
Abstract
Documentation of intraspecific genetic lineages and their evolutionary history can provide insight for current and future conservation and management actions. The Blanding’s Turtle, Emys (= Emydoidea) blandingii, is a long-lived species with a relatively narrow latitudinal distribution centered around the Great Lakes, but extending from Nebraska to Nova Scotia. It is listed as endangered or threatened throughout most of its range mainly due to habitat loss. Microsatellite loci have been predominantly used to test and generate hypotheses concerning the number of evolutionarily significant units and the history of lineage diversification in this species. Here we describe haplotypes from two mitochondrial and three nuclear loci generated from 32 localities across the species’ range to provide an additional perspective on existing patterns. Haplotype and nucleotide diversity were low in both sets of loci, with mitochondrial polymorphism comparable to the lowest found in any North American freshwater turtle. Spatial analyses of population differentiation supported the presence of two groups with a boundary in eastern Ontario that is roughly associated with the Appalachian Mountains as proposed by Mockford et al. (Conserv Gen 8:209–219, 2007). We suggest that the low diversity in these loci is likely related to periodic range contractions and expansions associated with glacial cycles and that the two groups recovered result from a deeper history of diversification. Our results are broadly consistent with previously identified range-wide structure and help to reconcile population structure found at smaller spatial scales, outcomes that will better inform conservation decision making for the species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phylogeographic analysis is a cornerstone of conservation genetics. With the goal of conserving genetic diversity within and among populations, the characterization of genetic lineages assists with guiding the release of captively bred individuals or the translocation of individuals from other populations. One challenge in realizing this goal is reconciling genetic information derived from different molecular markers and sample localities. Adding new datasets to previously studied species often yields differing, but ultimately deeper, perspectives on the level, distribution, and history of intraspecific genetic variation.
Turtles have proven to be an interesting group for molecular analyses. Rates of DNA sequence evolution are slow (Avise et al. 1992; Shaffer et al. 2013) and this should limit the rate of diversification. Limited diversification is expected to be further prevalent in species that occur in formerly glaciated regions (Lenk et al. 1999; Weisrock and Janzen 2000; Starkey et al. 2003; Spinks and Shaffer 2005; Rosenbaum et al. 2007; Amato et al. 2008; Rödder et al. 2013). Following range expansion, it is expected that there will be a broad distribution of few genetic variants due to a limited number of founders and/or recolonization from a bottlenecked population isolated in a refugium.
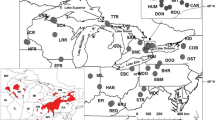
Blanding’s Turtle, Emys (= Emydoidea) blandingii, has been the subject of several conservation genetic studies over the past decade. It has a northern distribution, ranging from Nebraska east through the Great Lakes region, with disjunct populations in the northeastern United States and Nova Scotia (Fig. 1). Blanding’s Turtle is listed as threatened or endangered throughout most of its geographic range and this is likely due to habitat loss, road mortality, and poaching (Congdon et al. 2008). Its geographic range has been impacted by Pleistocene glaciations and the current distribution may have been established from one or more glacial refugia (Mockford et al. 2007; Rödder et al. 2013). An early microsatellite analysis suggested the presence of three evolutionarily significant units: (1) populations of the Great Lakes region west of the Appalachian Mountains, (2) disjunct populations in New York and New England east of the Appalachians and (3) an isolated population in Nova Scotia (Mockford et al. 2007). More recently, microsatellite analyses conducted with additional loci have revealed finer population structure on regional scales including: the western portion of the geographic range from Nebraska to Illinois (Sethuraman et al. 2014), within Ontario (Davy et al. 2014), within Illinois (Anthonysamy et al. 2017), and within Wisconsin (Reid et al. 2017). Further analyses are necessary to better understand these patterns.

Map of the geographic range of E. blandingii and sample localities of this study. Locality numbers are described in the text and are identified in Online Resource 1. The maximum of extent of the Wisconsin glaciation (18,000 ybp) is shown
To date, microsatellites have provided the bulk of population genetic information in Blanding’s Turtle. This is likely due in part to low levels of sequence polymorphism found in mitochondrial (mtDNA) and nuclear (nuDNA) loci in the species (Spinks and Shaffer 2009) which can constrain resolution of population structure. However, analyses of mtDNA and nuDNA sequences are better suited to understanding deeper intraspecific history due to their relatively slower overall mutation rates, and this information can be synthesized with more rapidly evolving markers to guide conservation decisions (Wang 2010; Weeks et al. 2011, Epps and Keyghobadi 2015). In addition, differences in structure among microsatellites, mtDNA, and nuDNA, can occur due to a range of processes, including variation in coalescence time, selective sweeps, and sex-biased dispersal (Zink and Barrowclough 2008; Karl et al. 2012; Toews and Brelsford 2012). Comparisons among markers have been well exemplified in the closely related Western Pond Turtle (Spinks and Shaffer 2005; Spinks et al. 2010, 2014) and European Pond Turtle (Lenk et al. 1999; Velo-Antón et al. 2008; Vamberger et al. 2015), but there is no geographically broad sample of sequence polymorphism in E. blandingii. Here we carry out a range-wide phylogeographic analysis of Blanding’s Turtle to: (1) characterize sequence variation in mitochondrial and nuclear loci and (2) use these data to evaluate phylogeographic patterns to inform conservation work.
