
Abstract
Blanding’s turtle, Emys blandingii, is a globally endangered species with a range centred on the Great Lakes of North America. Several disjunct populations also occur along the East Coast of North America. Previous studies suggest that the Great Lakes portion of the species’ range exhibits panmixia. However, E. blandingii is restricted to relatively small populations in many areas around the Great Lakes. Therefore, panmixia across large geographic distances in this area is unlikely. Here, we apply Bayesian analyses of population structure to samples collected across southern Ontario (N = 97) to test a null hypothesis of panmixia and assess possible management units (MUs), and to estimate rates of gene flow across the study area. Sampled sites in Ontario represent a minimum of four distinct genetic clusters of E. blandingii, which we recommend should be considered as independent MUs. Preliminary evidence suggests that further structure may be present in less robustly sampled areas, which deserve further consideration. Genetic diversity at sampled sites is comparable to that reported for other freshwater turtles. Our comparison between this study and previous work confirms that genetic diversity in E. blandingii is reduced in disjunct eastern populations compared to populations centred on the Great Lakes. Genetic diversity in E. blandingii is not correlated with latitude, and instead may reflect post-glacial dispersal of this species from multiple Pleistocene glacial refugia.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Blanding’s turtle (Emys blandingii, Holbrook 1938) is a moderately sized freshwater species found in the north-eastern United States and southern Canada. The main portion of its range is centred on the Great Lakes region. Small, disjunct populations occur in New York, Massachusetts, and Nova Scotia (Fig. 1; Ernst and Lovich 2009). Mean age of maturity in a well-studied Michigan population is 17.5 years, generation time is ~37 years, and longevity exceeds 75 years (Congdon and van Loben Sels 1991; Congdon et al. 1993; Brecke and Moriarty 1989). One consequence of this life history is that populations are sensitive to any increase in the mortality rate of reproductive adults (Congdon et al. 1993), and even small increases in adult mortality (or collection of adults) can cause significant population declines. A number of factors including road mortality, illegal collection, and habitat degradation are currently causing such declines (COSEWIC 2005). Therefore, E. blandingii was recently up-listed from Least Concern to Endangered by the International Union for Conservation of Nature (IUCN; van Dijk and Rhodin 2011).

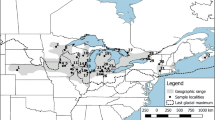
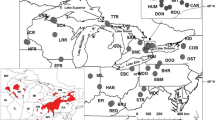
Approximate location of collection areas for E. blandingii sampled across southern Ontario. Black rectangle in top right inset indicates location of study area, and red indicates the species’ range in North America. Sampling focused on sites indicated with green squares: LE Lake Erie, GH Golden Horseshoe, PSD Parry Sound District, KAW Kawartha Lakes, EO Eastern Ontario. Sample sizes are included in each site marker. Open triangles indicate extra samples included opportunistically (each triangle represents an individual turtle): LHsouth south shore of Lake Huron, LHnorth north shore of Lake Huron, ALG Algonquin Provincial Park. Variation in sample sizes results from differential sampling effort; differences in sample sizes is not reflective of variation in actual population sizes. (Color figure online)
Using random amplified polymorphic DNA (RAPD) markers and microsatellites, Mockford et al. (1999, 2005, 2007) and Rubin et al. (2001) quantified genetic variation in E. blandingii across the species’ range. Band-sharing analyses of RAPD data showed that the disjunct Nova Scotian population differed genetically from central populations (Mockford et al. 1999; Rubin et al. 2001). Within Nova Scotia, analyses of microsatellite data based on FST values suggested significant differentiation among three subpopulations despite separation by <30 km. However, very little population structure was detected in the main portion of the range based on samples from Minnesota, Wisconsin, Illinois, Michigan, and Ontario (Mockford et al. 2007). Based on these data, Mockford et al. (2007) proposed that E. blandingii comprised three evolutionarily significant units (ESUs): (1) the population in Nova Scotia; (2) isolated populations in Massachusetts and New York; and (3) populations centred on the Great Lakes. The ESU concept does not apply to the legal conservation of turtles in either the USA or Canada, where protection is based on the concepts of distinctive population segments (Waples 1991; Rosen 2007) and designatable units (DUs; Green 2005), respectively. In Canada, the Committee on the Status of Endangered Wildlife in Canada (COSEWIC) recognizes E. blandingii populations in Nova Scotia and around the Great Lakes as two DUs.
