
Abstract
Blanding’s turtle (Emys blandingii) has declined substantially in North America due to anthropogenic activities, leaving populations smaller and increasingly fragmented spatially. We sampled 212 turtles to evaluate variation at eight microsatellite loci within and among 18 populations of E. blandingii across its primary range in the midwestern United States (Illinois, Iowa, Minnesota, and Nebraska). All loci and populations were highly polymorphic. Our analyses also detected considerable genetic structure within and among the sampled localities, and revealed ancestral gene flow of E. blandingii in this region north and east from an ancient refugium in the central Great Plains, concordant with post-glacial recolonization timescales. The data further implied unexpected ‘links’ between geographically disparate populations in Nebraska and Illinois. Our study encourages conservation decisions to be mindful of the genetic uniqueness of populations of E. blandingii across its primary range.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Barriers to migration of individuals can isolate populations over evolutionary time and even elicit speciation (Byrne et al. 2008). In the interim, displaced populations can establish distinct clades within species, as indicated by fossil and genetic evidence (Amato et al. 2008; Sommer et al. 2009; Ursenbacher et al. 2006). Range size changes, eliciting diversification and population bottlenecks, are greatly influenced by abiotic, often geological, events and yield distinct genetic patterns. More recently, anthropogenic forces have disturbed habitats and displaced taxa (Hoffmann et al. 2010; Stuart et al. 2004; Sutherland et al. 2012). This contemporary uptick of anthropogenic pressures and recognition of the relatively sharp decline in species numbers has elicited an increase in population genetic studies of organisms of conservation concern to illuminate evolutionarily significant units and provide added guidance for management to preserve unique genetic lineages (reviewed in Avise 2010; Frankham et al. 2002).
Fossil records of relict vertebrate populations in the Great Plains of North America suggest that repeated glacial recession and drastic climate change after glacial advance displaced various species (Smith 1957). Termed ‘xerothermic’ or interglacial periods, these ages, the last one from ~9000 to 4000 YBP, were marked by aridity and warming that Smith (1957) conjectured should have forced heat-intolerant species to alter their geographic ranges. Smith (1957) also hypothesized, based on the presence of current disjunct relicts of vertebrate species and evidence from skeletal remains, that additional eastern disjunct populations of several species once existed as post-glacial relicts during the latest xerothermic period, but are now extinct. Since Smith’s proposals, advances in molecular phylogeography and paleogeography have yielded more concrete evidence of the establishment of these relict populations due to glacial activity in the midwestern United States (e.g., Amato et al. 2008; Janzen et al. 2002; Weisrock and Janzen 2000; Wooding and Ward 1997). Such work is particularly important for vulnerable taxa like turtles, as recent and drastic habitat changes have extirpated multiple chelonian species over the last century (Shaffer 2009). Understanding the current genetic diversity and historical distribution patterns of turtles is, thus, essential to making more informed conservation and management decisions; indeed, identifying genetic discontinuities across the landscape is one of three crucial needs in studies of turtle conservation genetics (Alacs et al. 2007).
Blanding’s turtle [Emys (formerly Emydoidea) blandingii—Fritz et al. 2011] is semi-aquatic, with populations in Nebraska, Iowa, Minnesota, Illinois, Wisconsin, Michigan, and Ontario, along with geographically disjunct populations in Nova Scotia and the eastern seaboard (Ernst and Lovich 2009). In the Pleistocene, E. blandingii occupied a much wider range across North America (Jackson and Kaye 1974; Mockford et al. 1999; Smith 1957; Van Devender and King 1975). Archaeological records reveal post-glacial fossils of E. blandingii from Michigan, Maine, New York, and Ontario, as well as Pliocene fossils in Nebraska, Kansas, Oklahoma, and Pennsylvania (Ernst and Lovich 2009), which coincides with the idea of disjunct midwestern and eastern populations ~8000–4000 YBP (Smith 1957). Subsequent glacial recession, establishment of waterways connecting to the Great Lakes, and, more recently, anthropogenic factors, have contributed to a unique spatial distribution (Fig. 1; see Congdon and Keinath 2006). Blanding’s turtle is listed as ‘endangered’ across its range and ‘threatened’ on the IUCN Red List (Rhodin and van Dijk 2011), with this imperiled status mainly attributable to the combined effects of delayed maturity (Congdon et al. 1993; Congdon and Van Loben Sels 1993) and habitat destruction (e.g., Rubin et al. 2001).

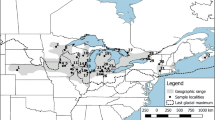
Localities and putative groupings of the 18 sites sampled for Emys blandingii in the midwestern United States. Gray areas are water bodies, the more southern dashed line indicates the extent of the pre-Wisconsinan glacial limit (Kansan/Nebraskan glaciations), and the more northern dashed line indicates the extent of the Laurentide Ice Sheet in the more recent Wisconsinan glaciation (top left). Representation of Model 1 (Table S1), with sites within 100 km linear distance from each other clustered to form groups (top right). Representation of Model 2 (Table S2), with sites in the same watershed clustered to form groups (middle left). Representation of Model 3 (Table S3), with sites in the same watershed and location with respect to the limit of the Laurentide Ice Sheet clustered to form groups (middle right). Representation of sample sizes (indicated by diameter of the pies) and of relative admixture distribution (indicated by shade of gray) inferred by STRUCTURE at K = 5 (bottom center)
Genetic studies have been conducted on populations of E. blandingii primarily in Nova Scotia to New York (Howes et al. 2009; Mockford et al. 2005), near Ann Arbor, Michigan (McGuire et al. 2013), and near Chicago, Illinois (Rubin et al. 2001). These studies reported little to no genetic structure among localities. However, at a broader scale, another study detected strong population genetic structuring between, but not so much within, the Great Lakes region and eastern North America in these turtles (Mockford et al. 2007; but see Spinks and Shaffer 2009). Even so, a large-scale population genetics study across the range of Blanding’s turtle in the midwestern United States (hereafter defined as west of Lake Michigan), where extensive anthropogenic landscape alterations have occurred over the past 150 years, has yet to be undertaken. In this study, we examined the population genetics of Blanding’s turtles sampled from Nebraska (NE), Iowa (IA), Minnesota (MN), and Illinois (IL) using microsatellite loci. First, we tested the hypothesis (sensu Mockford et al. 2007) that E. blandingii largely lacks much spatial genetic structure in this region. Second, we examined the alternative hypothesis (sensu Smith 1957) that current levels of genetic variation reflect post-glacial colonization (along watersheds in the Mississippi-Missouri River basins) of northern locales from refugia in Kansas and Missouri (Holman 1995; Van Devender and King 1975). Paleo-hydro-geological data indicate that such watersheds (see Table S3) formed after recession of the Laurentide Ice Sheet. At its peak, the Des Moines Lobe of this glacier separated our sampling locales into populations that are inside and outside this region (Stiff and Hansel 2004). We further assessed these two hypotheses by estimating ancestral gene flow and coalescent times (Hey and Nielsen 2004) and recent migration rates (Wilson and Rannala 2003). Finally, we provide some perspective on how our findings might impinge on conservation and management activities involving Blanding’s turtle in the midwestern United States.
