
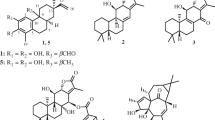
A new labdane-type diterpenoid, hedychiumin (1), has been isolated from the aerial part of Hedychium coronarium, together with five known compounds, (+)-coronarin A (2), (E)-15,16-bisnorlabda-8(17),11-dien-13-one (3), calcaratarin A (4), β-sitostenone (5), and stigmasta-4,22-dien-3-one (6). The structure of the new compound 1 was determined through spectroscopic and MS analyses. Among the isolates, hedychiumin (1) exhibited cytotoxicity, with IC50 value of 4.74 μg/mL, against P388D1 cell line.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hedychium coronarium J. Koenig (Zingiberaceae) is a perennial herb distributed in India, Southeast Asian countries, southern China, Japan, and Taiwan [1]. It is used as a folk medicine for treatment of stomach disorders, insomnia, headache, and contusion in China [2, 3]. Farnesane-type sesquiterpenes [2], labdane-type diterpenes [3,4,5,6,7,8,9,10], and their derivatives were isolated from this plant in previous studies. Many of these compounds were found to exhibit antiallergic [2], cytotoxic [3,4,5], hepatoprotective [6], and anti-inflammatory [7,8,9] activities. In a preliminary screening, the methanolic extract of the aerial part of this species showed cytotoxic activities in vitro. The current phytochemical investigation of the aerial part of this plant has led to the isolation of a new labdane-type diterpenoid, hedychiumin (1), along with five known compounds. The structural elucidation of 1 and the cytotoxic activities of all isolates are described herein.
Extensive fractionation of the n-hexane-soluble portion of a MeOH extract of the aerial part of Hedychium coronarium using silica gel column chromatography (CC) and preparative TLC afforded compounds 1–6.

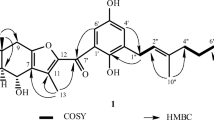
Compound 1 was isolated as colorless needles. The HR-ESI-MS gave an [M + Na]+ ion at m/z 299.1985 (calcd for C18H28O2Na, 299.1987), consistent with a molecular formula of C18H28O2. IR absorption bands for hydroxyl (3382 cm–1) and carbonyl (1668 cm–1) functions were observed. The 1H and 13C NMR data of 1 were similar to those of (+)-coronarin A (2) [11], except that the 12-acetyl group [δH 2.28 (3H, s); δC 27.3 (C-14), 197.9 (C-13)] of 1 replaced the furan-3-yl group [δH 6.55 (1H, br.s, H-14), 7.36 (2H, br.s, H-15, 16); δC 107.6 (C-14), 124.5 (C-13), 139.6 (C-16), 143.2 (C-15)] of (+)-coronarin A (2) [11]. This was supported by the HMBC correlations between H-14 (δ2.28) and C-12 (δ 133.6), C-13 (δ 197.9); and the NOESY correlations between H-14 (δ 2.28) and H-12 (δ 6.08), H-11 (δ 6.87). The coupling constant (J = 16.0 Hz) between H-11 and H-12 suggested the 11E-configuration of 1. The relative stereochemistry of 1 at chiral centers was based on analysis of the NOESY spectrum, which displayed NOESY correlations between H-5/H-7, H-5/H-9, H-5/H3-18, H-11/H3-20, H-11/H3-14, and H3-19/H3-20 and suggested that H-5, H-7, H-9, and H3-18 are α-oriented, and H3-19, H3-20, 7-OH, and the (E)-3-oxobut-1-enyl group are β-oriented. The full assignment of 1H and 13C NMR resonances was supported by 1H–1H COSY, DEPT, HSQC, NOESY, and HMBC (Fig. 1) spectral analyses. On the basis of the above data, the structure of 1 was elucidated as (E)-4-((1R,3S,4aS,8aS)-3-hydroxy-5,5,8a-trimethyl-2-methylenedecahydronaphthalen-1-yl)but-3-en-2-one, named hedychiumin.

Significant HMBC correlations of 1.
The known isolates were readily identified by a comparison of physical and spectroscopic data (UV, IR, 1H NMR,[α]D, and MS) with corresponding authentic samples or literature values, and this included (+)-coronarin A (2) [11], (E)-15,16-bisnorlabda-8(17),11-dien-13-one (3) [12], calcaratarin A(4) [13], β-sitostenone (5) [14], and stigmasta-4,22-dien- 3-one (6) [15, 16].