Recent studies highlight concerns with determining population structure based on FST. For example, Jost (2008) demonstrated that FST and related measures of diversity do not necessarily measure actual population differentiation. He proposed an alternative, more accurate measure (Dest). Jost (2008) also pointed out that statistical significance of FST was primarily a factor of sample size and may be biologically meaningless. Howes et al. (2009) summarized further concerns with analyses based on FST, including that assumptions of these analyses often were not met in natural populations (Whitlock and McCaughley 1999) and that FST does not reflect contemporary gene flow (Paetkau et al. 2004). FST and related measurements can provide information about historical migration rates, but they are not appropriate measures of population differentiation or structure (Jost 2009).
Howes et al. (2009) applied Bayesian methods that do not rely on FST to the data of Mockford et al. (2005), to study population connectivity in E. blandingii in Nova Scotia. They demonstrated moderate historic and current gene flow among the three known subpopulations, and clustered the two nearest subpopulations together as a genetically continuous unit. Bayesian analysis of three E. blandingii populations separated by <60 km in Illinois also detected no evidence of population structure (Banning-Anthonysamy 2012). Despite these results, panmixia across the main portion of the species’ range is unlikely due to potential dispersal barriers such as long, high escarpments, or large, upland areas containing only unsuitable habitat. E. blandingii is dependent on wetland habitats but has high dispersal ability, and individuals may travel >10 km overland between wetlands or to find a suitable nest site (Power 1989). Thus, population structure in this species may occur on a relatively large geographic scale (>100 km).
Although COSEWIC currently considers the “Great Lakes/St. Lawrence population,” comprised of all E. blandingii in Ontario and Quebec, as a single unit for management and recovery purposes (COSEWIC 2005), the hypothesis of panmixia in E. blandingii in Ontario has not been tested. A large portion of the core range of E. blandingii occurs in Ontario, but previous studies included only 11 samples from St. Lawrence Islands National Park (Mockford et al. 2007), in south-eastern Ontario on the U.S./Canadian border. Presence-absence data show that the distribution of E. blandingii in Ontario is not continuous (Ontario Nature Herpetofaunal Atlas, http://www.ontarionature.org/protect/species/reptiles_and_amphibians/map_blandings_turtle.html). Gaps in occurrence records may reflect historic or current barriers to gene flow, and further suggest that populations may not be panmictic across the province. Isolated, single records of occurrence may also represent individuals translocated from other populations, or released illegal pets.
Emys blandingii reaches the northern limit of its range in Ontario. Founder effects during recolonization of northern areas following the retreat of ice sheets can cause populations reaching their northern limits in recently glaciated areas to exhibit lower genetic diversity than more southern populations (e.g. Galbraith 2008). As a result, Ontario populations of E. blandingii may show lower genetic variation than populations at lower latitudes. Mockford et al. (2007) documented reduced genetic diversity in isolated eastern populations of E. blandingii in Nova Scotia, presumably also caused by founder effects and subsequent isolation during the last Ice Age.
Here, we apply three Bayesian analyses and a principal coordinates analysis to investigate population structure in E. blandingii across >500 km in southern Ontario. We investigate the level of population structure and genetic diversity at sampled sites to test the hypothesis of panmixia in Ontario populations of E. blandingii. We also compare genetic variation (heterozygosity) among populations in Ontario and the populations studied by Mockford et al. (2007) to test two hypotheses: (a) that variation will be lower in disjunct eastern populations than in populations around the Great Lakes, as suggested by Mockford et al. (2005); and (b) that variation will decrease with proximity to the northern limit of the species’ range.
Methods
We collected DNA from E. blandingii across southern Ontario between 2008 and 2011, with additional samples contributed by other researchers and government biologists (Fig. 1). Exact locations are withheld at the request of the Ontario Ministry of Natural Resources, to prevent an increase in collection pressure on populations. Turtles were captured by hand or in hoop traps baited with sardines at sites Lake Erie (LE), Golden Horseshoe (GH), Eastern Ontario (EO), Algonquin Park (ALG), Lake Huron South (LHsouth) and Lake Huron North (LHnorth). Blood was sampled by caudal venipuncture with a sterile syringe and blotted onto FTA cards (Whatman Inc., Clifton, NJ) for storage. Sampled individuals were released at their initial capture site. Extraction of DNA from FTA cards followed Smith and Burgoyne (2004).
In Parry Sound District (site PSD), muscle tissue was collected from road-killed individuals. At sites near the Kawartha Lakes (KAW), blood samples were taken from turtles injured on local highways and rehabilitated at the Kawartha Turtle Trauma Centre (Peterborough, Ontario). These blood samples were stored in heparin before analysis. Extraction of DNA from muscle and heparinised blood followed the phenol–chloroform procedure of Sambrook et al. (1989) and extracted DNA was cleaned with EtOH precipitation. Four additional blood samples were collected from captive E. blandingii at Scales Nature Park (Orillia, ON) that were from Ontario, but whose exact locations of origin were unknown.