Materials and methods
Fieldwork
We caught 212 Blanding’s turtles in 18 locations in the midwestern United States (Fig. 1). We trapped several wetlands per location for at least 20 trap-nights. From our 12 most productive localities, we sampled 16 individuals per population on average (range 6–61; Table 1). Of the remaining six localities, five yielded only a single individual and one yielded two individuals. These six sites were not included in the heterozygosity, population differentiation, and ancestral and recent migration analyses (see below), being used only for inferring population genetic structure and admixture using STRUCTURE (Pritchard et al. 2000). Tissue samples were either tail tips taken and stored in 95 % ethanol or blood extracted from the cranial sinus or caudal vein using a 28-gauge syringe, placed in lysis buffer or EDTA, and stored at −80 °C.
Genetic data generation
We extracted genomic DNA from each sample using either High Pure PCR Template Preparation Kit (Roche Laboratory) following the protocol outlined by the Roche Applied Science Chelex (Walsh et al. 1991), or phenol–chloroform extraction. For genotyping, we used eight tetra-nucleotide repeat microsatellite markers previously developed for a related turtle (Glyptemys muhlenbergii) (King and Julian 2004; GmuD21, GmuD55, GmuD87, GmuD88, GmuD90, GmuD93, GmuD95, and GmuD121). Detailed amplification procedures can be found elsewhere (Howeth et al. 2008); we genotyped PCR products on an Applied Biosystems 3100 Genetic Analyzer at the Iowa State University DNA facility using the Rox size standard (FAM and HEX dye sets). We genotyped negative and positive controls for each locus to assess false positives or negatives. We visualized and sized the results using GenoProfiler v. 2.1 (You et al. 2007) and PeakScanner v.1.0 (Applied Biosystems), and we manually determined the diploid allele sizes to identify unique alleles. We noted indeterminate genotypes, possibly due to amplification errors, as missing alleles. We could not resolve 76 diploid genotypes (4.5 %), which we then classified as missing data. Of the 212 genotyped individuals, 209 contained adequate information to be included in further analyses.
Genetic data analysis
We concentrated the majority of our population genetic analyses on the 12 well-sampled populations (Table 1). For additional analyses that do not require a priori specification of population of origin, we included the six other populations with minimal samples.
General population genetic analysis
We used Genepop v.4.0.10 (Raymond and Rousset 1995) and GDA v.1.1 (Lewis and Zaykin 2008; Weir 1996) to estimate allele frequencies, observed and expected heterozygosities, and pair-wise tests of linkage disequilibrium. We set dememorization numbers at 10,000 and performed 100,000 iterations for all permutation tests in Genepop.
We tested deviance from Hardy–Weinberg equilibrium (hereafter, HWE) at each locus using FSTAT v.2.9.3.2 (Goudet 1995). Because we evaluated HWE per locus, per population (8 loci × 12 populations), we used sequential Bonferroni to correct for multiple comparisons on the expected and observed heterozygosities (Rice 1989). We also performed a test for null alleles using Microchecker v.2.2.3 (van Oosterhout et al. 2006) because the observed heterozygosities and deviance from HWE suggested the presence of null alleles that could possibly skew the population genetics results. In so doing, we placed limits on allele sizes (repeat lengths) at each locus based on those reported in King and Julian (2004) for E. blandingii. Analyses at the population level detected the presence of excessive homozygosity in some loci (Table S6), but overall GmuD95 was the most problematic locus and was identified by Microchecker as likely having null alleles. Therefore, we hereafter conducted all data analyses with and without the most homozygous locus (i.e., GmuD95) as an assessment of the potential impact of null alleles.
Differentiation and population structure
We estimated F st between all pairs of the 12 well-sampled populations and calculated statistical significance (Weir and Cockerham 1984) using 1,000,000 genotypic permutations in Arlequin (Excoffier and Lischer 2010) followed by sequential Bonferroni correction for the 66 pairwise population comparisons (Rice 1989).
We also calculated other population-wide F-statistics (F is, F it, F st) with 95 % confidence intervals, after bootstrapping across all loci with 10,000 replicates in GDA v.1.1 (Lewis and Zaykin 2008). As defined by GDA, F is describes average genetic differentiation between 202 individuals within their sampling locations; F it quantifies genetic differentiation between 202 individuals in the total population, and F st measures differentiation between the sampled locations and the total population. We then analyzed population structure in several ways. First, we analyzed genetic differentiation due to linear geographic distance for the 12 well-sampled populations (Rousset 1997). This isolation-by-distance analysis regressed estimates of pairwise F st/1 − F st (Slatkin’s linearized F st distance) against the linear distance separating pairs of populations. We calculated this regression using a Mantel Test in GenAlEx v.6.2 (Peakall and Smouse 2006), with 1,000 permutations to assess statistical significance.
Second, linear distance is not always the best predictor of genetic differentiation, as different geographic and historical forces may contribute to large genetic differentiation even over very small spatial scales (reviewed in Avise et al. 1987). To ascertain a potentially better phylogeographic predictor of genetic variance, we addressed multiple hypotheses using AMOVAs and comparing AICc values for each model to assess fit (Excoffier et al. 1992; performed with 16,000 permutations across and within the sampled loci in Arlequin). We constructed three models that reflect putatively different genetic structures across the landscape: (1) linear geographical distance, (2) clustering of populations into groups based on current river basins/watersheds, and (3) clustering of populations into groups based on location inside or outside of the Des Moines Lobe of the Laurentide Ice Sheet.