Isolates from the aerial part of Hedychium coronarium were tested in vitro against DLD-1, CCRF-CEM, HL-60, and P388D1 cell lines; the cytotoxicity data are shown in Table 1. The anticancer agent doxorubicin was used as a positive control. From the results of our cytotoxicity tests, the following conclusions can be drawn: (a) (+)-coronarin A (2) and calcaratarin A (4) exhibited cytotoxicities, with IC50 values of 12.5 ± 1.9 and 12.3 ± 2.3 μg/mL, respectively, against the DLD-1 cell line. (b) (+)-Coronarin A (2) and calcaratarin A (4) exhibited cytotoxicities, with IC50 values of 13.1 ± 2.2 and 9.58 ± 1.14 μg/mL, respectively, against the CCRF-CEM cell line. (c) Calcaratarin A (4) exhibited cytotoxicity, with IC50 value of 12.8 ±1.4 μg/mL, against the HL-60 cell line. (d) Hedychiumin (1) and calcaratarin A (4) exhibited cytotoxicities, with IC50 values of 4.74 ± 0.53 and 8.56 ± 0.67 μg/mL, respectively, against the P388D1 cell line.
Experimental
General Experimental Procedures. Ultraviolet (UV) spectra were obtained on a Jasco UV-240 spectrophotometer. Infrared (IR) spectra (neat or KBr) were recorded on a PerkinElmer 2000 FT-IR spectrometer. Nuclear magnetic resonance (NMR) spectra, including correlation spectroscopy (COSY), nuclear Overhauser effect spectroscopy (NOESY), heteronuclear multiple-bond correlation (HMBC), and heteronuclear single-quantum coherence (HSQC) experiments, were recorded on a Varian Unity 400 spectrometer operating at 400 MHz (1H) and 100 MHz (13C), respectively, with chemical shifts given in ppm (δ) using tetramethylsilane (TMS) as an internal standard. Electrospray ionization (ESI) and high-resolution electrospray ionization (HR-ESI)-mass spectra were recorded on a Bruker APEX II mass spectrometer. Silica gel (70–230, 230–400 mesh) (Merck) was used for column chromatography (CC). Silica gel 60 F-254 (Merck) was used for thin-layer chromatography (TLC) and preparative thin-layer chromatography (PTLC).
Plant Material. The aerial part of H. coronarium was collected from Taitung District Agricultural Research and Extension Station, Taitung County, Taiwan, in June 2009 and identified by J. F. Chen. A voucher specimen (Chen 3011) was deposited in the Department of Pharmacy, Tajen University, Pingtung, Taiwan.
Extraction and Separation of Compounds. The dried aerial part (1.3 kg) of H. coronarium was pulverized and extracted three times with MeOH (10 L each) for 3 days. The methanol extract (128 g) was partitioned between n-hexane and H2O (1:1) to afford n-hexane-soluble (fraction A, 56 g) and H2O-soluble (fraction B, 71 g) fractions. The n-hexane-soluble fraction (56 g) was chromatographed on silica gel (70–230 mesh, 2.6 kg), eluting with n-hexane, gradually increasing the polarity with EtOAc to give nine fractions (A1–A9). Fraction A3 (5.2 g) was separated by column chromatography on silica gel (230–400 mesh, 240 g), eluting with CH2Cl2–acetone (12:1–1:1) to yield 10 fractions (A3-1–A3-10). Part (127 mg) of fraction A3-4 was purified by preparative TLC (silica gel, n-hexane–EtOAc, 10:1) to afford β-sitostenone (5) (6.3 mg). Fraction A4 (5.7 g) was separated by column chromatography on silica gel (230–400 mesh, 265 g), eluting with CH2Cl2–acetone (10:1–0:1) to yield eight fractions (A4-1–A4-8). Part (152 mg) of fraction A4-2 was purified by preparative TLC (silica gel, CHCl3–MeOH, 20:1) to afford stigmasta-4,22-dien-3-one (6) (5.4 mg). Part (145 mg) of fraction A4-5 was purified by preparative TLC (silica gel, n-hexane–acetone, 10:1) to yield (E)-15,16-bisnorlabda-8(17),11-dien-13-one (3) (5.5 mg). Fraction A5 (6.1 g) was separated by column chromatography on silica gel (230–400 mesh, 280 g), eluting with CH2Cl2–MeOH (15:1–0:1) to yield 11 fractions (A5-1–A5-11). Part (145 mg) of fraction A5-3 was purified by preparative TLC (silica gel, n-hexane–acetone, 8:1) to afford calcaratarin A (4) (4.8 mg). Part (140 mg) of fraction A5-6 was purified by preparative TLC (silica gel, CH2Cl2–MeOH, 8:1) to give hedychiumin (1) (4.4 mg). Part (195 mg) of fraction A5-10 was purified by preparative TLC (silica gel, n-hexane–EtOAc, 6:1) to obtain (+)-coronarin A (2) (7.2 mg).