Materials and methods
Samples and genetic loci
Samples were primarily acquired from tissues collected as part of previous studies (Mockford et al. 2007; Davy et al. 2014; Sethuraman et al. 2014; McCluskey et al. 2016; Reid et al. 2017), but also included novel collections and museum specimens. A total 81 turtles representing 32 localities were included in the analysis (Fig. 1; Online Resource 1). The largest sample originated from a locality in Nebraska (n = 10) while no samples were available from a disjunct region in the states of Maine, New Hampshire, and Massachusetts. Although many samples consisted of previously extracted DNA, some were available as tissues or blood. DNA was extracted using the Qiagen DNAeasy Blood and Tissue Kit for both tissue types.
Taq-mediated polymerase chain reactions (PCR) were used to amplify loci in a volume of 25 µl. Partial sequences of two mtDNA loci [control region (cr) and cytochrome b (cytb)] that were expected to show intraspecific polymorphism based on prior studies on related species (Lenk et al. 1999; Spinks and Shaffer 2005; Fritz et al. 2007) were used (Online Resource 2). Thermocycling for the control region followed Spinks and Shaffer (2005). For both cytb segments a touchdown protocol was employed (initial denaturation at 94 °C for 2 min; 40 cycles denaturing at 94 °C for 30 s, annealing at 60 °C decreasing 0.5 °C each cycle to 50 °C for 30 s, extension at 72 °C for 60 s; followed by a final extension at 72 °C for 10 min). A sample of ten nuDNA loci characterized in E. marmorata by Spinks et al. (2014) were evaluated for amplification and polymorphism (AMY1A, FSHR, NB10179, NB00504, NB12303, NB17367, NB10015, TRAF6, PTGER4, and NB10005) using the abovementioned touchdown protocol but with hot start (initial denaturation at 94 °C for 10 min). Due to inconsistent amplification or a lack of polymorphism, only three of the original ten nuDNA markers were retained for analysis: AMY1A, NB10005 and NB17367. The former two loci are anonymous while the third locus (NB17367) is a partial proteasome gene. All sequencing was carried out by Beckman Coulter Genomics (http://www.beckmangenomics.com) using both forward and reverse primers for each individual and sequences were submitted to GenBank (Online Resource 2).
Statistical analysis
Sequences were verified, edited, trimmed, and aligned in Geneious v. 7.1.2 (Kearse et al. 2012). We inspected aligned sequences by eye to identify single nucleotide polymorphisms (SNP). For nuDNA markers, we reconstructed haplotypes using PHASE v. 2.1 (Stephens et al. 2001; Stephens and Donnelly 2003) implemented in DnaSP v. 6.12.01 (Rozas et al. 2017). For each locus we used 500 steps for burn-in, 1 thinning interval, and 1000 iterations in Markov chain-Monte Carlo to estimate posterior probabilities of haplotypes under the default model of recombination. A threshold probability of ≥ 0.7 was used to include a haplotype call (c.f. Garrick et al. 2010) and three runs of the model were compared to verify haplotype assignments. Descriptive statistics, nucleotide diversity, number of haplotypes, and haplotype diversity were calculated in DnaSP. Samples missing data for one or more locus were excluded from multi-locus analyses. In all analyses involving more than one locus, mitochondrial loci were concatenated while nuclear loci were treated as independent samples of the genome.
Phylogeographic structure
We visualized the evolutionary relationship among samples using the Neighbor-Net algorithm (Bryant and Moulton 2004) employed in SplitsTree v. 4.14.6 (Huson and Bryant 2006). Such phylogenetic networks allow description of the ambiguity in phylogenetic datasets due to sampling error or conflicting signal among loci. Networks for mtDNA and nuDNA were constructed from uncorrected p-distances on concatenated and multiple loci, respectively. We used POFAD v. 1.07 (Joly and Bruneau 2006) to calculate distance matrices for both sets of loci, an approach that was chosen because it is able to account for allelic variation in nuDNA. Due to limited polymorphism and a lack of resolution, networks for individual loci are not presented.
We assessed the presence and nature of phylogeographical structure using analyses included in SPADS 1.0 (Dellicour and Mardulyn 2014). Global variation among localities was tested using ɸST based on Euclidean distance of haplotypes (AMOVA, Excoffier et al. 1992), and the level of phylogeographic signal was tested using the difference between NST and GST (Hardy and Vekemans 2002). For statistical inference, randomizing individuals across localities is used for ɸST while the difference between NST and GST relies on randomizing haplotypes across localities (Dellicour and Mardulyn 2014). We also used SPADS to evaluate the phylogeographic structure hypothesized by Mockford et al. (2007). Sample localities were classified into three groups: Midwestern United States (localities 1–26), Ontario and New York (localities 27–31), and Nova Scotia (locality 32). This classification assumes that the Nebraska sample locality, unsampled in the earlier study (Mockford et al. 2007), would cluster with other Midwest localities. It should also be noted that our dataset lacked samples from one of the northeastern regions (Massachusetts, New Hampshire, and Maine) and had limited coverage in New York. These analyses tested the hierarchical levels of variation among hypothesized groups (ɸCT), variation across localities range-wide (ɸST), and variation across localities within groups (ɸSC) permuted following Excoffier et al. (1992).