We amplified samples at four microsatellite loci developed for E. blandingii (Eb09, Eb11, Eb17 and Eb19; Osentoski et al. 2002). These loci were used by Mockford et al. (2007) and we included them to facilitate direct comparison of within-population genetic diversity between studies. We amplified an additional 13 loci from Glyptemys muhlenbergii that cross-amplified in E. blandingii (GmuB08, GmuD16, GmuD21, GmuD28, GmuD55, GmuD70, GmuD87, GmuD88, GmuD89, GmuD90, GmuD93, GmuD107 and GmuD121; King and Julian 2004). Amplification and allele scoring followed Davy et al. (2012), using the locus-specific annealing temperatures listed in Table 1. Genotyping error was assessed by including positive controls with each PCR reaction and re-amplifying ~6 % of the samples.
Evidence for null alleles and long allele drop-out was assessed with Micro-Checker (van Oosterhout et al. 2004) using 1,000 iterations. Frequency of null alleles was calculated with the method of Brookfield (1996). We calculated the number of alleles per locus, observed heterozygosity (HO) and expected heterozygosity (HE) in Genalex (Peakall and Smouse 2006). Allelic richness was rarefacted to correct for unequal sample sizes in Hp-Rare (Kalinowski 2004, 2005). Linkage disequilibrium and deviations from Hardy–Weinberg equilibrium (HWE) were tested in Genepop v.4.0.1 (Raymond and Rousset 1995; Rousset 2008). Significance levels were corrected for multiple comparisons following Rice (1989).
We assessed genetic differentiation among sample sites by calculating absolute differentiation (Dest, Jost 2008) of all sites with N ≥ 15 in smogd (Crawford 2010). For purposes of comparison with previous studies we also used fstat (Goudet 1995) to calculate pairwise FST and assessed significance with 10,000 randomizations. Isolation by distance (IBD, a significant correlation between geographic and genetic distance, Wright 1943) was assessed using ibdws (Jensen et al. 2005) with an input matrix of Dest and pairwise distances (km) between sites with N ≥ 15. We estimated average historical number of migrants per generation between populations (Nm) based on FST following Barton and Slatkin (1986). We also estimated recent directional historic migration rates (m) between pairs of sampled sites using a Bayesian approach in bayesass v.1.3 (Wilson and Rannala 2003), considering this an estimate of migration rates across approximately the past 1–5 generations, or 185 years, following Chiucchi and Gibbs (2010). bayesass analysis involved 106 generations with a burn-in of 10 %, sampling the chain every 1,000 generations. We considered migration to be significantly directional if the 95 % confidence intervals of the estimates for each direction did not overlap, following Howes et al. (2009).
Within-population heterozygosity (HO) from Ontario sites was compared to HO values reported in Mockford et al. (2007) using an independent-samples t test in SPSS v.20.0 (IBM-SPSS, Chicago, IL, USA) after testing normality of the data. Comparisons were made for each of the two loci successfully amplified in both studies, and across all sampled loci. We compared heterozygosity between sampled sites in Ontario and the “western” sites from Mockford et al. (2007); (all sampled sites west of the Appalachian Mountains). We also compared sites east of the Appalachian Mountains to western populations, combining study sites from Ontario with western sites from Mockford et al. (2007). Pearson’s correlation coefficient was used to test for significant relationships between latitude and HO among sites surrounding the Great Lakes.
Population structure was assessed by Bayesian inference in structure v.2.3.4 (Pritchard et al. 2000) and tess v.2.3.1 (Chen et al. 2007). structure considered possible K values (number of genetically distinct populations) from one to six with eight independent runs at each value of K. We used the LOCPRIOR function to consider the sample site of each individual in the analysis. Each run involved 750,000 generations with a burn-in of 75,000 generations. The model assumed correlated allele frequencies (Falush et al. 2003) and historical admixture between populations (Pritchard et al. 2000).
