -Cinnamoyl)- α -L-rhamnopyranoside (1). White amorphous powder, mp 199–201°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –47.9_ (c 0.5, MeOH). IR (KBr, νmax, cm–1): 3390 (hydroxyl group), 1635 and 1525 (phenyl group). UV (MeOH, λmax, nm): 235, 285. R f 0.57 (solvent A) and 0.19 (solvent B). Positive FAB-MS m/z [M + H]+ at 649, [M + K]+ at 687, and [M + Na]+ at 671, indicating a molecular weight of 648 calculated for C36H40O11. For 1H (400 MHz) and 13C (100 MHz) NMR spectroscopic data in DMSO-d6, see Table 1.
References
L. K. Fu, N. Li, and R. M. Robert, Flora of China, 4, 11 (1999).
C. L. Si, J. Z. Jiang, S. C. Liu, H. Y. Hu, X. D. Ren, G. J. Yu, and G. H. Xu, Holzforschung, 67, 357 (2013).
J. Z. Wang, X. Y. Wang, and G. D. Zhao, J. Northeast Forestry Univ., 25, 72 (1997).
M. L. Liu, J. Liu, J. N. Li, and W. Zhao, J. Anhui Agri. Sci., 39, 15420 (2011).
M. Phelan, S. A. Aherne, A. Wong, and N. M. O′Brien, J. Med. Food, 12, 1245 (2009).
A. Pichette, F. X. Garneau, F. I. Jean, and B. Riedl, J. Wood Chem. Technol., 18, 427 (1998).
C. W. Beninger and M. M. Abou-Zaid, Biochem. Syst. Ecol., 25, 505 (1997).
C. Nozzolillo, P. Isabelle, O. M. Andersen, and M. Abou-Zaid, Can. J. Bot., 80, 796 (2002).
C. L. Si, G. H. Xu, X. F. Huang, Z. G. Du, L. Wu, and W. C. Hu, Chem. Nat. Compd., 52, 132 (2016).
C. L. Si, X. D. Ren, Z. G. Du, X. F. Huang, and L. Wu, Chem. Nat. Compd., 51, 1059 (2015).
J. Harmatha and L. Dinan, Phytochem. Rev., 2, 321 (2003).
H. Matsushita and T. Miyase-Uneo, Phytochemistry, 30, 2025 (1991).
V. V. Velde, D. Lavie, H. E. Gottlieb, G. W. Perold, and F. Scheinmann, J. Chem. Soc. Perkin Trans. 1, 1159 (1984).
T. Yahagi, A. Daikonya, and S. Kitanaka, Chem. Pharm. Bull., 60, 129 (2012).
M. A. R. Maiada, M. D. Paul, E. J. David, and A. L. John, Phytochemistry, 29, 1971 (1990).
D. K. Kim, J. P. Lim, J. W. Kim, H. W. Park, and J. S. Eun, Arch. Pharm. Res., 28, 39 (2005).
C. H. Gao, X. X. Yi, W. P. Xie, Y. N. Chen, M. B. Xu, Z. W. Su, L. Yu, and R. M. Huang, Mar. Drugs, 12, 4353 (2014).
C. L. Si, L. L. An, D. N. Xie, C. Y. Liu, X. Q. Chen, G. H. Wang, D. Huo, Q. L. Yang, and Y. M. Hong, Wood Sci. Technol., 50, 645 (2016).
D. Zhang, A. J. Deng, L. Ma, X. F. You, Z. H. Zhang, Z. H. Li, J. D. Jiang, and H. L. Qin, Phytochem. Lett., 12, 320 (2016).
Z. G. Tai, F. M. Zhang, L. Cai, J. Shi, Q. E. Cao, and Z. T. Ding, Chem. Nat. Compd., 48, 221 (2012).
D. D. Khac, S. Tran-Van, A. M. Campos, J. Y. Lallemand, and M. Fetizon, Phytochemistry, 29, 251 (1990).
D. J. Kwon and Y. S. Bae, Chem. Nat. Compd., 47, 636 (2011).
Acknowledgment
The authors would like to acknowledge Jiangsu Province Biomass Energy and Materials Laboratory in the Institute of the Chemical Industry of Forest Products, CAF (JSBEM201601), and Foundation (No. 201531) of Jiangsu Provincial Key Laboratory of Pulp and Paper Science and Technology, Nanjing Forestry University, P. R. China, Introduction of Overseas and Technical and Managerial Personnel Program (20173600003) of State Administration of Foreign Experts Affairs, P. R. China, for financial support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 6, November–December, 2017, pp. 870–873.
Rights and permissions
About this article

Cite this article
Liu, CY., Xie, DN., An, LL. et al. Structural Elucidation of a New Flavolignan Acylglycoside from Fallen Needles of Pinus banksiana . Chem Nat Compd 53, 1020–1024 (2017). https://doi.org/10.1007/s10600-017-2192-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-017-2192-z