Hedychiumin (1). Colorless needles, mp 156–158°C (MeOH). [α] 25D +24.3° (c 0.16, CHCl3). UV (MeOH, λmax, nm) (log ε): 225 (4.01). IR (KBr, v max, cm–1): 3382 (OH), 1668 (C=O). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.85 (3H, s, H-19), 0.86 (3H, s, H-20), 0.91 (3H, s, H-18), 1.05 (1H, ddd, J = 13.2, 12.4, 4.4, H-1α), 1.16 (1H, dd, J = 12.4, 2.4, H-5), 1.18 (1H, ddd, J = 13.6, 13.2, 4.4, H-3α), 1.35 (1H, ddd, J = 12.8, 12.4, 11.2, H-6β), 1.45 (1H, br.d, J = 13.2, H-3β), 1.46 (1H, m, H-2α), 1.51 (1H, br.d, J = 12.4, H-1β), 1.53 (1H, m, H-2β), 2.10 (1H, ddd, J = 12.8, 5.2, 2.4, H-6α), 2.28 (3H, s, H-14), 2.37 (1H, d, J = 10.0, H-9), 4.05 (1H, dd, J = 11.2, 5.2, H-7), 4.73 (1H, br.s, H-17), 5.14 (1H, br.s, H-17), 6.08 (1H, d, J = 16.0, H-12), 6.87 (1H, dd, J = 16.0, 10.0, H-11). 13C NMR (100 MHz, CDCl3, δ, ppm): 15.0 (C-20), 19.1 (C-2), 21.8 (C-19), 27.3 (C-14), 32.6 (C-6), 33.3 (C-4), 33.4 (C-18), 40.2 (C-1), 42.0 (C-3), 52.4 (C-5), 60.2 (C-9), 73.3 (C-7), 107.5 (C-17), 133.6 (C-12), 146.6 (C-11), 151.3 (C-8), 197.9 (C-13). ESI-MS m/z 299 [M + Na]+. HR-ESI-MS m/z 299.1985 [M + Na]+ (calcd for C18H28O2Na, 299.1987).
(+)-Coronarin A (2). Colorless needles, mp 100–102°C (n-hexane). [α] 25D +26.5° (c 0.14, CHCl3). UV (MeOH, λmax, nm) (log ε): 232 (4.06). IR (KBr, v max, cm–1): 3418 (OH), 3087, 1652, 898 (exo-methylene bands). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.85 (3H, s, H-18), 0.85 (3H, s, H-19), 0.93 (3H, s, H-20), 1.05 (1H, dd, J = 12.8, 4.2, Hα-1), 1.10–1.60 (5H, overlapping peaks, H β -1, H2-2, 3), 1.16 (1H, dd, J = 13.0, 2.2, H-5), 1.35 (1H, ddd, J = 13.0, 12.0, 11.6, H β -6), 2.11 (1H, ddd, J = 11.6, 5.6, 2.2, Hα-6), 2.36 (1H, d, J = 9.4, H-9), 4.10 (1H, dd, J = 12.0, 5.6, H-7), 4.74 (1H, s, H-17), 5.13 (1H, s, H-17), 5.98 (1H, dd, J = 15.6, 9.4, H-11), 6.55 (1H, br.s, H-14), 6.62 (1H, d, J = 15.6, H-12), 7.36 (1H, br.s, H-15), 7.37 (1H, br.s, H-16). ESI-MS m/z 323 [M + Na]+.