We compiled the output of the structure runs with structure harvester v.0.6.92 (Earl and vonHoldt 2012) and used two methods to estimate K, the most probable number of genetically distinct populations represented in the data. The increase in pr(X|K), the probability of the data given a particular value of K, typically plateaus at the most likely value of K (Pritchard et al. 2000). The ad hoc ΔK method (Evanno et al. 2005) implemented in structure harvester was also used to calculate the second-order rate of change in log likelihood between successive values of K, which typically peaks at the appropriate value of K. We used the Greedy and LargeKGreedy algorithms in clumpp v.1.1.2 (Jakobsson and Rosenberg 2007) to permute and combine results from independent runs. Genetic clusters identified by structure were visualized with distruct v.1.1 (Rosenberg 2004).
tess analyses assumed an admixture model (Durand et al. 2009) and included the specific geographic location (GPS coordinates) of each sampled individual. tess considered possible Kmax values (maximum possible number of populations represented by the data) from two to eight, with ten independent runs at each Kmax. Runs included 50,000 sweeps with a burn-in of 10,000 sweeps. Data were assessed to ensure convergence and individual assignments were visualized with clumpp and distruct. We considered the most likely K to occur at the point of inflection of the decreasing deviance information criterion (DIC) values and at which the number of distinct clusters stabilized (Chen et al. 2007).
Assignment tests were conducted in Geneclass v.2.0 (Piry et al. 2004) using the Bayesian method of Rannala and Mountain (1997), with 100,000 iterations and a Type I error level of 0.05. This duplicates the analyses conducted by Howes et al. (2009), allowing a reasonable level of comparison between studies. Assignment tests considered only sampling areas with six or more samples. Individual samples from other sites and samples of unknown origin were then assessed by the program as “unknown”, and assigned to the most similar sampling area. Population structure was also visualized with a principal coordinate analysis (PCoA) in Genalex, based on Dest for sampled sites and on Nei’s unbiased genetic distance for individuals.
Results
Loci Eb09, Eb11, GmuD70, GmuD89, and GmuD90 either did not amplify, or could not be scored consistently despite multiple adjustments of PCR conditions. Thus, 12 loci were used for analyses. In total, 116 samples were collected but several yielded degraded DNA and were successfully amplified at only 5 or 6 loci. These samples were excluded and a total of 97 individuals (91 individuals from known locations) were genotyped at >10 loci and included in the final analysis.
Duplicate amplification of positive controls yielded identical genotypes, and the influence of genotyping error on our dataset was therefore considered negligible. micro-checker found evidence for potential null alleles at three loci (Eb19, GmuD93 and GmuD107). However, when the four largest samples were tested independently, the results were inconsistent among sites; only EO and GH showed evidence for potential null alleles, and only at locus Eb19.
Deviations from HWE were detected at locus Eb19 in PSD, GH, and EO, but not in KAW or LE. Evidence for LD was detected across the entire dataset between two pairs of loci: GmuD55–GmuD107 and GmuD28–GmuD107. However, LD was not detected when testing sampled areas independently and we therefore accepted the null hypothesis of linkage equilibrium. PI and PIsibs decreased to <0.01 with the inclusion of three and six loci, respectively. The 12 loci exhibited 3–16 alleles (mean 8.917, SD = 4.187). Observed heterozygosity (HO) ranged from 0.253 at locus GmuD21 to 0.845 at locus GmuD28 (Table 1).
Summary statistics for all sampling sites are shown in Table 2. Pairwise values of Dest ranged from 0.010 to 0.156 (mean = 0.083, SD = 0.044, Table 3). Values of FST ranged from 0.039 to 0.099 (mean 0.072, SD = 0.021). Based on FST, Nm among sites averaged 2.432, and Nm between each pair of sites ranged from 0.095 (GH–PSD) to 3.380 (PSD–EO; Table 3). Recent gene flow (m) among sites estimated with bayesass averaged 0.0363. Pairwise m ranged from 0.011 (EO to KAW) to 0.084 (KAW to PSD; Table 3). Only one pair of sites (KAW and PSD) had significantly asymmetrical estimates of gene flow, inferred from non-overlapping confidence intervals: estimated migration from KAW to PSD over 14 times greater than migration from PSD to KAW. No significant IBD was detected among the four sites with N > 12 (Z = 194.900, r = 0.233, p = 0.301).
The first axis of the PCoA of sampling sites accounted for 60.19 % of total variation. This axis separated sites LE and GH from PSD, KAW, and EO (Fig. 2). When the PCoA was conducted at the individual level, individuals clustered by site. Overlap between the edges of these clusters indicated that differentiation in this dataset may be occurring along clines rather than along sharply defined boundaries.

Principal coordinates analysis (PCoA) for 91 E. blandingii sampled from across southern Ontario based on 12 microsatellite loci). PCoA of sampling areas (a, b) supports the population structure inferred by Bayesian clustering analyses (Fig. 3). PCoA of individuals (c) shows that individuals largely cluster by sampling site (inferred population) in coordinate space
Geneclass assigned individuals from LE, GH, PSD, KAW, and EO to their area of origin with 69 % accuracy (Table 4). Samples from KAW were assigned to PSD (N = 5) or GH (N = 1). When KAW was removed from assignment tests, overall accuracy increased to 79 %. The two samples from the north shore of Lake Huron were assigned to PSD. The two samples from ALG and the sample from the south shore of Lake Huron were not assigned to any sampled clusters (p < 0.01).
