
Abstract
The effective design of species conservation programs is reliant on information such as extant geographic distribution, taxon-specific life-history characteristics, and the relative influence of historic processes and contemporary environmental parameters in shaping population genetic diversity. Seahorses are weak swimmers and have a brooded young, limiting their dispersal potential. They live in sheltered locations, which are physically isolated from each other. Therefore panmixia across their geographic range is unlikely. Hippocampus guttulatus, a seahorse inhabiting European waters, has a geographic range spanning a number of contemporary oceanographic features that are proposed barriers to gene flow. Thus this fish is well-placed to test the contributions of environment and life-history factors in shaping population structuring. This study found that mitochondrial DNA and nuclear DNA (microsatellite) genotype data are concordant in suggesting that, like many other small fishes in European waters, H. guttulatus extant populations expanded from at least one southern European refugial population. Subsequent population differentiation of four geographic lineages reflects contemporary oceanographic barriers to gene flow. Demographic analyses suggest a northward, and long-term isolation between Black Sea and Mediterranean Sea populations. Moreover H. guttulatus contemporary population distribution and population structure are predominately explained by historic and oceanographic influences. These findings suggest that conservation of genetic diversity in H. guttulatus may be aided by a network of marine protected areas (MPAs), implemented to conserve coastal habitats, but the species’ unusual life history and gamete retaining behaviours should be considered as part of management decisions including MPA design and fisheries management plans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Marine conservation can be enhanced when species-specific biological, ecological and genetic data are considered in conjunction with environmental parameters. For example, estimations of genetic connectivity can help resolve the relative influence of historical versus current environmental features and processes on contemporary population structuring. In marine species, many examples have shown the utility of genetic data in establishing the importance of life-history traits (Galindo et al. 2010), environmental factors (e.g. hydrodynamics, Schunter et al. 2011) and historic processes (Maggs et al. 2008; Patarnello et al. 2007) in driving contemporary population diversity and structuring. Such studies have been used to inform species management and conservation strategies (Planes et al. 2009).
Marine species have been predicted to have little genetic structure due to propagule-dispersing factors, such as long larval phases and dispersal by oceanographic currents (Ward et al. 1994). Whilst some species show close fit to expected patterns of high genetic connectivity (e.g. the coral Astroides calycularis, Casado-Amezua et al. 2012), many species that have potential for high gene flow do not exhibit panmictic populations (e.g. European anchovy, Zarraonaindia et al. 2012; bluefin tuna, Riccioni et al. 2010), and some species predicted to have substantial population substructure, such as the marbled goby, a lagoon-dwelling fish, show widespread genetic homogeneity (Merjri et al. 2011). Thus the population structures of marine species may or may not be easily predicted, as they are determined by complex interaction of factors.
Environmental factors are regularly shown to influence genetic differentiation of marine species. Factors include coastal topography (Nicastro et al. 2008), oceanic currents (Quinteiro et al. 2007), bathymetric profile (Hoarau et al. 2002), habitat availability (Astolfi et al. 2005), and temperature and salinity discontinuities (Jorgensen et al. 2005). For example, water temperature changes seasonally in temperate zones, but currents (e.g. Gulf stream) or depth (e.g. thermoclines) can also maintain distinct long-term temperature discontinuities. Historic, events such as bottlenecks in population size (Bargelloni et al. 2005), species range expansion (Wilson 2006) and vicariance (Arnaud-Haond et al. 2007), occur in response to environmental or anthropogenic factors, and may also influence contemporary population connectivity, species geographic range, and the distribution of genetic diversity.
Both historic and extant environmental conditions in European waters of the Northeast Atlantic Ocean, Mediterranean Sea and Black Sea are known to have influenced their different faunal compositions. Within and between these waters there are a number of recognised barriers to individual dispersal that define population divergence of marine species, such as the Straits of Sicily and the Almeria-Oran frontal system in the Mediterranean Sea (Galarza et al. 2009; Patarnello et al. 2007; Schunter et al. 2011).
Seahorses have distinct life-history characteristics that have led researchers to hypothesize limited connectivity among patchily distributed populations, as well as making them more vulnerable to habitat destruction and overexploitation (reviewed by Vincent et al. 2011). Predictions of low individual dispersal capacity result from traits, such as internal fertilisation and brooding of young, a short planktonic juvenile phase (Boisseau 1967), small home range (Curtis and Vincent 2006) and weak swimming ability (Blake 1976). However seahorses are known to move up to 150 m daily, within a lagoon system (Caldwell and Vincent 2013) and have the potential for occasional migration events by rafting (Luzzatto et al. 2013; Perante et al. 2002; Vandendriessche et al. 2005). In addition, most seahorse species are socially (Foster and Vincent 2004) and genetically serially monogamous (Woodall et al. 2011a), which could result in a lower effective population size due to limited parental crossings. Seahorses therefore exhibit characteristics that suggest highly structured genetic populations.
Hippocampus guttulatus, Cuvier 1829, is distributed along coasts of the North-East Atlantic from the English Channel in the north to Northwest Africa in the south, and throughout the Mediterranean Sea and Black Sea (Lourie et al. 2004). Like many seahorses, H. guttulatus is a shallow coastal and estuarine dweller, often associated with seagrass beds (Curtis and Vincent 2005), potentially limiting dispersal across deep open water. Throughout its range these habitats are disjunct (Green and Short 2003) and as such this species exhibits non-continuous populations, which can reduce the chance of nearby populations mixing. Seahorses such as H. guttulatus are thus well placed to elucidate the relative influence of life history traits versus environmental and historical climatic factors in determining population connectivity.