We further used SPADS to cluster sample localities without designating a priori groups using spatial analysis of molecular variance (SAMOVA) (Dupanloup et al. 2002) and Monmonier’s algorithm implemented in BARRIER v. 2.2 (Manni et al. 2004). SAMOVA seeks to maximize differentiation among population clusters while accounting for relative geographic position, whereas BARRIER directly identifies genetic breaks and therefore provides indirect evidence of population structure. Both analyses rely on a network of sample localities generated by Delaunay triangulation and Voronoi tessellation (described in Manni et al. 2004). SAMOVA criteria were set as the number of hypothesized populations, K, ranging from 2 to 10 with 10,000 iterations (the number of repetitions of steps 5–9 of the SAMOVA algorithm) applied in ten replicate runs. When interpreting the SAMOVA results, the best value for K was chosen using the highest ɸCT value provided there was more than one individual in the cluster. For nuDNA loci, the multi-locus ɸ-statistics are a weighted average of the individual loci but for mtDNA loci we analyzed a concatenated sequence to account for the non-independent inheritance of cytb and cr. The BARRIER analysis was conducted using interindividual genetic distance matrices generated in SPADS (IID2, an uncorrected p-distance derived from Miller 2005; Miller et al. 2006). Following the recommendations of Manni et al. (2004), we tested for the presence of isolation by distance since algorithm may construct barriers due to large geographic distances among a few localities. Alleles in Space (Miller 2005) was used to test for the relationship between IID2 and geographic distance, and if a correlation was found a matrix of IID2 residuals was used to identify barriers. Finally, the GDisPAL function described in SPADS was employed to visualize the interpolation of interindividual genetic distances across the geographic range. We ran the function in R version 3.4.1 (R Core Team 2018) using a raster of the Blanding’s Turtle range, the geographic coordinates of the sample localities, and residual IID2. The resulting interpolation surface was saved as a raster file and projected as a map in QGIS version 2.18.10 (Team 2009).
Demographic analysis
To investigate the hypothesis of population growth following glacial retreat, we calculated neutrality statistics derived from expectations under coalescent theory (reviewed in Ramos-Onsins and Rozas 2002) in DnaSP v. 6.12.01 (Rozas et al. 2017). Statistics included were Tajima’s D (Tajima 1989), Fu’s Fs (Fu 1997), and Ramos-Onsins and Rozas’ R2 (Ramos-Onsins and Rozas 2002), each having different effectiveness under various demographic scenarios, levels of recombination, and sample sizes (Ramírez-Soriano et al. 2008). We employed coalescent simulations, assuming a null hypothesis of constant population size over time and an infinite allele model of mutation (Standard Neutral Model in DnaSP), to calculate 95% confidence intervals and assess whether the observed metrics deviated from neutrality. The simulations were run with 1000 replicates and employed values of θ estimated from the data.
Results
Genetic diversity
Between 44 and 67 individuals were genotyped for each locus, with a combined total of 42 for mtDNA loci and 55 for nuDNA loci (Table 1; Online Resource 1). Mitochondrial cr and cytb had similar levels of polymorphism with four segregating sites each, all of which were substitutions except for one deletion in cr. The number of haplotypes (cr = 6, cytb = 7) and haplotype diversity were similar, but nucleotide diversity was slightly lower in cytb. The number of segregating sites ranged from 2 to 6 in each of the three nuDNA loci (Table 1) and all were substitutions. We found a total of 14 haplotypes across nuDNA loci, with haplotype number varying from 3 (NB17367) to 6 (AMY1A) (Online Resource 3). A relatively low haplotype diversity was observed for NB10005 and this is due to most haplotypes being rare in the sample. Levels of nucleotide diversity were comparable to those found in mtDNA loci but with a relatively high value found in AMY1A.
Phylogeographic structure
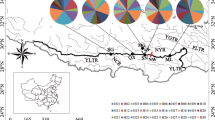
Descriptive relationships among samples in split networks suggested some geographic separation but with conflicting partitions as indicated by parallel edges (Fig. 2). The mtDNA network (Fig. 2a) was simpler and relatively less ambiguous. Except for one individual from western Ontario, samples from Nova Scotia, New York, and eastern Ontario were divided from other samples. Nebraska individuals and one individual from eastern Iowa also appeared to be separated from other samples. The nuDNA network (Fig. 2b) was much less resolved with multiple parallel edges and samples from across geographic range occupying similar splits. However, there is some suggestion that samples from Nova Scotia sort together.