To test these models, we employed several grouping schemes (Fig. 1). For the first model, we used Geographic Distance Matrix Generator v.1.2.3 (Ersts 2010) to calculate linear distances between populations from GPS coordinates for the collected specimens (Table 1). We clustered populations within ~100 linear km (based on geographic distribution of our sampling) of each other (see Table S1—Model1) to assess isolation by distance. Populations that fell into two clusters were resolved by grouping them with other populations that were the closest linear distance to them. For the second model, we grouped populations into watersheds (Midwest Natural Resources Group, www.epa.gov/Region5/mnrg/), yielding four groups spanning the Missouri River Watershed, Upper Mississippi River Watershed, Minnesota River Watershed, and Illinois River Watershed—Southern Lake Michigan Crescent Watershed (see Table S2—Model2). We designed this model knowing that these semi-aquatic turtles can migrate several kilometers terrestrially (Ernst and Lovich 2009). Individuals trapped from locations separated by small terrestrial areas were counted as part of the same watershed. We constructed the third model to assess population structure relative to current watersheds, as in the second model, but also incorporated separation by the last glacial maximum (LGM) limit of the Laurentide Ice Sheet (Ehlers and Gibbard 2008). This model sorted our sampled locations into five groups, essentially similar to the second model but with the Upper Mississippi River Watershed divided into regions located inside (or north) of the LGM limit and outside (or south) of it (see Table S3—Model3). For each model, we calculated pairwise F-statistics and made comparisons with an exact test of population differentiation (Goudet 1995; Raymond and Rousset 1995), where ‘populations’ are the defined groups for each model. This test determines the probability that ‘k’ genotypes are distributed among ‘r’ populations by using an r x k contingency table. We explored potential states of the contingency table using a Markov chain with 16,000 permutations of genotypes among populations. We compared Weir and Cockerham (1984) estimates of F-statistics and AMOVA results from these latter two population differentiation models to determine the better predictor of genetic variance using AICc (Burnham and Anderson 1998).
Lastly, we included all 209 individuals with adequate genotype information from all 18 populations to explore population structure with STRUCTURE v.2.2 (Pritchard et al. 2000; STRUCTURE is not sensitive to sample size per population) using the admixture model and specifying no a priori models of subpopulation structure. We allowed the Dirichlet parameter for the degree of admixture (α) to be inferred from the data, with an initial value of 1.0 and uniform priors for all populations. To determine correlated allele frequencies and to compute probability of the data to estimate K (the most likely number of putative populations), we performed 20 runs for each value of K (1–18) with 10,000 MCMC repetitions. In each case, we allowed a burn-in period of 10,000 for K from 1 to 18, running models with and without GmuD95. We first plotted the mean and variance in likelihood per K using STRUCTURE HARVESTER v.0.6.92 (Earl and vonHoldt 2012) and implemented the Evanno et al. (2005) method. We extracted and visualized the Q value tables from the results of STRUCTURE using Distruct v.1.1 (Rosenberg 2004).
Historical population parameters
We next traced ancestral patterns of gene flow among our sampled populations. We used coalescent reconstructions with IM (Hey and Nielsen 2004) to evaluate pairwise maximum likelihood estimates of ancestral gene flow and time since splitting between the five groups identified by STRUCTURE (see below). This method yielded an approximate timeline of historic genetic differentiation events among these groups based on a rate of 0.0005 mutations per locus per generation (Howes et al. 2009), a generation time of 37 years (Congdon et al. 2003), and a stepwise mutation model suitable for microsatellites (Kimura and Ohta 1978). We imposed uninformative prior distributions on the upper-bound values of migration rates and effective population sizes, depending on their convergence in the results (see Hey and Nielsen 2004). We performed multiple independent runs each with a burn-in period of 100,000, and observed the shape of the posterior distribution after every 30-min run post-burn-in. We assessed mixing of the MCMC by plotting marginal histograms and trend lines of the parameter estimates to confirm that the MCMC sufficiently explored the parameter space and approached stationarity. Most chains converged within five 30-min runs. As expected of microsatellite data, the Effective Sampling Size values were low (<50) for most runs, but since the results were congruent over multiple independent runs, we take this as sufficient evidence that the runs had converged (J. Hey pers. comm.). Having excluded GmuD95, we performed five separate sets of runs of five pairs of clustered populations each, incrementally changing the priors depending on their convergence and the completion of the posterior distributions. These clustered populations derived from the 12 localities grouped into five populations as identified by STRUCTURE in Table S4, but with the population comprised of Carroll-IL, Will-IL, and Grant-NE (hereafter, we refer to populations with a county-state designation) split into Illinois and Nebraska clusters to determine the time since split between these two groups. We then used the estimates of migration rates and population-scaled mutation rates to calculate demographic parameters (with 95 % confidence intervals), such as the effective population sizes, time since splitting in years, and migration rates per generation between pairs of the clustered populations (Hey and Nielsen 2004).
We also estimated relatively recent (roughly several generations) bidirectional migration rates between the same clusters (see Table S4) with BayesAss v.1.3 (Wilson and Rannala 2003). This method uses an MCMC method applied to diploid data to determine recent migration rates and to assign ancestries to individuals. We performed multiple initializations of MCMCs and analyzed the trace files of logarithmic probabilities using Tracer v.1.5.0 (Rambaut and Drummond 2007) to ensure good mixing and effective sampling from the posterior distribution. We constructed ~95 % confidence intervals around mean recent migration rates as mean ± 1.96 * standard deviation. For each initialization, we utilized 10 million iterations of the MCMC, with a burn-in of 1 million iterations, sampling from every 1,000 iterations.