A recent study assessed genetic variation in H. guttulatus across a small part of the species’ geographic range (NW Iberian Peninsula), and found no significant barriers to gene flow (Lopez et al. 2015), but studies of three other syngnathid species from areas more representative of the geographic range of H. guttulatus suggest more defined population differentiation in such species. Contemporary population structure of the pipefish Syngnathus typhle has been shown to be influenced by Pleistocene glaciations and post glacial recolonisation (Wilson and Eigenmann Veraguth 2010). Another European pipefish (S. abaster) displays significant post-glacial fragmentation and differentiation (Sanna et al. 2013). Similarly, both contemporary (i.e. oceanographic barriers) and historic factors (i.e. Pleistocene glaciation) were identified as shaping the population structure across European waters in Hippocampus hippocampus (Woodall et al. 2011b). However the finer population structure predicted by the species life-history characteristics was not present.
In this study mtDNA and nuclear DNA (microsatellite) markers were applied to samples from the entire geographic range of H. guttulatus to: investigate contemporary genetic population structure; identify potential barriers to gene flow; infer demographic history, including times of population divergence and range expansion; and propose conservation and management practices in the light of data from this and other European seahorse species.
Materials and methods
Sample acquisition and DNA extraction
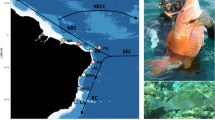
Specimens were collected from 17 locations across the NE Atlantic Ocean, northern Mediterranean Sea and Black Sea, covering over 6000 km of coastline and with a range of 60–1200 km between neighbouring sites (Fig. 1; Table 1). Since seahorses generally live in low densities, are cryptic, and are not commercially targeted in Europe, they are particularly difficult to sample. As a result at some sites it was necessary to re-sample over successive days and/or consecutive years (sites PUK, TPO, MSP and SFR). Tissue samples were collected from each individual in situ underwater and non-lethally to minimise impacts on individuals and populations (Woodall et al. 2012). Genomic DNA was isolated from 3–4 mm2 of dorsal fin tissue using a standard cetyltrimethyl ammonium bromide (CTAB) chloroform/isoamyl alcohol DNA extraction method (after Winnepenninckx et al. 1993).

Map of Hippocampus guttulatus sample sites and potential oceanographic barriers along the European coastline including the regional groupings assigned to populations (in brackets). Proposed oceanographic barriers to effective dispersal/gene flow in H. guttulatus 1 Brittany, 2 Cape Finisterre, 3 Gibraltar straits, 4 Almeria-Oran front, 5 Siculo-Tunisian front, 6 Bosphorus straits
Mitochondrial DNA sequencing
Fragments of mitochondrial DNA in the hypervariable 5′ end of the control region (CR) and the cytochrome b gene (cytb) were amplified for a maximum of 29 specimens from each of the 17 range-wide locations (Table 1). The CR was amplified using seahorse-specific primer HCAL2 (Teske et al. 2003) and H. guttulatus-specific primer HIPPCONR (5′AAG CCG AGC GTT CTC TCC3′). The cytb was amplified using primer SHORSE 5.3L (Casey et al. 2004) and H. guttulatus-specific primer GUTTCYTB-R (5′AGG GGG TTC TAC AGG CAT TAC3′). Each 50 µl PCR reaction contained: 5 µl 10 × manufacturer provided buffer, 2.5 µl MgCl2 (50 mM—Bioline, UK), 5 µl deoxynucleotide triphosphate mix (dNTP) (1.25 nM), 1.2 µl of each primer (10 µM), 0.25 µl Taq polymerase (5 U/µl—Bioline, UK), 14.25 µl H2O and 20 µl template DNA (10–50 ng). The PCR profile was composed of an initial denaturation step (2 min at 94 °C), followed by 35 cycles of denaturation (30 s at 94 °C), annealing (30 s at 50 °C) and extension (60 s at 72 °C), and a final extension step (2 min at 72 °C).
Amplified products were purified, using either PCR purification kit (Qiagen) or Exonuclease 1-Shrimp Alkaline phosphatase protocols, sequenced in both directions by Macrogen (Korea), then deposited in Genbank (Accession numbers: KM061952–KM062016).
Amplification and screening of microsatellites
Twenty-five previously developed seahorse-specific microsatellite primers (Galbusera et al. 2007; Pardo et al. 2006) were tested for amplification, allelic variation, null alleles and stutter bands. Five polymorphic microsatellite loci were selected for final screening (Hgu4, Hgu12, Hcaµ11, Hcaµ25 & Hcaµ27); the other loci either failed to amplify or were monomorphic. In total 313 specimens were genotyped from ten locations for a minimum of 15 individuals per site (Table 2). Loci were amplified separately in 10 µl reactions containing 2 µl template DNA (1–5 ng), 1 µl manufacturer-provided buffer, 0.6 µl MgCl2 (50 mM—Bioline), 1 µl dNTP mix (1.25 mM), 0.25 µl of each primer (10 µM, one being Cy5′ labelled), 0.05 µl Taq polymerase (5U/µl-Bioline). The thermocycle profile comprised an initial denaturing step (3 min at 95 °C), followed by 35 cycles of denaturing (30 s at 95 °C), annealing (30 s at 50 °C (Hgu12), 53 °C (Hcaµ11 and Hcaµ25) or 55 °C (Hgu4 and Hcaµ27), and extension (30 s at 72 °C), with a final extension step (3 min at 72 °C)). PCR products were run on 6 % denaturing polyacrylamide gels in an ALFexpressII automated DNA sequencer (Amersham Pharmacia) and allele sizes scored using Fragment Manager v2.9 (Amersham Pharamcia).