Split networks of uncorrected p-distances from concatenated mitochondrial (a) and multi-locus nuclear (b) loci using the NeighborNet algorithm. Nodes are labelled with the state or province of the sample with numbers in parentheses representing more than one individual. Asterisk indicates samples from western Ontario (locality 27)
We found more support for differentiation in statistical inference of haplotype variation. The null hypothesis of panmixia across the geographic range (global ɸST) was rejected in all loci, but only concatenated mtDNA loci suggested a strong signal of phylogeographic structure (NST–GST, Table 2). Although the data tended to support the presence of three phylogeographic groups (ɸCT) hypothesized by Mockford et al. (2007), there was a lack of structure in mitochondrial CR and nuclear AMY1A in AMOVA. The SAMOVA analysis supported the presence of three phylogeographic clusters in concatenated mtDNA data and two phylogeographic clusters in multi-locus nuDNA data. Sample localities in the three mtDNA clusters included: Grant County, Nebraska (locality 1), the midwestern US east to Parry Sound, Ontario (localities 1–2, 4–7, 9, 11–17, 21–27), and eastern Ontario to Nova Scotia (localities 28–29, 31–32). The two clusters in nuDNA loci included only Nova Scotia samples in one (locality 32) and all other sample localities in the other (localities 1, 3, 5–10, 12, 14–17, 19–26, 27–29). When the clusters were used to define groups in AMOVA, there was an overall increase in ɸCT values relative to the a priori grouping hypothesized by Mockford (Table 2). This was particularly the case with mtDNA data where each locus and the concatenated loci increased by at least 0.25. Individual nuDNA data appeared to be less resolved, but the multi-locus ɸCT of increased from 0.29 in a priori groups to 0.46 in SAMOVA clusters.
We found a positive correlation between interindividual geographic distance and genetic distance in both sets of markers (mtDNA r = 0.62, P < 0.0001; nuDNA r = 0.41, P < 0.0001). Residuals from these matrices (residual IID2) resulted in similar clustering in BARRIER as those found in SAMOVA (not shown). When barriers were sequentially added in the mtDNA dataset, the first barrier split off localities in eastern Ontario, New York and Nova Scotia (localities 28–29, 31–32) from the rest of the range before the second barrier split off Nebraska samples. In the nuDNA dataset, the one barrier led to the clustering of Jefferson, New York (locality 29) with Nova Scotia. These results were represented in the interpolation of the residuals of both datasets using the GDisPAL function (Fig. 3).

Interpolation of interindividual genetic distances for mitochondrial (a) and nuclear (b) DNA sequences across the geographic range of E. blandingii. Genetic distances are based on residuals of the relationship between genetic and geographic distance calculated in SPADS 1.0. Lightly colored regions indicate areas of relatively greater genetic distance
Demographic analysis
Overall, we did not find support for a historical bottleneck followed by range expansion in the neutrality tests (Table 3). Only the nuclear locus NB10005 had statistical support for expected negative values in Tajima’s D under this scenario.
Discussion
Low genetic variation
The difference in levels of genetic variation among species provides useful context for considering diversification history within Blanding’s Turtle (Table 4). We found that sequence polymorphism in E. blandingii is low in the loci of both genomes, confirming data collected at a more limited scale in an interspecific analysis (Spinks and Shaffer 2009). The low variability is particularly evident in mtDNA, loci which have a longer history of use in phylogeography and are better characterized. Taxa closely related to E. blandingii have more segregating sites regardless of sample size for mtDNA loci. For example, the number of segregating sites is seven- to ten-fold higher in E. marmorata for control region and cytochrome b, respectively. Meanwhile, only G. muhlenbergii (Rosenbaum et al. 2007) has levels of genetic variation comparable to E. blandingii. Therefore, although mtDNA sequence variation is low in turtles overall (Avise et al. 1992), species such as E. blandingii and G. muhlenbergii represent the lower bound of this variability. Meanwhile, levels of nuDNA locus polymorphism are similar to E. marmorata but the sample size used for E. mamorata was an order of magnitude lower.
Geographic range expansion and contraction associated with glacial cycles during the Pleistocene could be an important contributor to this pattern (Hewitt 2000). North America experienced three such cycles in the past 320,000 years, with the most recent glaciation (Wisconsin) ending ~ 20,000 years ago. In species with northern distributions, genetic bottlenecks can occur as distributions contract during cold periods leaving minimal genetic variation among future colonizers. If colonization is rapid following deglaciation, the limited polymorphism is expected to be distributed over a large geographic area. The Blanding’s Turtle distribution appears to be particularly sensitive to glaciation (Rödder et al. 2013). In an analysis of 59 North American species, climate–based models of the paleogeographic range of E. blandingii show it to have among the smallest refugia and that these refugia occur for extended time periods during glacial events. Other turtle species with northern distributions such as G. insculpta and G. muhlenbergii suggest similar correspondence between variability in geographic range size (Rödder et al. 2013) and reduced genetic variation (Table 4). Meanwhile, the same models applied to the more genetically diverse E. marmorata indicate a more stable distribution and the potential for larger refugial populations.