Results
Microsatellite characteristics and deviations from HWE
The number of alleles per locus and size of the alleles for all loci were largely within the ranges reported by King and Julian (2004). The average number of alleles per locus (all eight loci were polymorphic) was 20.1 across all populations. The average observed heterozygosity among the 12 well-sampled populations across all loci was 0.54 (range 0.25–0.82; Table S5) and 0.52 across all 18 populations, indicating high levels of polymorphism. We detected significant heterozygote deficiency on average across all loci in the Grant-NE, Bremer-IA, Muscatine-IA, Clinton-IA, Scott-MN, Carroll-IL, and Will-IL populations (Table 1; but see also Table S6 for a per locus analysis). We detected significant (P < 0.05) heterozygote deficiency in individuals from multiple populations at various loci (Table S6). However, after correction for multiple comparisons, only Grant-NE, Muscatine-IA, Bremer-IA, Carroll-IL, McHenry-IL, and Will-IL at GmuD95, Muscatine-IA at GmuD90, and Will-IL at GmuD93 remained out of HWE, primarily due to heterozygote deficiency (Table S6). Microchecker revealed the possibility of null alleles at GmuD95, and hence many further analyses were performed both with and without this locus. Finally, we detected no evidence of linkage disequilibrium between any pair of loci (all P > 0.05), suggesting random assortment among the eight loci (Table S7).
Population structure
The average frequency of private alleles in the sampled populations was fairly low at 0.0777. Pairwise F st values fell between 0.01 and 0.47 (Table 2), indicating low to moderate levels of genetic differentiation between the 12 well-sampled populations (55 of 66 comparisons were significant after Bonferroni correction; Table 2). The highest significant F st was 0.469 between McHenry-IL and Grant-NE, which, not surprisingly, is the second most geographically-distant population-pair sampled (~1,102 km). Still, significant pairwise F st values were detected even over short distances. For instance, the F st of 0.287 between Clinton-IA and Carroll-IL fell in the upper half of our 66 comparisons, but are two of the geographically-closest populations sampled (~15 km, though separated by the Mississippi River).
Other comparisons exhibited low F st values. Notably, Winnebago-IA showed non-significant F st values with both Jones-IA and Clinton-IA (F st = 0.045, P = 0.097; F st = 0.048, P = 0.116, respectively) even though these populations are ~253 and ~294 km linear distance apart. Pairwise comparisons between these three populations from eastern Iowa and two other eastern Iowa populations (Worth-IA and Bremer-IA) all yielded F st values <0.058 that are not significantly different from zero, indicating little genetic differentiation within the drainages of the Winnebago/Shell Rock/Cedar, Wapsipinicon, and Maquoketa Rivers. Also of note, the Grant-NE population, located in western Nebraska, had comparatively low (albeit, significantly different from zero) F st values with Carroll-IL in western Illinois and Will-IL in eastern Illinois (F st = 0.187, P < 0.0001; F st = 0.135, P = 0.001, respectively), considering that our largest F st value (0.469) was between Grant-NE and McHenry-IL, which is only 84 km north of Will-IL.
Overall, estimates of Weir and Cockerham F-statistics involving the 12 well-sampled populations revealed a greater signature of inbreeding than expected (F is = 0.136 (0.027–0.275, 95 % CI) with GmuD95 and F is = 0.075 (0.010–0.165, 95 % CI) without GmuD95). Furthermore, global F st values suggested considerable genetic differentiation, and ranged from 0.263 (0.184–0.357, 95 % CI) with GmuD95 to 0.270 (0.178–0.379, 95 % CI) without GmuD95 (see Table S8).
Geographic and genetic distances exhibited a positive correlation (R 2 = 0.179; Fig. 2). Thus, while geographic distance among populations likely contributes to genetic differentiation, other factors play a role as well. We thus performed AMOVAs to estimate the amount of variance in multilocus genotypes explained by each of three models (Model 1—populations grouped by linear geographic distance, Model 2—populations grouped by current watershed distributions, and Model 3—populations grouped by current watershed distributions plus relative location inside or outside the Laurentide Ice Sheet—see Tables S1, S2 and S3; Fig. 1). AMOVA revealed that most of the genetic variation occurred within populations in all three models (68.8–70.8 %), with much smaller amounts occurring among populations (24.1–26.3 %) or among clusters of populations (4.8–5.1 %) as identified above (Table S9). A smaller number of groups (four, as hypothesized in Model 2—see Table S2) better explained the genetic data than did a larger number of groups (six, as hypothesized in Model 1—Table S1) (∆AICc >42). A comparison of the model of clustering by watersheds alone (i.e., four groups—see Table S2) to one clustering by watersheds and the Laurentide Ice Sheet (five groups, as hypothesized in Model 3—see Table S3) yielded a ∆AICc = 2.22, indicating that both models have substantial support (see Table S9).

Plot of linear geographic distance between sampled populations of Emys blandingii vs. Slatkin’s linearized F st genetic distance between these same sampled populations to estimate the presence of isolation by distance. This plot was derived from a Mantel test performed in GenAlex v.6.2 with 1000 permutations
Population structure was estimated for all 209 individuals from all 18 populations without using any prior geographic information in STRUCTURE. Across all eight loci, the most likely population structure was K = 4, but excluding GmuD95 from the STRUCTURE analyses yielded K = 5 regions (Figs. 1, 3, Fig. S1).

Estimates of admixture proportions in sampled populations of Emys blandingii. Twenty runs of STRUCTURE were performed for each value of K, under the admixture model, and the Dirichlet parameter ‘alpha’ was inferred from the data. Each run was performed using the 209 genotyped individuals from all 18 localities with a burn-in period of 10000 and 10000 MCMC reps. Left all loci. Right all loci except GmuD95
Historical population parameters
We detected considerable diversity in N e among five clusters of populations (the four from STRUCTURE (with adequate sample sizes) split into five clusters to resolve divergence time between Grant-NE and Carroll-IL, Will-IL) (Table S10.1). Median pairwise N e estimates ranged from 750 (95 % CI = 466–1326; Grant-NE vs. Carroll-IL, Will-IL) to 1,681,177 (95 % CI = 1,682,883–1,684,589; IA populations vs. Carroll-IL, Will-IL). Although N e in this latter case and for Scott-MN, Muscatine-IA (Group 2, Table S3) vs. Grant-NE derived from analyses that failed to converge, all other estimates came from analyses that converged within five runs.