Genetic diversity
Sequence chromatographs were manually checked for errors and edited unambiguously in BIOEDIT v7.2.5 (Hall 1999). Consensus sequences were then aligned using CLUSTAL X (Thompson et al. 1997). Genetic diversity indices of haplotype diversity (h) and nucleotide diversity (π) were calculated in ARLEQUIN v3.5.1.3 (Excoffier and Lisher 2010). Diversity was calculated for all sample locations and regions with a sample size of 15 or larger. Genealogy networks created in TCS v1.21 (Clement et al. 2000), were used to visualise nucleotide sequence divergence and to illuminate the genetic relationship between haplotypes.
For microsatellite loci, number of alleles, conformity with Hardy–Weinberg (HW) expectation and linkage disequilibrium were computed in GENEPOP v4.0 (Rousset 2008). Observed and expected heterozygosity were calculated in ARLEQUIN. Due to small samples sizes, sites CGR and KGR were pooled to form a single Greek sample (GRE), after testing for allele frequency conformity between the individual samples.
Power analysis
No evidence of null alleles was detected within any microsatellite locus using FreeNA (Chapuis and Estoup 2007), and a <1 % genotyping error was established by re-scoring five separate gels of each locus and comparing allele sizes with the original scoring. POWSIM v4.0 (Ryman and Palm 2006) was used to test the power of the microsatellite data to detect signals of genetic differentiation with current sample sizes, using different levels of genetic divergence ranging from F ST = 0.005 to 0.200.
Genetic differentiation
Populations were combined into seven regions (Table 1) for testing for differentiation, based on geographic distance between sites and biogeographic provinces. Genetic structuring was assessed in ARLEQUIN with Analysis of Molecular Variance (AMOVA), using ΦST (mtDNA) and F ST (microsatellites) to test for significant differences within and between regions across Europe. To determine which pairwise comparisons contributed to the genetic structure inferred in AMOVA, two measures of genetic differentiation were used, the fixation index F ST implemented in ARLEQUIN for mtDNA and FSTAT v2.9.2.3 (Goudet 2001) for microsatellites. Estimates of gene flow between regions were made using the maximum likelihood method (ML), implemented in MIGRATE 3.2.1 (Beerli and Felsenstein 2001). The estimates in MIGRATE were based on MCMC simulations using ten long chains and five short chains, of 150,000 and 11,250 genealogies respectively, with a burn-in of an additional 10,000, data recorded every 20 reconstructed genealogies. The mutation model was derived by calculating the gamma distribution (alpha) in PAUP* v4.0b10 (Swofford 2003).
Subpopulation assignment tests were performed on population level microsatellite data in STRUCTURE v2.3.4 (Pritchard et al. 2000) using both admixture and no admixture models with a burn-in of 5 × 105 and 1 × 106 MCMC chains. Both models were tested, as some regions contained nearby unsampled populations (southern Portugal). These populations are more likely to have admixture than geographically distant/isolated ones (i.e. Black Sea). All possible numbers of populations (K) were tested (1–9), using 20 replicates, and the most parsimonious were assessed according to ΔK (Evanno et al. 2005) using STRUCTURE HARVESTER web v0.6.92 (Earl and vonHoldt 2012).
The Mantel test, which tests the correlation between genetic and geographic distance, was implemented in IBDWS v 3.23 (Jensen et al. 2005) using 30,000 randomisations on concatenated sequences and microsatellite genotypes separately. Distances between sampled sites were calculated using minimum sea distances (Table S1).
Historical processes
To infer the probability of demographic parameters we used an approximate Bayesian computation (ABC) approach in the program DIYABC v.2.03 (Cornuet et al. 2010, 2008), wherein molecular data are condensed into summary statistics and then compared to simulated data using a coalescent population model. For our model, we simulated four major regions of H. guttulatus distribution: 1) UK+BISCAY, 2) SPORT+MSP, 3) WMED+EMED, and 4) BLACK. This regional grouping was selected based on concordance of population differentiation estimates from both mtDNA and microsatellite analyses (see previous methods and Table 3 as well as Figs. 3, 4) and inferred oceanographic regions. The posterior distributions of parameters were calculated based on 1 million simulations using a total of 48 summary statistics. The fit of summary statistics to the model and chosen prior distributions were evaluated by locating the observed value and each summary statistic within a principal component analysis of 5000 simulated data sets. Microsatellite summary statistics included Mean size variance, two-sample F ST, and (du)2. Mitochondrial summary statistics included Mean pairwise differences, Variance of pairwise differences, Tajima’s D, Private segregating sites, and Mean numbers of rarest segregating sites. Between-population statistics included Mean of pairwise differences and F ST (Hudson et al. 1992). Simulations were based on a complete dataset of 214 individuals. Mutation rates for mtDNA and microsatellites were uniformly distributed with an upper and lower bound of 8.00E−9 to 1.3E−8 and 1.00E−005 to 1.00E−004 (in units of per site/per generation/per lineage) respectively. Uniform priors for effective population size ranged from (Ne) of 10 × 102 to 15 × 106, and divergence time 10 × 102 to 10 × 105 scaled to a generation time of 1 year. Euclidean distances between the observed and simulated data set were computed using a local linear regression, and 5000 of the closest simulated to the observed datasets were retained to estimate posterior distributions of 18 parameters, which included divergence times, effective populations sizes, and timing and magnitude of size change within each region (Table 4) (Beaumont et al. 2002; Cornuet et al. 2008).