The DIC in the tess analysis decreased gradually from Kmax = 2 with no clear point of inflection (mean ΔDIC = 54.2, Fig. 3a). Individual q-matrices stabilized at Kmax = 2. No clearly defined new clusters appeared at higher values of Kmax, although potential admixture from a third population became apparent in site EO at Kmax = 3. The first resolved population included LE, GH, and LHsouth (mean q = 0.969, SD = 0.094). The second population included all other samples (mean q = 0.709, SD = 0.377). One sample from PSD was assigned with approximately equal probability to both populations (0.493 vs. 0.507).

Population structure inferred by Bayesian inference for 91 E. blandingii collected across southern Ontario. a Tess results showing decreasing deviance information criterion (DIC) with increasing values of Kmax. b Structure results, mean estimated ln probability of the data [L(K)] for increasing values of K, and ΔK, the second order rate of change of L(K) following Evanno et al. (2005). Site abbreviations are explained in Fig. 1
structure resolved the same two populations as tess at K = 2 (Fig. 3b). At K = 3, LE and LHsouth (mean q = 0.939, SD = 0.049) separated from population GH (mean q = 0.821, SD = 0.160) with evidence of admixture remaining between the two clusters. At K = 4, EO (mean q = 0.832, SD = 0.1) separated from a final population consisting of PSD, KAW, LHnorth and ALG (mean q = 0.776, SD = 0.071).
Heterozygosity data were normally distributed and Levene’s test indicated equal variances (F = 0.09, p = 0.927). Data from the two loci used both in this study and by Mockford et al. (2007); (Eb17 and Eb19) were combined for comparison. Observed heterozygosity in the Great Lakes portion of the species’ range was significantly higher at locus Eb17 (t = −3.621, d.f. = 15, p = 0.003) but not at locus Eb19 (t = −1.823, d.f. = 15, p = 0.088). When mean heterozygosity across all loci used in both studies was compared, HO was significantly higher in western populations (t = −3.749, d.f. = 15, p = 0.002) than in the disjunct eastern populations. No difference in heterozygosity occurred between the western populations sampled by Mockford et al. (2007) and the populations sampled in this study (t = −0.413, df = 10, p = 0.688). Latitude and HO/HE were not correlated (Pearson’s correlation = 0.056, N = 11, p = 0.869).
Discussion
Our analyses identify a minimum of two genetic populations and four subpopulations of E. blandingii in southern Ontario, and refute the hypothesis of panmixia. Intensive urban development and expanding road networks make current migration between these four subpopulations nearly impossible, but our results indicate that historic migration rates were also low enough for genetic drift to cause differentiation among populations. As a result, assignment tests can identify individuals to their area of origin with relatively high accuracy, especially considering the small sample sizes available for this study. Diversity does not decrease with latitude in this species. Our data further support the finding of Mockford et al. (2007) that the isolated populations of E. blandingii east of the Appalachian Mountains show reduced diversity relative to western populations.
Management units (MUs) are typically delimited based on both genetic and demographic data (Moritz 1994; Palsbøll et al. 2006). We propose four tentative MUs for E. blandingii in Ontario: LE, GH, Georgian Bay-Parry Sound District, and EO. structure analyses and assignment tests show that these units are genetically differentiated, and distribution data (Ontario Nature Reptile and Amphibian Atlas) imply that these four proposed MUs may be isolated demographically. These four areas are unlikely to qualify as DUs under Canadian law (Green 2005) because we are not aware of empirical evidence that they experience significantly different risks of extinction. However, both the LE and GH MUs are increasingly isolated in declining patches of habitat, and DU designations may become appropriate in the future. The Georgian Bay-Parry Sound District MU is widely distributed and probably represents a large population. The same is likely true in EO, although it is currently not possible to estimate population size across these larger units. Our sampling was limited, and further sampling is required to clarify the exact boundaries of these MUs.
Assignment tests and clustering analyses provide preliminary evidence about connectivity between the proposed MUs and the four under-sampled sites (LHsouth, LHnorth, KAW and ALG). The northern extreme of the species’ range (LHnorth) may be genetically continuous with the population in PSD. Clustering analyses and strong assignment of individuals from KAW to PSD implies similar genetic continuity between these two areas (Table 4; Fig. 3), which are connected by large remaining areas of suitable habitat. The samples from LHsouth and ALG were not strongly associated with any of the four proposed MUs and also represent isolated occurrences (Ontario Nature Reptile and Amphibian Atlas). None of the undersampled sites can be assumed to be part of the four tentative MUs until further evidence is available.