(E)-15,16-Bisnorlabda-8(17),11-dien-13-one (3). Colorless needles, mp 145–147°C (CH2Cl2–MeOH). [α] 25D +6.2° (c 0.15, CHCl3). UV (MeOH, λmax, nm) (log ε): 227 (4.02). IR (KBr, v max, cm–1): 1673 (C=O). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.85 (3H, s, H-19), 0.90 (3H, s, H-18), 0.90 (3H, s, H-20), 2.28 (3H, s, H-14), 4.41 (1H, br.s, H-17), 4.80 (1H, br.s, H-17), 6.08 (1H, d, J = 16.0, H-12), 6.87 (1H, dd, J = 16.0, 10.0, H-11). ESI-MS m/z 283 [M + Na]+.
Calcaratarin A (4). Colorless oil. [α] 25D +14.2° (c 0.15, CHCl3). UV (MeOH, λmax, nm) (log ε): 233 (4.04). IR (neat, v max, cm–1): 1685 (C=O), 3084, 1641, 891 (exo-methylene bonds). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 0.75 (3H, s, H-20), 0.82 (3H, s, H-19), 0.89 (3H, s, H-18), 1.09 (1H, ddd, J = 13.6, 12.8, 3.8, Hα-1), 1.14 (1H, dd, J = 12.6, 2.4, H-5), 1.19 (1H, ddd, J = 13.8, 13.2, 4.2, Hα-3), 1.34 (1H, dddd, J = 13.2, 12.6, 12.6, 4.2, H β -6), 1.42 (1H, br.d, J = 13.2, H β -3), 1.51 (1H, m, Hα-2), 1.58 (1H, dddd, J = 13.8, 13.6, 3.6, 3.3, H β -2), 1.75 (1H, br.d, J = 12.8, H β -1), 1.75 (1H, dddd, J = 13.2, 5.4, 2.4, 2.4, Hα-6), 1.90 (1H, br.d, J = 10.8, H-9), 2.03 (1H, ddd, J = 13.2, 12.6, 5.4, Hα-7), 2.41 (1H, ddd, J = 13.2, 4.2, 2.4, H β -7), 2.46 (1H, ddd, J = 17.4, 10.8, 6.0, H-11), 2.57 (1H, dd, J = 13.2, 5.4, H-14), 2.60 (1H, dd, J = 13.2, 5.4, H-14), 2.64 (1H, ddd, J = 17.4, 6.0, 3.0, H-11), 3.35 (6H, s, MeO-15 × 2), 4.42 (1H, d, J = 1.3, H-17), 4.44 (1H, t, J = 5.4, H-15), 4.84 (1H, d, J = 1.3, H-17), 6.55 (1H, t, J = 6.0, H-12), 9.33 (1H, s, CHO). ESI-MS m/z 371 [M + Na]+.
β-Sitostenone (5). Colorless needles, mp 87–89°C (MeOH). [α] 24D +85.7°(c 0.22, CHCl3). UV (MeOH, λmax, nm) (log ε): 243 (4.19). IR (KBr, v max, cm–1): 1679 (C=O). ESI-MS m/z 435 [M + Na]+.
Stigmasta-4,22-dien-3-one (6). Colorless needles, mp 125–127°C (MeOH). [α] 24D +58.5° (c 0.18, CHCl3). UV (MeOH, λmax, nm) (log ε): 243 (4.19). IR (KBr, v max, cm–1): 1680 (C=O). ESI-MS m/z 433 [M + Na]+.
Cytotoxic Assay. The cell lines used in this study were DLD-1 cells (human colorectal carcinoma), CCRF-CEM cells (human lymphoblastic leukemia), HL-60 cells (human myeloid leukemia), and P388D1 cells (murine macrophage-like lymphoma). The above cell lines were purchased from the Bioresource Collection and Research Center (BCRC), Food Industry Research and Development Institute (FIRDI), Hsinchu 300, Taiwan.