Results
Population description
A total of 236 individuals were genotyped for both CR and cytb and concatenated to give a sequence of 991 bp. The concatenated sequences revealed 70 haplotypes, with the most common haplotype seen in 28 % of individuals across all regions. Total haplotype diversity was high (h = 0.91) and nucleotide diversity was low (π = 0.003) (Table 1). High haplotype diversity was found across all locations and regions, with the exception of the UK and southern France. Nucleotide diversity was low across all populations and regions. The maximum parsimony network of concatenated sequences resembles a shallow star-like pattern (Fig. 2). Little geographic structuring can be seen in the network. The most common haplotype is represented in all regions and almost all other common haplotypes are found in multiple regions. In addition all regions display multiple private haplotypes. However the percentage of private haplotypes present differed considerably between populations; the UK has none and the Black Sea 81 %, whereas the others have between 40 and 55 %.

Hippocampus guttulatus mtDNA haplotype network based on concatenated partial CR and cytochrome b sequences. Haplotypes are shown with size proportional to observed frequency, and segments represent the four proposed regional metapopulations. Lines indicate single mutations and black squares unobserved intermediate haplotypes
All 313 individuals sampled were genotyped at five microsatellite loci, with all loci displaying no significant overall departures from Hardy–Weinberg expectations of genotype frequencies or linkage disequilibrium. Moderate to low levels of genetic variability were observed at all loci (Table 2), but private alleles were present at each locus and all sampled locations. Observed and expected heterozygosity (Table 2) did not display geographic patterns, and no indication of inbreeding (FIS) was significant following Bonferroni correction. Power analysis based on sample size and screened microsatellite loci suggested that genetic divergence can be detected with >93 % confidence for F ST of 0.005, 98 % confidence for F ST of 0.007 and >99.9 % confidence for F ST ≥ 0.010. An expected F ST of zero estimates α to be 0.060–0.078, indicating expected levels of type I error. These results suggest that the five loci have the power to detect low levels of genetic differentiation (down to F ST of 0.005).
Genetic differentiation
The global F ST (ΦST 0.089 p < 0.0001 mtDNA; F ST = 0.087, p < 0.0001 microsatellites) indicated that there was significant population genetic differentiation across the sampled range. The AMOVA indicated that the greatest proportion of variation at both mtDNA and nDNA loci is among individuals within sample sites, although both marker types detected significant variation among regions (mtDNA: 10.4 %, ΦCT 0.104, p < 0.0001) (nDNA; 7.28 %, F CT 0.157, p < 0.005), with marginally significant variation between locations within regions for the microsatellite data (Table S2).
When samples were grouped and tested by geographical region, widespread significant genetic structuring was shown in both mtDNA and microsatellite data across the range of H. guttulatus (Table 3). The majority of pairwise F ST tests were significant even after sequential Bonferroni correction, the two exceptions being UK v BISCAY and MSP v SPORT. Gene flow estimates calculated in Migrate reveal a complex population structure (Fig. 3) that shares aspects of the pattern revealed in pairwise differentiation tests (Table 3), with high values within UK-BISCAY and SPORT-MSP but much lower values elsewhere. The UK-BISCAY estimates are bidirectional but unequal, with substantially more gene flow southwards, whereas the SPORT-MSP estimates are bidirectional and symmetrical. The Black Sea displays zero gene flow between it and all other regions. However the EMED populations do have genetic exchange with populations from WMED, MSP and SPORT. The STRUCTURE analysis with both admixture and non-admixture models indicated highest support for three genetic clusters among the sampled locations, which are UK-BISCAY, SPORT-MSP-WMED-EMED and BLACK (Fig. 4). Subsequent analysis of just the SPORT-MSP-WMED-EMED cluster shows clear of support for divergent clustering of SPORT-MSP and WMED-EMED (Fig. 4) resulting in four overall clusters. Further combinations of regional groups were tested in STUCTURE and results were consistent with the groups given above (data not shown). As a precautionary analysis to comply with the conservation management aims of the study, four metapopulations were chosen for demographic coalescent model analysis. Henceforth these metapopulations are referred to as N. ATLANTIC (UK and BISCAY), SW. IBERIA (SPORT and MSP), MED (WMED and EMED) and BLACK.