Bayesian assignment of individuals to Ontario populations had higher general accuracy (0.67–0.80) than that reported for the three Nova Scotia subpopulations (0.37–0.73; Howes et al. 2009), probably due to increased population structure and greater geographic distances in Ontario. This difference may also be an artefact of the different loci, and different number of loci, used in the two studies.
Interestingly, results from both structure and tess suggest possible past translocations of individuals between some subpopulations (Fig. 3; individuals with an ~50 % probability of membership to two populations may be first-generation offspring of migrants who mated with residents). This result may also be due to other causes (for example, natural migration from an unsampled population), but incidental and unrecorded translocations of individual turtles by members of the public occurs regularly in Ontario (F. Ross, pers. comm.; S. Gillingwater, pers. comm.; C. Davy, unpublished data). This is a separate issue from collection for the commercial pet trade, which also threatens E. blandingii (van Dijk and Rhodin 2011). There are no data on the frequency of these casual translocations and we cannot be certain that the observed admixture resulted from translocations, but the possibility should not be discounted.
The range of Nm values calculated for Ontario (0.952–3.380) is slightly lower than that reported from three sites in Nova Scotia (1.76–5.80; Mockford et al. 2005), but our estimates of recent gene flow (0.011–0.163) are comparable (0.012–0.16 in Nova Scotia; Howes et al. 2009). In both regions, no strong, overall directionality of gene flow among populations is apparent. Further comparative analyses of gene flow based on data from E. blandingii and other, co-occurring species could explicitly test hypotheses about potential barriers to gene flow on the current and historic landscape.
A negative correlation between genetic diversity and latitude is expected in turtles in post-glacial landscapes, due to founder effects during northward range expansions following the last ice age (Galbraith 2008). Diversity in E. blandingii does not support this prediction, possibly because this species has a compressed latitudinal range and may have used East–West re-colonization routes as well as moving northward. Fossil evidence places E. blandingii in southern Indiana 15–14 ka bp, and fossils are also known from Indiana and Michigan 6–4 ka bp (Holman 1992). Some populations might have used Pleistocene refugia in the southern Atlantic plain (Bleakney 1958). Others may have persisted near the Great Lakes throughout the Wisconsonian ice age, rapidly recolonizing the area from multiple directions as the ice sheets retreated (Holman 1992). Reduced genetic diversity (HO) in the disjunct populations east of the Appalachian Mountains remains evident when our samples from Ontario are considered, further supporting the hypothesis that these represent relict populations stranded in glacial refugia during the last ice age (Mockford et al. 2005, 2007).
Our data suggest that loss of genetic diversity is not an immediate threat to E. blandingii. We detected private alleles at several sites, but no alleles are fixed at any loci. Heterozygosity is comparable to that reported for other populations of turtles (summarized by Vargas-Ramirez et al. 2012). Long generation times may have slowed the loss of genetic diversity in declining populations of E. blandingii. However, this species continues to experience significant population fragmentation and decline across its range (van Dijk and Rhodin 2011). If this trend continues, the impacts of reduced genetic diversity will eventually become unavoidable.
Current conservation efforts for E. blandingii should mitigate demographic threats by reducing adult mortality (Congdon et al. 2008) and increasing recruitment of juveniles (and eventually mature adults) to populations. Increasing population size can prevent further loss of genetic diversity in threatened populations (Frankham et al. 2002), and simultaneously targets demographic threats to a population. Measures that can increase habitat and population connectivity should explicitly consider genetic structure. For example, anthropogenic features that fragment habitat and cause mortality (e.g. highways, urban development) also reduce gene flow among population fragments. This effect can be mitigated using tools such as wildlife underpasses, or corridors of suitable habitat. Conversely, actions such as translocations that involve moving individuals across the landscape should also explicitly consider genetic population structure. Mixing of genetic populations can have serious consequences for fitness if locally adapted genes or co-adapted gene complexes are disrupted (outbreeding depression, Templeton 1986). There is no empirical evidence to suggest local genetic adaptations in E. blandingii, but minimal genetic data exist for this species and the possibility should not be discounted.
We have clearly demonstrated that E. blandingii is not panmictic across Ontario, even on a relatively small scale (approximately 500 km between the most distant sites). Future analyses may reveal similar levels of population structure across the rest of the central range of E. blandingii, or even further ESUs beyond those already identified by Mockford et al. (2007). Such information would be crucial to effective conservation and recovery of this endangered species. Further analyses of current and historic gene flow in E. blandingii can also clarify the post-glacial colonization routes of turtles into Canada, and test the role of landscape features in shaping the current distribution of E. blandingii across its range.