The oldest ‘split’ events were estimated to have occurred well into the Pleistocene, while the youngest probably transpired in the recent past (Table S10.2). In the former case, the IA populations (Group 1, Table S3) and McHenry-IL (Group 4, Table S3) apparently split ~353,250 YBP (95 % CI = 185,250–410,750 YBP), with strong subsequent unidirectional gene flow from east to west (median m 2 = 10.65 individuals/generation, 95 % CI = 6.75–23.35; median m 1 = 1.05, 95 % CI = 0.85–2.65). Also, the IA populations and the Grant-NE population were estimated to have split ~231,750 YBP (95 % CI = 187,250–495,250 YBP), with little subsequent gene flow between the two localities (median m 1 = 0.15, 95 % CI = 0.15–1.35; median m 2 = 2.95, 95 % CI = 1.95–6.85). At the other extreme, the most recent split occurred around a median of 850 YBP (95 % CI around mean = 950–1150 YBP) between Carroll-IL, Will-IL and the IA populations, with strong bi-directional gene flow of 13.45 individuals/generation (95 % CI = 11.75–16.95) from IA into Carroll-IL, Will-IL and 22.65 individuals/generation (95 % CI = 17.05–26.25) from Carroll-IL, Will-IL into IA. These analyses also accorded with a puzzling result obtained from the F st and STRUCTURE analyses. That is, despite considerable geographic distance, splits between Grant-NE and Carroll-IL, Will-IL, and Grant-NE and McHenry-IL, were estimated to have occurred relatively recently (~22,550 YBP, 95 % CI = 16,550–90,650 YBP and ~ 22,250 YBP, 95 % CI = 20,550–95,450 YBP, respectively). Subsequent gene flow from Grant-NE to Carroll-IL, Will-IL also appears to be non-trivial (median m 2 = 8.25, 95 % CI = 3.65–21.35).
In contrast to these analyses of long-term gene flow, BayesAss detected no substantial recent migration between any of the four clusters defined by STRUCTURE (Table S11, Group 5 was excluded due to small sample size, as with IM analyses above). The highest unidirectional migration rate was estimated for Muscatine-IA, Scott-MN into Grant-NE, Carroll-IL, Will-IL at only 0.03 individuals/generation. By comparison, much higher migration rates were detected by BayesAss relative to IM, but were restricted within their respective STRUCTURE groups (m > 0.94, 95 % CI = 0.896–0.993).
Discussion
Overall, our genetic results accord with a classic biogeographic scenario (sensu Smith 1957; see below) that populations of Blanding’s turtle (Emys blandingii) across the midwestern United States (i.e., west of Lake Michigan) are significantly differentiated from each other. We identify 4–5 unique genetic groups of Blanding’s turtles in this region, which do not necessarily conform to their present geography. Indeed, although separated by >1,000 km, western Nebraska and eastern Illinois populations exhibit unexpectedly close population genetic structure. Regardless, our results also indicate strong support for the post-Pleistocene distribution of these turtles along watersheds in the Mississippi-Missouri River basins and along aquatic corridors established after the LGM, with limited gene flow more recently (Fig. 1).
Post-Pleistocene distribution of herpetofauna, including Blanding’s turtle, in the Great Lakes Region is thought to have occurred in two main phases. Species first migrated south and west during glacial advances, then colonized northern and eastern regions (and re-adjusted ranges in the south and west) during recession of the Laurentide Ice Sheet, with subsequent declines in population size and connectivity in the new locales (Smith 1957). Our molecular analyses of E. blandingii populations in the midwestern United States are consistent with this scenario, which invokes classic conditions of bottlenecks, reduced population sizes, and limited gene flow that promote among-population differentiation via random genetic drift at neutral genetic loci. Other molecular studies of post-glacial colonization and phylogeography of amphibians, snakes, and other turtle taxa in this general region comport with our findings (e.g., Austin et al. 2002; Fontanella et al. 2008; Janzen et al. 2002; Lee-Yaw et al. 2008; Placyk et al. 2007; Starkey et al. 2003; Weisrock and Janzen 2000). Unlike in our system, however, most nuclear and mitochondrial phylogeographic studies of herpetofauna in the Great Plains report little genetic variation, particularly among more northerly populations. These authors typically attribute this result to combined effects of population bottlenecks during glacial displacement and subsequent rapid northward colonization (e.g., Amato et al. 2008), but this pattern of genetic depauperation also may derive from slower rates of molecular evolution in those loci compared to the hypervariable microsatellite loci used in our study.
In general, Blanding’s turtle populations sampled in our study exhibit low to moderate genetic differentiation and considerable molecular phylogeographic structure. Linear distance among localities explains some of the genetic differentiation, but the signals of watershed and LGM distribution are also detectable and significantly explain a larger fraction of the genetic data. These geographic groupings of populations (Missouri River watershed, Minnesota River watershed, Mississippi River watershed inside the Des Moines Lobe of the Laurentide Ice Sheet, Mississippi River watershed outside the Des Moines Lobe of the Laurentide Ice Sheet, and Southern Lake Michigan Crescent watershed) notably, although not completely, correspond with the 4–5 unique genetic groups independently identified by the STRUCTURE analyses (Figs. 1, 3, Fig. S5).
These phylogeographic results further accord, in general, with the spatiotemporal dynamics of Pleistocene glacial advances and retreats in the midwestern United States. The peak of the Illinoisan glacial period occurred ~300,000–150,000 YBP (Mickelson and Colgan 2003), during which glaciers extended into Kansas, Missouri, and southern Illinois (Stiff and Hansel 2004). Our genetic data, through multiple phylogeographic and migration analyses, suggest that it is around this time that the NE, IA, and IL groups began to differentiate, potentially from an ancestral source population in the south-central Great Plains, which would accord with fossil evidence (summarized in Ernst and Lovich 2009). Subsequent glacial advances and retreats included those involving the Laurentide Ice Sheet during the Wisconsinan glacial period (~100,000–4,000 years ago) (Stiff and Hansel 2004), which reached as far south as south-central Iowa (i.e., the Des Moines Lobe). These non-uniform advance-retreat dynamics by glaciers presumably further created isolated aquatic corridors for northward and eastward colonization of regions by E. blandingii and other water-linked herpetofauna during our current Holocene interglacial period (e.g., Amato et al. 2008; Austin et al. 2002; Lee-Yaw et al. 2008; Starkey et al. 2003; see Fig. 1), which are reflected in the phylogeographic and gene flow relationships among population clusters.