Given the patterns of range dynamics and overall levels of genetic variation among species, we expected neutrality tests within E. blandingii to deviate from a null model of constant population size and neutral evolution. Demographic expansion following glaciation should increase the number of recent mutations leading to higher haplotype diversity relative nucleotide diversity. For the majority of the loci and tests, this expectation was not met (Table 3), tempering our ability to make a strong conclusion concerning post-glacial expansion of effective population size.
Other explanations for low genetic variation in turtle species include: slow rates of molecular evolution related to long generation time (Avise et al. 1992; Shaffer et al. 2013), recent history of diversification (Weisrock and Janzen 2000), and selective sweeps (Rosenbaum et al. 2007; Amato et al. 2008). Although each mechanism may have an important role in particular species, or turtles in general, none appear to account for the observed data. Blanding’s Turtle does have a relatively long generation time (estimated at 37 years, Congdon et al. 1993), but it is comparable to Wood Turtle (36–47 years, van Dijk and Harding 2011), a species with an order of magnitude higher level of cytb polymorphism (Table 3). The divergence time hypothesis suggests evolutionary rates may not be slow but rather that recently evolved species have not had sufficient time to develop sequence polymorphism. Although estimates of divergence dates within Emys encompass a wide range (9–35 mybp), the most extensive phylogenetic analysis of the group to date provides no evidence that E. blandingii has a much more recent origin than any of its congeners (Spinks et al. 2016). Finally, the selective sweep hypothesis is often invoked to explain low levels of mitochondrial polymorphism since directional selection on a particular locus will lead to correlated evolution across the mitochondrial genome due to linkage. Such selection is not expected to impact variation in unlinked nuDNA or microsatellite loci. The broadly concordant population structure we observe in range-wide analyses among the three sets of loci (discussed below) is more consistent with neutral evolutionary processes rather than selection acting in parallel in two or more lineages. Clearly, characterization of many more loci is needed to resolve questions surrounding the low genetic variation in the species and develop a more detailed understanding of population history (c.f. Spinks et al. 2014).
Phylogeographic structure
Despite the limited polymorphism we observed, the presence of two genetic groups with a likely boundary located in eastern Ontario was supported by both sets of loci (Table 2; Fig. 3). This pattern generally corresponds with the structure found by Mockford et al. (2007) but with some notable differences that likely contribute to the incomplete sorting of haplotypes (Fig. 2) and the better performance of the SAMOVA analysis. Mockford et al. (2007) identified a primary boundary through eastern Ontario and northern New York, and a secondary boundary within the eastern region that divided Massachusetts and Nova Scotia populations from Ontario and New York populations. Our mitochondrial dataset supported the presence of the first boundary but did not recover separation within the eastern region. Additionally, mtDNA separated the Nebraska locality and grouped Parry Sound, Ontario near Lake Huron (not sampled by Mockford) with midwestern US populations. Nuclear data led to the identification of a boundary in the eastern part of the range but phylogeographic resolution was weaker (Table 2) with the SAMOVA analysis and the interpolation of interindividual distances differing in the position of the putative boundary (Nova Scotia singly isolated, or Nova Scotia clustered with Jefferson, New York). Overall, our results point to the Appalachian Mountains as a fundamental barrier within the geographic range, with possible colonization to eastern Ontario of eastern lineages through the Hudson River valley (Mockford et al. 2007).
Two explanations have been proposed for the history of E. blandingii populations that could explain the observed phylogeographic structure. In one scenario (Schmidt 1938; Smith 1957), climate warming following glacial retreat during the Hypsithermal (9000–4000 ybp) led to the expansion of prairie habitat eastward into the Atlantic region. Blanding’s Turtle populations are thought to have followed this expansion only to retreat later as the climate cooled, leaving relictual populations in the eastern part of the geographic range. An alternative explanation is that refugia may have occurred both along the Atlantic coastal plain and in the southern Great Plains regions during glaciations (Bleakney 1958a, b), therefore suggesting a periodic history of isolation. Blanding’s Turtle fossils occur predominantly in the Great Plains and Midwest, dating as early as the late Hemiphillian (5 mybp), (Parmley 1992; Holman 1995), but there is one specimen from eastern Pennsylvania from the late Irvingtonian (0.75 mybp) (Parris and Daeschler 1995). The possibility of an Atlantic refuge is supported by this fossil observation and is concordant with molecular lineages found within Chrysemys picta (Starkey et al. 2003) and G. insculpta (Amato et al. 2008). The slow rate of molecular evolution in turtles in general (Avise et al. 1992; Shaffer et al. 2013), combined with these observations, suggests to us that the population structure represents ancient polymorphism from an earlier history of divergence. An effort to model the number and distribution of past refugia, based on ecological niche models and past climate, for comparison with genetic patterns (e.g. Reid et al. 2018) would be useful in further resolving the history of the species.