Map of Hippocampus guttulatus migration rates estimated using MIGRATE. The thicker the line the larger the migration rate and the dashed line shows no migrate exchange is suggested in any direction

Hippocampus guttulatus population structure inferred by STRUCTURE analysis; for the whole geographic region (a) and for the central regions (SW. IBERIA and MED) (b)
Mantel tests to assess correlation of genetic and geographic distance gave a positive and significant relationship among all Atlantic and Mediterranean samples (mtDNA: r = 0.6910, p < 0.01; microsatellites: r = 0.5267, p < 0.05). Subdivision of the sample sets indicated that the significant relationship was maintained across the samples from the Atlantic Ocean to Malaga site (MSP) (r = 0.5352 p < 0.001), but that no correlation existed across the Mediterranean samples (r = 0.3033, p > 0.05).
Historical processes
The DIYABC coalescent analysis indicated large values for contemporary effective population size in all four regional metapopulations (NE of ~730 to 1130 K—Table 4). Estimates of divergence times between the four populations were all relatively recent, ranging from 18 Kya between N.ATLANTIC and SW.IBERIA up to 66 Kya between SW.IBERIA and MED (Table 4). Estimates of time since population expansion are even more recent, ranging from 2.4 to 9.5 Kya (Table 4). However, because our models do not include divergence with gene flow, divergence times should be considered as approximations, allowing for the possibility of lineage divergence with gene flow taking place over a longer period of time.
Discussion
Genetic variability
Levels of genetic diversity within species, are important to consider in conservation and management plans, where maintenance of genetic diversity is recommended (Kenchington et al. 2003). The high haplotype number combined with low nucleotide diversity observed in H. guttulatus is indicative of recent population expansion (Grant and Bowen 1998) across the species range. There are two exceptions to this range-wide pattern: the most northern population in the UK and a population located in the Thau lagoon. The lower diversity in the UK can be explained by Hewitt’s (2000) model of colonisation of geographically peripheral range edge sites. However the Thau lagoon has extremely limited water exchange with the Mediterranean Sea, therefore its lower diversity more likely to be a result of inbreeding (Frankham 2005). These patterns are common in marine species (Astolfi et al. 2005; Gysels et al. 2004; Teske et al. 2003) both at the extreme limits of the species’ range and in isolated sites. Such differences in diversity, however, were not observed in H. hippocampus (Woodall et al. 2011b), which may result from differences in habitat preference between these two seahorse species, with H. hippocampus more often found along open coasts whereas H. guttulatus is more frequently found in discontinuous habitats such as estuaries and lagoons (Woodall 2009).
The range-wide ubiquity of a common mtDNA haplotype combined with many closely related haplotypes, and regional population groups with differing proportions of private haplotypes, in H. guttulatus is congruent with the distribution of microsatellite genotype variation. This pattern is common to other seahorses and is thought to reflect post-bottleneck expansions from a single refugium with ongoing contemporary gene flow (Saarman et al. 2010; Woodall et al. 2011b). Other syngnathids species, however display different distributions of genetic diversity, so species-specific characteristics need to be discussed and taken into account in management. The Mediterranean lagoon-dwelling pipefish Syngnathus abaster has a more complex haplotype network but no shared haplotypes between populations, and similar nucleotide diversity to seahorses (Sanna et al. 2013). This suggests that the fragmented habitat and life-history characteristics of the species have resulted in population isolation and breakdown of gene flow following the initial post-glacial expansion. On a larger geographical scale the seahorse H. erectus also demonstrates regionality and genomic divergence, with little connectivity between northern and southern populations occupying waters with very different environmental conditions (Boehm et al. 2015).
Population structuring
Genetic analysis revealed a complex pattern of subpopulations and connectivity with the initial regional assignments (Table 1), with four geographically defined lineages: UK to northern Spain; Portugal to Malaga on the Mediterranean coast of Spain; the rest of the Mediterranean; and the Black Sea. There was some evidence for divergence between western and eastern regions of the Mediterranean Sea, but this was to an extent much less than the other divisions and not supported by all analyses.
Subpopulation genetic divergence revealed in H. guttulatus appears to partially reflect that found in the congeneric and co-distributed short snouted seahorse H. hippocampus (Woodall et al. 2011b). It may be expected that both species would have a similar pattern of population differentiation as they can co-occur and have very similar life-history characters. However they show differences in micro-habitat preference (Curtis et al. 2007) and macro-habitat distribution (Woodall 2009). Hippocampus hippocampus also has a greater southern latitudinal range and is thought to have undergone more recent range expansion than H. guttulatus (Boehm et al. 2013; Teske et al. 2007) as H. guttulatus is considered to have diverged much earlier than H. hippocampus. The apparent greater structuring of H. guttulatus suggests that a different combination of historical and contemporary processes may have contributed to these species’ population structure.
Impact of life history on genetic diversity
In contrast to expectations of very limited dispersal and gene flow predicted from species biology and life history, the pattern of limited genetic differentiation observed within and among geographical regions across the species range suggests that H. guttulatus dispersal, although limited in places, is sufficient to maintain long-term gene flow across relatively large distances. The apparent isolation of the Black Sea population, signified by significant in-breeding and genetic differentiation, and breakdown of gene flow across several regions (NW Iberia and Mediterranean coast of southern Spain) illustrates the potential for this species to form segregated populations. The overall genetic structure across large areas suggests that unsampled stepping-stone populations could be the conduit for genetic exchange between sampled populations, as supported by isolation-by-distance effects across large parts of the range (Palumbi 2003).