References
Banning-Anthonysamy WJ (2012) Spatial ecology, habitat use, genetic diversity, and reproductive success: measures of connectivity of a sympatric freshwater turtle assemblage in a fragmented landscape. Dissertation, University of Illinois at Urbana-Champaign
Barton NH, Slatkin M (1986) A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 56:409–416
Bleakney JS (1958) A zoogeographic study of the amphibians and reptiles of eastern Canada. Natl Mus Can Bull 155:1–119
Brecke BJ, Moriarty JJ (1989) Emydoidea blandingii (Blanding’s turtle). Longevity. Herpetol Rev 20:53
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Chiucchi JE, Gibbs HL (2010) Similarity of contemporary and historical gene flow among highly fragmented populations of an endangered rattlesnake. Mol Ecol 19:5345–5358
Congdon JD, van Loben Sels RC (1991) Growth and body size in the Blanding’s turtles (Emydoidea blandingii): relationships to reproduction. Can J Zool 69:239–245
Congdon JD, Dunham AE, van Loben Sels RC (1993) Delayed sexual maturity and demographics of Blanding’s turtles (Emydoidea blandingii): implications for conservation and management of long-lived organisms. Conserv Biol 7:826–833
Congdon JD, Graham TE, Herman TB, Lang JW, Pappas MJ, Brecke BJ (2008) Emydoidea blandingii (Holbrook 1838)—Blanding’s turtle. In: Rhodin AGJ, Pritchard PCH, van Dijk PP, Saumure RA, Buhlmann KA, Iverson JB (eds) Conservation biology of freshwater turtles and tortoises: a compilation project of the IUCN/SSC Tortoise and Freshwater Turtle Specialist Group. Chelon Res Monogr 5:015.1–015.12. doi:10.3854/crm.5.015.blandingii.v1.2008. http://www.iucn-tftsg.org/cbftt/. Accessed 4 Feb 2013
COSEWIC (2005) COSEWIC assessment and update status report on the Blanding’s Turtle Emydoidea blandingii in Canada. Committee on the Status of Endangered Wildlife in Canadal, Ottawa, viii +40 pp. www.sararegistry.gc.ca/status/status_e.cfm. Accessed 10 Jan 2012
Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Res 10:556–557
Davy CM, Leifso AE, Conflitti IM, Murphy RW (2012) Characterization of 10 novel microsatellite loci and cross-amplification of two loci in the snapping turtle (Chelydra serpentina). Conserv Genet Resour 4:695–698
Durand E, Jay F, Gaggiotti OE, François O (2009) Spatial inference of admixture proportions and secondary contact zones. Mol Biol Evol 26:1963–1973
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ernst CH, Lovich JL (2009) Turtles of the United States and Canada, 2nd edn. Johns Hopkins University, Baltimore
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stevens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1615
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University, Cambridge
Galbraith DA (2008) Population biology and population genetics. In: Steyermark AC, Finkler MS, Brooks RJ (eds) The biology of the snapping turtle. Johns Hopkins University, Baltimore, pp 168–180
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Green DM (2005) Designatable units for status assessment of endangered species. Conserv Biol 19:1813–1820
Holman JA (1992) Late quaternary herpetofauna of the central Great Lakes region, U.S.A.: zoogeographical and paleoecological implications. Quat Sci Rev 11:345–351
Holbrook JE (1838) North American herpetology; or, a description of the reptiles inhabiting the United States. In: Dobson J (ed), vol 3, 1st edn. Philadelphia, pp 122
Howes BJ, Brown JW, Gibbs HL, Herman TB, Mockford SW, Prior KA, Weatherhead PJ (2009) Directional gene flow patterns in disjunct populations of the black ratsnake (Pantherophis obsoletus) and the Blanding’s turtle (Emydoidea blandingii). Conserv Genet 10:407–417
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13. v.3.23. http://ibdws.sdsu.edu/. Accessed 10 Jan 2011
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Jost L (2009) D vs. GST: response to Heller and Siegismund (2009) and Ryman and Leimar. Mol Ecol 18:2088–2091
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5:539–543
Kalinowski ST (2005) HP-Rare: a computer program for performing rarefaction on measures of allelic diversity. Mol Ecol Notes 5:187–189
King TL, Julian SE (2004) Conservation of microsatellite DNA flanking sequences across 13 Emydid genera assayed with novel bog turtle (Glyptemys muhlenbergii) loci. Conserv Genet 5:719–725
Mockford SW, Snyder M, Herman TB (1999) A preliminary examination of genetic variation in a peripheral population of Blanding’s turtle, Emydoidea blandingii. Mol Ecol 8:323–327