The broad pattern of phylogeographic structure that we observed provides an interesting contrast with the consistent population genetic clustering found in microsatellite analyses conducted at smaller geographic scales (Mockford et al. 2005; Davy et al. 2014; Sethuraman et al. 2014; McCluskey et al. 2016; Anthonysamy et al. 2017; Reid et al. 2017). It is clear from these studies that the addition of more microsatellite loci increases the power to resolve finer genetic differences among populations (pairwise FST < 0.2). This differentiation has been linked most often to the history of post-glacial dispersal and the boundaries of large watersheds (Davy et al. 2014; Sethuraman et al. 2014; McCluskey et al. 2016), but recent work has begun to find subtle structure related to fragmentation in contemporary landscapes (Anthonysamy et al. 2017; Reid et al. 2017). The levels of genetic variation in microsatellite loci found in these studies is also moderately high, an observation that is likely due to the maintenance of alleles during bottlenecks in long-lived species with overlapping generations (Kuo and Janzen 2004; Willoughby et al. 2013; Davy et al. 2014) and the relatively high mutation rate of these markers. We therefore suggest that the contrast between sequence and microsatellite datasets is likely to be one of temporal scale, where the latter is more appropriate for detecting processes operating more recently within and among populations (Wang 2010, Epps and Keyghobadi 2015).
Management for genetic populations
The conditions that have made Blanding’s Turtle populations at risk of continued decline persist. Genetic analyses are fundamental to determining which populations are at the greatest risk, how habitat connectivity can lead to gene flow, and where individuals might be drawn from when population augmentation becomes necessary. The observation that contemporary gene flow is nearly non-existent among Blanding’s Turtle populations in most regions (Davy et al. 2014; Sethuraman et al. 2014; Anthonysamy et al. 2017; Reid et al. 2017) and apparent inbreeding depression (Sethuraman et al. 2014) makes the possibility of such interventions more acute.
Our generally concordant results between mitochondrial and nuclear loci are consistent with the range-wide differentiation described by Mockford et al. (2007). Management at the scale of the geographic range should seek to maintain populations representing this diversity while also accounting for finer scale differentiation that has been found in the regional analyses discussed above. The synthesis of information derived from phylogeographic and landscape genetic analyses provides an opportunity to conduct such management more effectively (Epps and Keyghobadi 2015). For example, conservation efforts where the two putative lineages meet in Ontario and New York (Fig. 3) should be considered carefully given that neighboring populations near the contact zone are expected to have a longer history of divergence than that found within groups. Although adaptive variation across the range has not been investigated in the species, translocations across this boundary could elevate the risk of outbreeding depression resulting from disruption of co-adapted gene complexes developed over longer time. Additionally, landscape genetic analyses that identify habitat connectivity associated with gene flow can benefit from controlling for such phylogeographic breaks. This overall approach can be adopted within the eastern and western portions of the range to identify more recent, but significant, histories of divergence to more effectively promote population connectivity and resilience through habitat restoration and translocation.
References
Amato ML, Brooks RJ, Fu J (2008) A phylogeographic analysis of populations of the wood turtle (Glyptemys insculpta) throughout its range. Mol Ecol 17:570–581
Anthonysamy WJB, Dreslik MJ, Douglas MR, Thompson D, Klut GM, Kuhns AR, Mauger D, Kirk D, Glowacki GA, Douglas ME, Phillips CA (2017) Population genetic evaluations within a co-distributed taxonomic group: a multi-species approach to conservation planning. Anim Conserv 21:137–147
Avise JC, Bowen BW, Lamb T, Meylan AB, Bermingham E (1992) Mitochondrial DNA evolution at a turtle’s pace: evidence for low genetic variability and reduced microevolutionary rate in the Testudines. Mol Biol Evol 9:457–473
Bleakney JS (1958a) A zoogeographical study of the amphibians and reptiles of eastern Canada. Natl Mus Can Bull 155:1–119
Bleakney S (1958b) Postglacial dispersal of the turtle Chrysemys picta. Herpetologica 14:101–104
Bryant D, Moulton V (2004) Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21:255–265
Congdon JD, Dunham AE, van Loben Sels R (1993) Delayed sexual maturity and demographics of Blanding’s Turtles (Emydoidea blandingii): implications for conservation and management of long-lived organisms. Conserv Biol 7:826–833