Contemporary barriers to gene flow in H. guttulatus
Cape Finisterre
A major barrier to gene flow between the northern Spanish and southeast Portuguese sites was supported in H. hippocampus (Woodall et al. 2011b) and a similar pattern is consistent with our analysis of H. guttulatus. Other studies have observed Cape Finisterre in northwest Spain as being associated with genetic differentiation of marine populations (Neiva et al. 2012; Piñeira et al. 2008; Quesada et al. 1998) although a small-scale study of H. guttulatus across this area did not find population differentiation to either side of the cape (Lopez et al. 2015). A more southerly barrier to gene flow, between Rio Mondego and Rio Sado in central Portugal, has been suggested for other marine species (Diekmann et al. 2005; Pascoal et al. 2009). Further small-scale research will be required to elucidate exactly where gene flow breaks between H. guttulatus populations around the northwestern Iberian Peninsula occur. However the interaction of current and upwelling systems along with fragmented habitat are likely to define the location of the barrier, which could be understood by biophysical oceanographic modelling (Nolasco et al. 2013).
Gibraltar Straits or Almeria/Oran front?
Genetic data indicate that the Malaga population, in the Alboran Sea, east of the Straits of Gibraltar but west of the Almeria-Oran front (AOF), is part of the southern Iberia metapopulation (Atlantic coast). Thus the AOF correlates with H. guttulatus population structure and is the likely barrier to genetic exchange between Atlantic Ocean and Mediterranean Sea populations. The Atlantic–Mediterranean biogeographic boundary has been analysed in over 70 studies of many different marine organisms, and both fish (Charrier et al. 2006; Domingues et al. 2007) and invertebrates (Baus et al. 2005; Perez-Losada et al. 2002) show genetic differentiation correlating with the AOF. A number of studies, reviewed in Patarnello et al. (2007), suggest that the AOF is a significant physical barrier to individual dispersal and gene flow.
The Siculo-Tunisian Straits
Although to a much lesser extent than other gene flow boundaries identified in the present study, there is some evidence for genetic differentiation between populations of the western and eastern basins of the Mediterranean. The shallow sill of the Siculo-Tunisian Straits disrupts local hydrodynamics and current flows, and so hinders genetic exchange between the two basins in a number of marine species (Merji et al. 2009; Serra et al. 2010) and is thought to be a biogeographic boundary (Bianchi and Morri 2000). The absence of isolation-by-distance effects in H. guttulatus across the Mediterranean suggest that the differentiation across the Siculo-Tunisian Strait is worthy of further investigation and decision as to its importance to management of this species.
The Bosporus Straits and the Black Sea
The Black Sea is geographically isolated with only a narrow connection to the Mediterranean Sea through the Bosporus Straits. The historic isolation of the Black Sea and its distinct present environmental parameters (Sorokin 2002) suggest that the observed seahorse population structure could be a result of both historic and contemporary conditions. Our coalescent analysis suggests that historically the Black Sea population diverged from that in the Mediterranean roughly 50 Kya, just prior to the last glacial maximum (LGM), followed by a more recent population expansion after the LGM. The present low but significant genetic differentiation of the Black Sea H. guttulatus population (Table 3) indicates that it has not yet achieved migration-drift equilibrium with the Mediterranean population since the LGM. Genetic differentiation of Black Sea from eastern Mediterranean fish populations has been reported previously (Debes et al. 2008; Durand et al. 2013; and see Patarnello et al. 2007), but by contrast so has genetic homogeneity in other fish species (Magoulas et al. 2006), including the confamilial pipefish (Wilson & Eigenmann Veraguth 2010). It is likely that H. guttulatus has experienced episodic colonisation, isolation and gene flow in the Black Sea during multiple glacial cycles, in common with other fish species such as shad (Faria et al. 2012), but at present the Black Sea appears to harbour a distinct subpopulation of this seahorse.
Historic demographic effects on diversity and distribution
The H. guttulatus mtDNA haplotype network is consistent with a past demographic process of population expansion following a bottleneck across the species range. Such a demographic signature of population bottleneck plus expansion is found in many other marine fishes across the same geographic range (e.g. Domingues et al. 2007), including other Syngnathids (Saarman et al. 2010; Wilson and Eigenmann Veraguth 2010; Woodall et al. 2011b). The DIYABC analyses suggest that isolation and divergence of populations of H. guttulatus across Europe occurred during the LGM (66–18 Kya), and that population expansions occurred in all sub-populations after the LGM to the present (<10 Kya). Similar demographic signatures of past glacial periods are commonly seen in European marine species, and in some populations of Syngnathidae (Maggs et al. 2008; Wilson and Eigenmann Veraguth 2010; Woodall et al. 2011b). In common with other temperate marine species, and in accord with Hewitt’s (2000) model, the presence of a common haplotype across the range and higher genetic diversity of the more southern populations (Iberian peninsula and Mediterranean in the present study) indicates that these areas harboured larger or refugial populations during previous glacial periods of the Pleistocene, and that the more northern populations of Biscay that exhibit the most extreme signals of expansion (Table 1) may have been extirpated and subsequently recolonized (at least during the more extreme glacial maxima before the LGM.