Our phylogeographic findings are intriguing in light of other studies that had previously detected relatively little genetic differentiation among E. blandingii populations west of Lake Michigan (Mockford et al. 2007; Rubin et al. 2001; Spinks and Shaffer 2009). These three studies employed various molecular (nuclear RAPDs and microsatellites, nDNA and mtDNA sequences) and analytical (ANOVA, F st , STRUCTURE, phylogenetic, etc.) approaches. West of Lake Michigan, Mockford et al. (2007) used five di- or tri-nucleotide nuclear microsatellite loci to examine one IL, one WI, and three MN populations. They detected low F st values (< 0.10 in all 10 pairwise comparisons), yet significant differentiation, between IL, WI, and two of the MN sites. We also found significant genetic differentiation between an eastern IL and MN population (Figs. S6 and S7), but our values of F st between IL and MN were substantially higher than those reported in Mockford et al. (2007). We confirmed this genetic differentiation with STRUCTURE using only our genotypic data for turtles from Will-IL and Scott-MN. [We performed another STRUCTURE analysis using the five loci from our study with heterozygosity similar to that of the Mockford et al. study and only our 12 best-sampled localities (Figs. S6 and S7). This analysis confirmed our findings using all eight tetra-nucleotide loci and 18 localities.] We expect that our comparatively higher F st values between IL and MN could be due in part to the fact that MN sites for Mockford et al. (2007) were located along the Mississippi River in one of the largest known populations. The MN site that we sampled may be more isolated, and thus be subject to stronger drift and subsequently exhibit more differentiation, compared to eastern IL populations.
Genetic differentiation west of Lake Michigan, albeit weak, has been found in other studies as well. Rubin et al. (2001) studied the same IL and WI populations (and two others outside the region) using nuclear RAPD markers and detected weak differentiation among the IL and WI populations. In a study with an interspecific phylogenetic focus, Spinks and Shaffer (2009) included four midwestern populations (WI, two from MN, and NE) from which one individual each was sequenced at one mtDNA locus and three nDNA loci, and noted that “intraspecific branch lengths were relatively short…”. In our case, we targeted more populations (18 vs. 5 (Mockford et al. 2007), 3 (Rubin et al. 2001), and 4 (Spinks and Shaffer 2009), respectively) over a larger fraction of this geographic region. We also chose tetra-nucleotide microsatellite loci that were likely to be hypervariable (average number of alleles per locus across all 18 midwestern populations was ~20 vs. <10 across five midwestern populations (Mockford et al. 2007), <3 across three midwestern populations (Rubin et al. 2001), and <4 across nine populations throughout the range (Spinks and Shaffer 2009), respectively). These multiple methodological considerations may have enhanced our capability to detect genetic structure in this geographic region of the United States compared to the three previous studies.
As discussed above, while our findings are generally congruent with those obtained in studies of other aquatic herpetofauna, spatiotemporal dynamics of glacial advances and retreats, and multiple analytical approaches, we nonetheless obtained some unexpected results. Of particular note is the apparent genetic similarity of microsatellites between western Nebraska and eastern Illinois populations despite a linear distance between these localities of >1,000 km. Had we not sampled intervening Minnesota and Iowa populations of E. blandingii, we might have inferred incorrectly that this species possesses negligible genetic variation and structure in the midwestern United States. As it is, we are left without a geologically plausible explanation for this puzzling similarity. Possible hypotheses include (1) humans transported Blanding’s turtles between the two sites (e.g., Mormons during their forced exodus in 1846 from Illinois to Utah), (2) Blanding’s turtles at both sites retain similar ancestral genetic polymorphisms (i.e., incomplete lineage sorting), and (3) alleles at the microsatellite loci for Blanding’s turtles at these two sites have converged independently (i.e., homoplasy). In the first case, we cannot rule out the possibility of translocations by humans, but this would seem an improbable explanation for our findings considering our abundant sampling in these regions. Consequently, we attempted to resolve this conundrum by analyzing variation in DNA sequences of flanking regions of the most homozygous microsatellite locus (GmuD95) and two of the most variable microsatellite loci (GmuD121 and GmuD21). Results (see Figs. S2, S3, S4, Tables S12 and S13) from this analysis suggest two patterns of molecular evolution at these three loci—(a) possible allele size homoplasy and flanking region SNP variation at GmuD95, and (b) no repeat size variation and little SNP variation in flanking regions at GmuD121 and GmuD21, indicative of microsatellite saturation at these two loci (longest recorded allele size was 154 bases for GmuD121 and 152 bases for GmuD21 in these populations). Regardless, analysis without GmuD121, GmuD21, and GmuD95 still shows low genetic differentiation (F st < 0.184, P < 0.0001) between Grant-NE and eastern Illinois, and higher genetic differentiation between McHenry-IL and Will-IL (F st = 0.325, P < 0.0001) and between Grant-NE and Iowa populations (F st > 0.23, P < 0.0001).
Regardless of the explanation for this unusual pattern, our extensive genetic study of Blanding’s turtle in the midwestern United States has significant conservation and management implications for this imperiled species. We identified a considerable pool of genetic variation across populations and substantial geographic structuring of this genetic variation with relatively little recent gene flow, possibly because of colossal loss of hospitable environments between essential terrestrial and aquatic habitats and between population localities (e.g., Beaudry et al. 2008). This current situation could be catastrophic for E. blandingii and other taxa with movement-heavy, biphasic natural histories. For example, Blanding’s turtle has not reproduced in any known western Iowa populations for at least 20 years and those populations are now limited to a few very old (possibly 50–100 years old) turtles (Christiansen 1998). Moreover, evidence consistent with excessive inbreeding (e.g., % inviable eggs) has been detected in other Iowa localities (53 %; JLC, unpublished) and in the McHenry-IL population (48 %; SH, unpublished), in contrast to Grant-NE where the population size remains large (21 %; FJJ et al. unpublished). Beyond the need for detailed demographic studies, our genetic evaluation of midwestern Blanding’s turtles makes clear that any management action, such as assisted translocation of E. blandingii between localities, would benefit from being conducted with an eye toward accounting for the genetically structured groups that we detected (reviewed in Alacs et al. 2007). Although we unfortunately have no evidence of local phenotypic adaptation in E. blandingii, as noted above most molecular phylogeographic studies of herpetofauna in this region have reported little genetic variation. Consequently, midwestern Blanding’s turtles exemplify a relatively unique outcome and should accordingly evince a vigilant management approach to ensure retention of genetic diversity. Still, intensification of changes to the regional landscape is further restricting natural gene flow and population size for E. blandingii, thus balancing genetic and demographic concerns, among other issues, will require challenging management decisions (see also McGuire et al. 2013).