Congdon JD, Graham TE, Herman TB, Lang JW, Pappas MJ, Brecke BJ (2008) Emydoidea blandingii (Holbrook 1838) - Blanding’s Turtle. In: Rhodin AGJ, Pritchard PCH, van Dijk PP, Saumure RA, Buhlmann KA, Iverson JB (eds) Conservation biology of freshwater turtles and tortoises: a compilation project of the IUCN/SSC Tortoise and Freshwater Turtle Specialist Group Chelonian Research Monographs No 5. p.^pp. 015.011–015.012
Davy CM, Bernardo PH, Murphy RW (2014) A Bayesian approach to conservation genetics of Blanding’s turtle (Emys blandingii) in Ontario. Canada Conserv Gen 15:319–330
Dellicour S, Mardulyn P (2014) SPADS 1.0: a toolbox to perform spatial analyses on DNA sequence data sets. Mol Ecol Res 14:647–651
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Epps CW, Keyghobadi N (2015) Landscape genetics in a changing world: disentangling historical and contemporary influences and inferring change. Mol Ecol 24:6021–6040
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochonridal DNA. Genetics 131:479–491
Fritz U, Guicking D, Kami H, Arakelyan M, Auer M, Ayaz D, Fernández CA, Bakiev AG, Celani A, Džukić G (2007) Mitochondrial phylogeography of European pond turtles (Emys orbicularis, Emys trinacris)–an update. Amphib-Reptilia 28:418–426
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Garrick RC, Sunnucks P, Dyer RJ (2010) Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation. BMC Evol Biol 10:118
Hardy OJ, Vekemans X (2002) SPAGeDI: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Holman JA (1995) Pleistocene amphibians and reptiles in North America. Oxford University Press, New York
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
Joly S, Bruneau A (2006) Incorporating allelic variation for reconstructing the evolutionary history of organisms from multiple genes: an example from Rosa in North America. Syst Biol 55:623–636
Karl SA, Toonen R, Grant W, Bowen B (2012) Common misconceptions in molecular ecology: echoes of the modern synthesis. Mol Ecol 21:4171–4189
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kuo C-H, Janzen FJ (2004) Genetic effects of a persistent bottleneck on a natural population of ornate box turtles (Terrapene ornata). Conserv Gen 5:425–437
Lenk P, Fritz U, Joger U, Wink M (1999) Mitochondrial phylogeography of the European pond turtle, Emys orbicularis (Linnaeus 1758). Mol Ecol 8:1911–1922
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol 76:173–190
McCluskey EM, Mockford SW, Sands K, Herman TB, Johnson G, Gonser RA (2016) Population genetic structure of Blanding’s Turtles (Emydoidea blandingii) in New York. J Herpetol 50:70–76
Miller MP (2005) Alleles In Space (AIS): computer software for the joint analysis of interindividual spatial and genetic information. J Hered 96:722–724
Miller MP, Bellinger MR, Forsman ED, Haig SM (2006) Effects of historical climate change, habitat connectivity, and vicariance on genetic structure and diversity across the range of the red tree vole (Phenacomys longicaudus) in the Pacific Northwestern United States. Mol Ecol 15:145–159
Mockford S, McEachern L, Herman T, Snyder M, Wright JM (2005) Population genetic structure of a disjunct population of Blanding’s turtle (Emydoidea blandingii) in Nova Scotia, Canada. Biol Conserv 123:373–380
Mockford SW, Herman TB, Snyder M, Wright JM (2007) Conservation genetics of Blanding’s turtle and its appliction in the identification of evolutionary significant units. Conserv Gen 8:209–219
Parmley D (1992) Turtles from the late Hemphillian (latest Miocene) of Knox County, Nebraska. Tex J Sci 44:339–348
Parris DC, Daeschler E (1995) Pleistocene turtles of Port Kennedy cave (late Irvingtonian), Montgomery County, Pennsylvania. J Paleontol 69:563–568
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Ramírez-Soriano A, Ramos-Onsins SE, Rozas J, Calafell F, Navarro A (2008) Statistical power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics 179:555–567
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Reid BN, Mladenoff DJ, Peery MZ (2017) Genetic effects of landscape, habitat preference and demography on three co-occurring turtle species. Mol Ecol 26:781–798
Reid BN, Kass JM, Wollney S, Jensen EL, Russello MA, Viola EM, Pantophlet J, Iverson JB, Peery MZ, Raxworthy CJ, Naro-Maciel E (2018) Disentangling the genetic effects of refugial isolation and range expansion in a trans-continentally distributed species. Heredity. https://doi.org/10.1038/s41437-018-0135-5
Rödder D, Lawing AM, Flecks M, Ahmadzadeh F, Dambach J, Engler JO, Habel JC, Hartmann T, Hörnes D, Ihlow F (2013) Evaluating the significance of paleophylogeographic species distribution models in reconstructing Quaternary range-shifts of Nearctic chelonians. PLoS ONE, 8:e72855
Rosenbaum PA, Robertson JM, Zamudio KR (2007) Unexpectedly low genetic divergences among populations of the threatened bog turtle (Glyptemys muhlenbergii). Conserv Gen 8:331–342
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Schmidt KP (1938) Herpetological evidence for the postglacial eastward expansion of the steppe in North America. Ecology 19:396–407