Conservation conclusions
Our data indicate substantial genetic diversity and connectivity across the European range of H. guttulatus, but also the effects of two substantial barriers to gene flow (and consequent genetic differentiation), at Cape Finisterre and the Bosporus Straits, and further differentiation across the AOF and between the eastern and western Mediterranean. These patterns reveal that both contemporary processes (life-history and oceanographic features) and historic (paleoclimatic) events influence present population structure of H. guttulatus. We suggest that following the initial speciation in the Miocene (Teske et al. 2007), contraction of the species range during Pleistocene glacial maxima to at least one southern European refugial population followed by recurrent expansion and re-colonisation from these sites has been mediated by the isolating mechanism of oceanographic features combined with the low dispersal potential of H. guttulatus.
Current genetic structuring and diversity suggests four main H. guttulatus metapopulations, with potential subdivision of the east and west Mediterranean, which should be recognised as management units (MU) (Palsboll et al. 2006). Further studies are required to confirm the status of the Mediterranean populations, to provide details of genetic differentiation across smaller geographic ranges (additional sub-structuring) and of specific genetic barriers. This data could be used to determine if particular priority should be given to specific populations (Volkmann et al. 2014). However current data suggest that the MU designation is robust and should be considered as the basis of a management strategy for this species, which would mean combining range-wide coastal habitat conservation and transboundary planning for protected areas.
Connectivity around the coastline is reliant on suitable habitat for H. guttulatus, which should be considered carefully in conservation plans. The observed population structure suggests that the sedentary nature of this fish is most likely partially offset by the dispersal of juveniles as zooplankton, occasional migration events by adults, and/or dispersal by rafting (Luzzatto et al. 2013).
Coastal ecosystems have many wildly varying environmental parameters, suggesting seahorses often experience non-ideal conditions, which in turn may cause demographic fluctuations (Curtis and Vincent 2006; Woodall 2009, 2012). These demographic decreases may be the drivers for the observed genetic differentiation. Additionally, reduced genetic diversity as a result of these localised bottleneck events is thought to be an indicator of extinction risk in threatened species (Frankham 2005). Care should therefore be taken not just to conserve H. guttulatus metapopulations but also to protect potential habitat. Indeed this ecosystem management approach is now popular (Pérez-Ruzafa et al. 2008), and an identified international fisheries policy goal (Veitch et al. 2012). The effects of contemporary climate change is also likely to result in changes to the population structure through habitat and hydrodynamic changes, and thus to the location and possibly the composition of the MUs suggested here. Hippocampus guttulatus is currently listed as Data Deficient on the IUCN Red List (Woodall 2012) with a suggestion that more information on population demographic changes is required before it can be categorized. Therefore long-term monitoring of known populations is required to determine population trends. There is no known targeted fishery for this species, but seahorses are threatened by anthropogenic activities in coastal ecosystems, such as habitat disturbance from aggregate dredging, coastal development, pollution and fishing activity (Vincent et al. 2011). As reported for H. hippocampus (Woodall et al. 2011b) there are no Europe-wide conservation measures in place for seahorses, but it is important for management agencies to work internationally due to the transboundary nature of the H. guttulatus’ range and proposed MUs. In summary, the design of any proposed international management strategies should be informed by the meta-populations elucidated in this study, but further monitoring of population structure and demography is recommended to ensure the long-term viability of European seahorse populations.
References
Arnaud-Haond S, Migliaccio M, Diaz-Almela E, Teixira S, Van de Vilet MS, Alberto F, Procaccini G, Duarte CM, Serrao EA (2007) Vicariance patterns in the Mediterranean Sea: east-west cleavage and low dispersal in the endemic seagrass Posidonia oceanica. J Biogeogr 34:963–976
Astolfi L, Dupanloup I, Rossi R, Bisol PM, Faure E, Congiu L (2005) Mitochondrial variability of sand smelt Atherina boyeri populations from north Mediterranean costal lagoons. Mar Ecol Prog Ser 297:233–243
Bargelloni L, Alarcon JA, Alvarez MC, Penzo E, Magoulas A, Palma J, Patarnello T (2005) The Atlantic-Mediterranean transition: discordant genetic patterns in two seabream species Diplodus puntazzo (Cetti) and Diplodus sargus (L.). Mol Phylogenet Evol 36:523–535
Baus E, Darrock DJ, Bruford MW (2005) Gene-flow patterns in Atlantic and Mediterranean populations of the Lusitanian sea star Asterina gibbosa. Mol Ecol 14:3373–3382
Beaumont MA, Zhang W, Balding DJ (2002) Approximate Bayesian computation in population genetics. Genetics 162:2025–2035
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98:4563–4568
Bianchi NC, Morri C (2000) Marine biodiversity of the Mediterranean Sea: situation, problems and prospects for future research. Mar Poll Bullet 40:367–376
Blake RW (1976) On seahorse locomotion. J Mar Biol Assoc UK 56:939–949