References
Alacs EA, Janzen FJ, Scribner KT (2007) Genetic issues in freshwater turtle and tortoise conservation. Chelonian Res Monogr 4:107–123
Amato ML, Brooks RJ, Fu J (2008) A phylogeographic analysis of populations of the wood turtle (Glyptemys insculpta) throughout its range. Mol Ecol 17:570–581
Austin JD, Lougheed SC, Neidrauer L, Chek AA, Boag PT (2002) Cryptic lineages in a small frog: the post-glacial history of the spring peeper, Pseudacris crucifer (Anura: Hylidae). Mol Phylogenet Evol 25:316–329
Avise JC (2010) Perspective: conservation genetics enters the genomics era. Conserv Genet 11:665–669
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE, Reeb CA, Saunders NC (1987) Intraspecific phylogeography—the mitochondrial DNA bridge between population genetics and systematics. Annu Rev Ecol Syst 18:489–522
Beaudry F, deMaynadier PG, Hunter ML Jr (2008) Identifying road mortality threat at multiple spatial scales for semi-aquatic turtles. Biol Conserv 141:2550–2563
Burnham KP, Anderson DR (1998) Model selection and inference: a practical information-theoretic approach. Springer-Verlag, New York
Byrne M, Yeates DK, Joseph L et al (2008) Birth of a biome: insights into the assembly and maintenance of the Australian arid zone biota. Mol Ecol 17:4398–4417
Christiansen JL (1998) Perspectives on Iowa’s declining amphibians and reptiles. J Iowa Acad Sci 105:109–114
Congdon JD, Keinath DA (2006) Blanding’s Turtle (Emydoidea blandingii): a technical conservation assessment. USDA Forest Service, Rocky Mountain Region. http://www.fs.fed.us/r2/projects/scp/assessments/blandingsturtle.pdf. Accessed Feb 2013
Congdon JD, Van Loben Sels RC (1993) Relationships of reproductive traits and body-size with attainment of sexual maturity and age in Blanding’s Turtles (Emydoidea blandingii). J Evol Biol 6:547–557
Congdon JD, Dunham AE, Van Loben Sels RC (1993) Delayed sexual maturity and demographics of Blanding’s turtles (Emydoidea blandingii)—implications for conservation and management of long-lived organisms. Conserv Biol 7:826–833
Congdon JD, Nagle RD, Osentoski MR, Kinney OM, Van Loben Sels RC (2003) Life history and demographic aspects of aging in the long-lived turtle (Emydoidea blandingii). In: Finch CE, Robine J-M, Christen Y (eds) Brain and longevity. Springer, Germany, pp 15–31
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ehlers J, Gibbard P (2008) Extent and chronology of Quaternary glaciation. Episodes 31:211–218
Ernst CH, Lovich JE (2009) Turtles of the United States and Canada, 2nd edn. Johns Hopkins, Baltimore
Ersts PJ (2010) Geographic Distance Matrix Generator (version 1.2.3). American Museum of Natural History, Center for Biodiversity and Conservation. http://biodiversityinformatics.amnh.org/open_source/gdmg/documentation.php. Accessed Feb 2011
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial-DNA restriction data. Genetics 131:479–491
Fontanella FM, Feldman CR, Siddall ME, Burbrink FT (2008) Phylogeography of Diadophis punctatus: extensive lineage diversity and repeated patterns of historical demography in a trans-continental snake. Mol Phylogenet Evol 46:1049–1070
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Fritz U, Schmidt C, Ernst CH (2011) Competing generic concepts for Blanding’s, Pacific and European pond turtles (Emydoidea, Actinemys and Emys)-which is best? Zootaxa 2791:41–53
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Hey J, Nielsen R (2004) Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167:747–760
Hoffmann M, Hilton-Taylor C, Angulo A et al (2010) The impact of conservation on the status of the world’s vertebrates. Science 330:1503–1509
Holman JA (1995) Pleistocene amphibians and reptiles in North America. Oxford Monographs on Geology and Geophysics No. 32, Oxford
Howes BJ, Brown JW, Gibbs HL, Herman TB, Mockford SW, Prior KA, Weatherhead PJ (2009) Directional gene flow patterns in disjunct populations of the black ratsnake (Pantherophis obsoletus) and the Blanding’s turtle (Emydoidea blandingii). Conserv Genet 10:407–417
Howeth JG, McGaugh SE, Hendrickson DA (2008) Contrasting demographic and genetic estimates of dispersal in the endangered Coahuilan box turtle: a contemporary approach to conservation. Mol Ecol 17:4209–4221
Jackson CJ, Kaye JM (1974) The occurrence of Blanding’s turtle, Emydoidea blandingii, in the Late Pleistocene of Mississippi (Testudines: Testudinae). Herpetologica 30:417–419
Janzen FJ, Krenz JG, Haselkorn TS, Brodie ED Jr, Brodie ED III (2002) Molecular phylogeography of common garter snakes (Thamnophis sirtalis) in western North America: implications for regional historical forces. Mol Ecol 11:1739–1751
Kimura M, Ohta T (1978) Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci USA 75:2868–2872
King TL, Julian SE (2004) Conservation of microsatellite DNA flanking sequence across 13 emydid genera assayed with novel bog turtle (Glyptemys muhlenbergii) loci. Conserv Genet 5:719–725
Lee-Yaw JA, Irwin JT, Green DM (2008) Postglacial range expansion from northern refugia by the wood frog, Rana sylvatica. Mol Ecol 17:867–884
Lewis P, Zaykin D (2008) GDA v1.1 http://www.eeb.uconn.edu/people/plewis/software.php. Accessed Feb 2010
McGuire JM, Scribner KT, Congdon JD (2013) Spatial aspects of movements, mating patterns, and nest distributions influence gene flow among population subunits of Blanding’s turtles (Emydoidea blandingii). Conserv Genet. doi:10.1007/s10592-013-0493-8
Mickelson DM, Colgan PM (2003) The southern Laurentide Ice Sheet. Develop Quat Sci 1:1–16