Sethuraman A, McGaugh SE, Becker ML, Chandler CH, Christiansen JL, Hayden S, LeClere A, Monson-Miller J, Myers EM, Paitz RT, Refsnider JM, VanDeWalle TJ, Janzen FJ (2014) Population genetics of Blanding’s turtle (Emys blandingii) in the midwestern United States. Conserv Gen 15:61–73
Shaffer H, Minx P, Warren D, Shedlock A, Thomson R, Valenzuela N, Abramyan J, Amemiya C, Badenhorst D, Biggar K, Borchert G, Botka C, Bowden R, Braun E, Bronikowski A, Bruneau B, Buck L, Capel B, Castoe T, Czerwinski M, Delehaunty K, Edwards S, Fronick C, Fujita M, Fulton L, Graves T, Green R, Haerty W, Hariharan R, Hernandez O, Hillier L, Holloway A, Janes D, Janzen F, Kandoth C, Kong L, de Koning A, Li Y, Literman R, McGaugh S, Mork L, O’Laughlin M, Paitz R, Pollock D, Ponting C, Radhakrishnan S, Raney B, Richman J, St John J, Schwartz T, Sethuraman A, Spinks P, Storey K, Thane N, Vinar T, Zimmerman L, Warren W, Mardis E, Wilson R (2013) The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol 14:R28
Smith PW (1957) An analysis of post-Wisconin biogeography of the prairie peninsula region based on distributional phenomena among terrestrial vertebrate populations. Ecology 38:205–218
Spinks PQ, Shaffer HB (2005) Range-wide molecular analysis of the western pond turtle (Emys marmorata): cryptic variation, isolation by distance, and their conservation implications. Mol Ecol 14:2047–2064
Spinks PQ, Shaffer HB (2009) Conflicting mitochondrial and nuclear phylogenies for the widely disjunct Emys (Testudines: Emydidae) species complex, and what they tell us about biogeography and hybridization. Syst Biol 58:1–20
Spinks PQ, Thomson RC, Shaffer HB (2010) Nuclear gene phylogeography reveals the historical legacy of an ancient inland sea on lineages of the western pond turtle, Emys marmorata in California. Mol Ecol 19:542–556
Spinks PQ, Thomson RC, Shaffer HB (2014) The advantages of going large: genome-wide SNPs clarify the complex population history and systematics of the threatened western pond turtle. Mol Ecol 23:2228–2241
Spinks PQ, Thomson RC, McCartney-Melstad E, Shaffer HB (2016) Phylogeny and temporal diversification of the New World pond turtles (Emydidae). Mol Phylogenet Evol 103:85–97
Starkey DE, Shaffer HB, Burke RL, Forstner MRJ, Iverson JB, Janzen FJ, Rhodin AGJ, Ultsch GR (2003) Molecular systematics, phylogeography, and the effects of Pleistocene glaciation in the painted turtle (Chrysemys picta) complex. Evolution 57:119–128
Stephens M, Donnelly P (2003) A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Team QD (2009) QGIS Geographic Information System. Open Source Geospatial Foundation, Chicago
Toews DP, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Vamberger M, Stuckas H, Sacco F, D’Angelo S, Arculeo M, Cheylan M, Corti C, Lo Valvo M, Marrone F, Wink M (2015) Differences in gene flow in a twofold secondary contact zone of pond turtles in southern Italy (Testudines: Emydidae: Emys orbicularis galloitalica. E. o. hellenica, E. trinacris). Zool Scr 44:233–249
van Dijk PP, Harding J (2011) Glyptemys insculpta (errata version published in 2016), The IUCN Red List of Threatened Species. https://dx.doi.org/10.2305/IUCN.UK.2011-1.RLTS.T4965A11102820.en. Accessed 18 April 2018
Velo-Antón G, García-París M, Rivera AC (2008) Patterns of nuclear and mitochondrial DNA variation in Iberian populations of Emys orbicularis (Emydidae): conservation implications. Conserv Gen 9:1263–1274
Wang IJ (2010) Recognizing the temporal distinctions between landscape genetics and phylogeography. Mol Ecol 19:2605–2608
Weeks AR, Sgro CM, Young AG, Frankham R, Mitchell NJ, Miller KA, Byrne M, Coates DJ, Eldridge MD, Sunnucks P (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725
Weisrock DW, Janzen FJ (2000) Comparative molecular phylogeography of North American softshell turtles (Apalone): implications for regional and wide-scale historical evolutionary forces. Mol Phylogenet Evol 14:152–164
Willoughby JR, Sundaram M, Lewis TL, Swanson BJ (2013) Population decline in a long-lived species: the wood turtle in Michigan. Herpetologica 69:186–198
Zink RM, Barrowclough GF (2008) Mitochondrial DNA under siege in avian phylogeography. Mol Ecol 17:2107–2121
Acknowledgements
This research was supported by the Indiana Academy of Science and the IPFW Honors Program. We thank Christina Davy, Mike Finkler, Rusty Gonser, Dave Mifsud, Brendan Reid, and Sasha Tetzlaff for sharing samples with us. The Field Museum and the Royal Ontario Museum were helpful in providing additional samples. We also appreciate the insights shared by Phil Spinks on choosing polymorphic nuclear loci.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jordan, M.A., Mumaw, V., Millspaw, N. et al. Range-wide phylogeography of Blanding’s Turtle [Emys (= Emydoidea) blandingii]. Conserv Genet 20, 419–430 (2019). https://doi.org/10.1007/s10592-018-01140-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-018-01140-6