Mockford SW, McEachern L, Herman TB, Snyder M, Wright JM (2005) Population genetic structure of a disjunct population of Blanding’s turtle (Emydoidea blandingii) in Nova Scotia, Canada. Biol Conserv 123:373–380
Mockford SW, Herman TB, Snyder M, Wright JM (2007) Conservation genetics of Blanding’s turtle and its application in the identification of evolutionarily significant units. Conserv Genet 8:209–219
Moritz C (1994) Defining ‘evolutionarily significant units’ for conservation. Trends Ecol Evol 9:373–375
Osentoski MF, Mockford S, Wright JM, Snyder M, Herman TB, Hughes CR (2002) Isolation and characterization of microsatellite loci from the Blanding’s turtle, Emydoidea blandingii. Mol Ecol Notes 2:147–149
Paetkau D, Slade R, Burden M, Estoup A (2004) Direct, real-time estimation of migration rate using assignment methods: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Palsbøll PJ, Bérubé M, Allendorf FW (2006) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol 6:288–295
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) GeneClass2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Power TD (1989) Seasonal movements and nesting ecology of a relict population of Blanding’s turtles (Emydoidea blandingii) in Nova Scotia. M.Sc. Thesis, Acadia University, Wolfville
Pritchard JK, Stephens M, Donnelly PJ (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9221
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rosen T (2007) The endangered species act and the distinct population segment policy. Ursus 18:109–116
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) Genepop’007: a complete reimplementation of the Genepop software for Windows and Linux. Mol Ecol Res 8:103–106
Rubin CS, Warner RE, Bouzat JL, Paige KN (2001) Population genetic structure of Blanding’s turtles (Emydoidea blandingii) in an urban landscape. Biol Conserv 99:323–330
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning—a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, New York
Smith LM, Burgoyne LA (2004) Collecting, archiving and processing DNA from wildlife samples using FTA® databasing paper. BMC Ecol 4:4. http://www.biomedcentral.com/1472-6785/4/4. Accessed 12 Nov 2010
Templeton AR (1986) Coadaptation and outbreeding depression. In: Soulé M (ed) Conservation biology: the science of scarcity and diversity. Sinauer, Sunderland, pp 105–116
van Dijk PP, Rhodin AGJ (2011) Emydoidea blandingii. In: IUCN 2011. IUCN red list of threatened species. Version 2011.2. http://www.iucnredlist.org. Accessed 15 Dec 2011
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vargas-Ramirez M, Stuckas H, Castaňo-Mora OV, Fritz U (2012) Extremely low genetic diversity and weak population differentiation in the endangered Colombian river turtle Podocnemis lewyana (Testudines: podocnemididae). Conserv Genet 13:65–77
Waples RS (1991) Pacific salmon, Oncorhynchus spp. and the definition of “species” under the endangered species act. Mar Fish Rev 53:11–22
Whitlock MC, McCaughley DE (1999) Indirect measures of gene flow and migration: FST ≠ 1/(4 Nm + 1). Heredity 82:117–125
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wright S (1943) Isolation by distance. Genetics 28:114–138
Acknowledgments
Laboratory analyses were funded by a Species at Risk Research Fund for Ontario grant from the Government of Ontario, and we thank B. Johnson and J. Philips of the Toronto Zoo for collaborating on this grant. Other funding was provided by a Canada Graduate Scholarship from the National Science and Engineering Research Council of Canada (NSERC) to CMD, a NSERC Canada Discovery Grant (A3148) to RWM and a Canada Collection grant from Wildlife Preservation Canada to CMD. Sample collection was possible thanks to the generous assistance of J. Baxter-Gilbert, S. Carstairs, B. Caverhill, S. Coombes, J. Crowley, J. Litzgus, M. Keevil, I. Macintosh, J. Paterson, J. Trottier, J. Riley, J. Rouse, D. Seburn, J. Urquhart and A. Whitear. J. Hathaway and J. Pierce allowed sampling of E. blandingii at Scales Nature Park. Jessica Hsiung assisted with figure preparation and provided an original illustration of a Blanding’s Turtle. We thank D. Currie, M. J. Fortin, D. McLennan, S. Lougheed, C. Wilson and two anonymous reviewers for comments on an earlier version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davy, C.M., Bernardo, P.H. & Murphy, R.W. A Bayesian approach to conservation genetics of Blanding’s turtle (Emys blandingii) in Ontario, Canada. Conserv Genet 15, 319–330 (2014). https://doi.org/10.1007/s10592-013-0540-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-013-0540-5