Mockford SW, Snyder M, Herman TB (1999) A preliminary examination of genetic variation in a peripheral population of Blanding’s turtle, Emydoidea blandingii. Mol Ecol 8:323–327
Mockford SW, McEachern L, Herman TB, Synder M, Wright JM (2005) Population genetic structure of a disjunct population of Blanding’s turtle (Emydoidea blandingii) in Nova Scotia, Canada. Biol Conserv 123:373–380
Mockford SW, Herman TB, Snyder M, Wright JM (2007) Conservation genetics of Blanding’s turtle and its application in the identification of evolutionarily significant units. Conserv Genet 8:209–219
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Placyk JS, Burghardt GM Jr, Small RL, King RB, Casper GS, Robinson JW (2007) Post-glacial recolonization of Michigan by the common gartersnake (Thamnophis sirtalis) inferred from mtDNA sequences. Mol Phylogenet Evol 43:452–467
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rambaut A, Drummond AJ (2007) Tracer v1.4. http://beast.bio.ed.ac.uk/Tracer. Accessed Feb 2010
Raymond M, Rousset F (1995) GENEPOP (Version-1.2)—population-genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rhodin AGJ, van Dijk PP (2011) Emydoidea blandingii. In: IUCN 2012. IUCN Red List of Threatened Species. Version 2012.1 www.iucnredlist.org. Accessed 15 Aug 2012
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rubin CS, Warner RE, Bouzat JL, Paige KN (2001) Population genetic structure of Blanding’s turtles (Emydoidea blandingii) in an urban landscape. Biol Conserv 99:323–330
Shaffer HB (2009) Turtles (Testudines). In: Hedges SB, Kumar S (eds) The timetree of life. Oxford University Press, Oxford, pp 398–401
Smith PW (1957) An analysis of post-Wisconsin biogeography of the prairie peninsula region based on distributional phenomena among terrestrial vertebrate populations. Ecology 38:205–218
Sommer RS, Lindqvist C, Persson A, Bringsoe H, Rhodin AGJ, Schneeweiss N, Siroky P, Bachmann L, Fritz U (2009) Unexpected early extinction of the European pond turtle (Emys orbicularis) in Sweden and climatic impact on its Holocene range. Mol Ecol 18:1252–1262
Spinks PQ, Shaffer HB (2009) Range-wide molecular analysis of the western pond turtle (Emys marmorata): cryptic variation, isolation by distance, and their conservation implications. Mol Ecol 14:2047–2064
Starkey DE, Shaffer HB, Burke RL, Forstner MRJ, Iverson JB, Janzen FJ, Rhodin AGJ, Ultsch GR (2003) Molecular systematics, phylogeography, and the effects of Pleistocene glaciation in the painted turtle (Chrysemys picta) complex. Evolution 57:119–128
Stiff BJ, Hansel AK (2004) Quaternary glaciations in Illinois. In: Ehlers J, Gibbard PL (eds) Quaternary glaciations: extent and chronology 2: Part II North America. Elsevier, Amsterdam, pp 71–82
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues ASL, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783–1786
Sutherland WJ, Aveling R, Bennun L et al (2012) A horizon scan of global conservation issues for 2012. Trends Ecol Evol 27:12–18
Ursenbacher S, Carlsson M, Helfer V, Tegelstrom H, Fumagalli L (2006) Phylogeography and Pleistocene refugia of the adder (Vipera berus) as inferred from mitochondrial DNA sequence data. Mol Ecol 15:3425–3437
Van Devender TR, King JE (1975) Fossil Blanding’s turtles, Emydoidea blandingii (Holbrook), and the late Pleistocene vegetation of western Missouri. Herpetologica 31:208–212
van Oosterhout C, Weetman D, Hutchinson WF (2006) Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol Ecol Notes 6:255–256
Walsh PS, Metzger DA, Higuchi R (1991) Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Weir BS (1996) Genetic data analysis II. Sinauer, Sunderland
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population-structure. Evolution 38:1358–1370
Weisrock DW, Janzen FJ (2000) Comparative molecular phylogeography of North American softshell turtles (Apalone): implications for regional and wide-scale historical evolutionary forces. Mol Phylogenet Evol 14:152–164
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wooding S, Ward RH (1997) Phylogeography and Pleistocene evolution in the North American black bear. Mol Biol Evol 14:1096–1105
You FM, Luo MC, Gu YQ, Lazo GR, Deal K, Dvorak J, Anderson OD (2007) GenoProfiler: batch processing of high-throughput capillary fingerprinting data. Bioinformatics 23:240–242
Acknowledgments
Thanks to Steve Dinkelacker, Robb Goldsberry, Chris Phillips, and Sara Ruane for assistance with collecting and to two anonymous reviewers for constructive criticisms. Turtles and tissues for this project were obtained under Iowa DNR Scientific Collecting permits to JLC and TJV, Illinois Endangered Species permits to FJJ and SH, Minnesota DNR permits to JMR, and a Nebraska Game & Parks Commission permit to Steve Dinkelacker. Funding for this research was provided in part by a contract from the McHenry County Conservation Foundation and National Science Foundation grants IBN-0080194, DEB-0089680, and DEB-0640932 to FJJ, and by a National Aeronautics and Space Administration Iowa Space Grant Consortium grant to FJJ and JLC. Additional funding for sequencing and bioinformatics support was provided by National Science Foundation DEB-0508665 to FJJ and EMM, as well as a grant from the EEOB Department at ISU and a James Cornette Fellowship to AS.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sethuraman, A., McGaugh, S.E., Becker, M.L. et al. Population genetics of Blanding’s turtle (Emys blandingii) in the midwestern United States. Conserv Genet 15, 61–73 (2014). https://doi.org/10.1007/s10592-013-0521-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-013-0521-8