The cytotoxic activities of the compounds against DLD-1, CCRF-CEM, HL-60, and P388D1 were assayed by a modification of the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric method [17]. To measure the cytotoxic activities of the purified compounds against the above tumor cells, each cell line was initiated at 5 ×105 cells/well in 96-well microtiter plates (Falcon). Eight concentrations (triplicate) of the test compounds (dissolved in 0.5% DMSO) encompassing a 128-fold range were added to each cell line. Each tumor cell was enumerated using MTT (Sigma) after exposure to the test compounds for 3 days. Fifteen microliters of 1 mg/mL MTT was added to each well, and the plates were incubated at 37°C for a further 4 h. The formazan crystals were redissolved in DMSO (Merck) for 10 min with shaking, and the plate was read immediately on a microtiter plate reader (Dynatech) at a wavelength of 570 nm. The IC50 value was defined as the concentration of the test compound necessary to inhibit the growth to 50% of the control in the MTT assay. The anticancer agent doxorubicin and 0.5% DMSO were used as the positive control and solvent control, respectively.
References
J. C. Wang, Zingeraceae in Flora of Taiwan, Vol. 5, 2nd Ed., Editorial Committee of the Flora of Taiwan, Taipei, Taiwan, 2000, pp. 707–724.
T. Morikawa, H. Matsuda, Y. Sakamoto, K. Ueda, and M.Yoshikawa, Chem. Pharm. Bull., 50, 1045 (2002).
N. Chimnoi, C. Sarasuk, N. Khunnawutmanotham, P. Intachote, S. Seangsai, B. Saimanee, S. Pisutjaroenpong, C. Mahidol, and S. Techasakul, Phytochem. Lett., 2, 184 (2009).
H. Itokawa, H. Morita, I. Katou, K. Takeya, A. J. Cavalheiro, R. C. B. de Oliveira, M. Ishige, and M. Motidome, Planta Med., 54, 311 (1988).
G. Suresh, P. P. Reddy, K. S. Babu, T. B. Shaik, and S. V. Kalivendi, Bioorg. Med. Chem. Lett., 20, 7544 (2010).
S. Nakamura, Y. Okazaki, K. Ninomiya, T. Morikawa, H. Matsuda, and M. Yoshikawa, Chem. Pharm. Bull., 56, 1704 (2008).
H. Matsuda, T. Morikawa, Y. Sakamoto, I. Toguchida, and M. Yoshikawa, Heterocycles, 56, 45 (2002).
H. Matsuda, T. Morikawa, Y. Sakamoto, I. Toguchida, and M. Yoshikawa, Bioorg. Med. Chem., 10, 2527 (2002).
J. J. Chen, C. W. Ting, Y. C. Wu, T. L. Hwang, M. J. Cheng, P. J. Sung, T. C. Wang, and J. F. Chen, Int. J. Mol. Sci., 14, 13063 (2013).
N. Nakatani, H. Kikuzaki, H. Yamaji, K. Yoshio, C. Kitora, K. Okada, and W. G. Padolina, Phytochemistry, 37, 1383 (1994).
I. Kumrit, A. Suksamrarn, P. Meepawpan, S. Songsri, and N. Nuntawong, Phytother. Res., 24, 1009 (2010).
H. Itokawa, S. Yoshimoto, and H. Morita, Phytochemistry, 27, 435 (1988).
L.Y. Kong, M. J. Qin, and M. Niwa, J. Nat. Prod., 63, 939 (2000).
J. J. Chen, P. J. Sung, I. S. Chen, M. J. Cheng, Y. P. Lim, H. R. Liao, T. H. Chang, D. C. Wei, and J. Y. Chen, Chem. Nat. Compd., 51, 302 (2015).
J. J. Chen, H. C. Hung, P. J. Sung, I. S. Chen, and W. L. Kuo, Phytochemistry, 72, 523 (2011).
A. Barrero, J. F. Sanchez, E. J. Alvarez-Manzaneda, M. M. Dorado, and A. Haidour, Phytochemistry, 32, 1261 (1993).
T. Mosmann, J. Immunol. Methods, 65, 55 (1983).
Acknowledgment
This research was supported by a grant from the Ministry of Science and Technology, Taiwan (NSC 101-2320-B-127-001-MY3), awarded to Prof. J.-J. Chen. This work was also supported by the grant from the Zuoying Branch of Kaohsiung Armed Forces General Hospital (ZBH 104-21).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2017, pp. 61–64.
Rights and permissions
About this article

Cite this article
Chen, LC., Wen, ZH., Sung, PJ. et al. New Labdane-Type Diterpenoid and Cytotoxic Constituents of Hedychium coronarium . Chem Nat Compd 53, 72–76 (2017). https://doi.org/10.1007/s10600-017-1914-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-017-1914-6