Boehm JT, Woodall LC, Teske PR, Lourie SA, Baldwin C, Waldman J, Hickerman M (2013) Marine dispersal and barriers drive Atlantic seahorse diversification. J Biogeogr 40:1839–1849
Boehm JT, Waldman J, Robinson JD, Hickerson MJ (2015) Population genomics reveals seahorses Hippocampus erectus of the western mid-Atlantic coast to be residents rather than vagrants. PLoS One 10:e0116219
Boisseau J (1967) Les régulations hormonales de l’incubation chez un vertèbre male: recherches sur la reproduction de l’hippocampe. PhD thesis, L’Université de Bordeaux, France
Caldwell IR, Vincent ACJ (2013) A sedentary fish on the move: effects of displacement on long-snouted seahorse (Hippocampus guttulatus Cuvier) movement and habitat use. Environ Biol Fish 96:67–75
Casado-Amezua P, Goffredo S, Templado J, Machordom A (2012) Genetic assessment of population structure and connectivity in the threatened Mediterranean coral Astroides calycularis (Scleractinia, Dendrophylliidae) at different spatial scales. Mol Ecol 21:3671–3685
Casey SP, Hall HJ, Stanley HF, Vincent ACJ (2004) The origin and evolution of seahorses (genus Hippocampus): a phylogenetic study using the cytochrome b gene of mitochondrial DNA. Mol Phylogenet Evol 30:261–272
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Charrier G, Chenel T, Durand JD, Girand M, Quiniou L, Laroche J (2006) Discrepancies in phylogeographical patterns of two European anglerfishes (Lophius budegassa and Lophius piscatorius). Mol Phylogenet Evol 38:742–754
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cornuet J-M, Santos F, Beaumont MA, Robert CP, Marin J-M, Balding DJ, Guillemaud T, Estoup A (2008) Inferring population history with DIY ABC: a userfriendly approach to approximate Bayesian computation. Bioinfomatics 24:2713–2719
Cornuet J-M, Ravigne V, Estoup A (2010) Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform 11:401
Curtis JMR, Vincent ACJ (2005) Distribution of sympatric seahorse species along a gradient of habitat complexity in a seagrass-dominated community. Mar Ecol Prog Ser 291:81–91
Curtis JMR, Vincent ACJ (2006) Life history of an unusual marine fish: survival, growth and movement patterns of Hippocampus guttulatus Cuvier 1829. J Fish Biol 68:707–733
Curtis JMR, Ribeiro J, Erzini K, Vincent ACJ (2007) A conservation trade-off? Interspecific differences in seahorse responses to experimental changes in fishing effort. Aquat Conserv 17:468–484
Debes PV, Zachos FE, Hanel R (2008) Mitochondrial phylogeography of the European sprat (Sprattus sprattus L., Clupeidae) reveals isolated climatically vulnerable populations in the Mediterranean Sea and range expansion in the northeast Atlantic. Mol Ecol 17:3873–3888
Diekmann OE, Coyer JA, Ferreira J, Olsen JL, Stam WT, Pearson GA, Serrao EA (2005) Population genetics of Zostera noltii along the west Iberian coast: consequences of small population size, habitat discontinuity and near-shore currents. Mar Ecol Prog Ser 290:89–96
Domingues VS, Faria C, Stefanni S, Santos RS, Brito A, Almada VC (2007) Genetic divergence in the Atlantic-Mediterranean Montagu’s blenny, Coryphoblennius galerita (Linnaeus 1758) revealed by molecular and morphological characters. Mol Ecol 16:3592–3605
Durand JD, Blel H, Shen KN, Koutrakis ET, Guinand B (2013) Population genetic structure of Mugil cephalus in the Mediterranean and Black Seas: a single mitochondrial clade and many nuclear barriers. Mar Ecol Prog Ser 474:243–261
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lisher HEL (2010) Arlequin ver. 3.5: a new series of programs to perform population genetics analysis under Linux and Windows. Mol Ecol Resour 10:564–567
Faria R, Weiss S, Alexandrino P (2012) Comparative phylogeography and demographic history of European shads (Alosa alosa and A. fallax) inferred from mitochondrial DNA. BMC Evol Biol 12:194
Foster SJ, Vincent ACJ (2004) Life history and ecology of seahorses: implications for conservation and management. J Fish Biol 65:1–61
Frankham R (2005) Genetics and extinction. Biol Conserv 126:131–140
Galarza JA, Carreras-Carbonell J, Macpherson E, Pascual M, Roques S, Turner GF, Rico C (2009) The influence of oceanographic fronts and early life-history traits on connectivity among littoral fish species. Proc Natl Acad Sci USA 106:1473–1478
Galbusera PHA, Gillemot S, Jouk P, Teske PR, Hellemans B, Volckaert AMJ (2007) Isolation of microsatellite markers for the endangered Knysna seahorse Hippocampus capensis and their use in the detection of a genetic bottleneck. Mol Ecol Notes 7:638–640
Galindo HM, Pfeiffer-Herbert AS, McManus MA, Chao Y, Chai F, Palumbi SR (2010) Seascape genetics along a steep cline: using genetic patterns to test predictions of marine larval dispersal. Mol Ecol 19:3692–3707
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). Universite de Lausanne, Lausanne
Grant WS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89:415–426
Green EP, Short FT (2003) World atlas of seagrasses. University of California Press, Berkeley
Gysels ES, Hellemans B, Patarnello T, Volckaerst FAM (2004) Current and historical gene flow of the sand goby Pomatoschistus minutus on the European Continental Shelf and in the Mediterranean Sea. Biol J Linn Soc 83:561–576
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hoarau G, Rijnsdorp AD, Van der Veer HW, Stam WT, Olsen JL (2002) Population structure of plaice (Pleuronectes platessa L.) in northern Europe: microsatellite revealed large-scale spatial and temporary homogeneity. Mol Ecol 11:1165–1176
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of gene flow from DNA sequence data. Genetics 132:583–589
Jensen JL, Bohonah AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13
Jorgensen HBH, Hansen MM, Bekkevold D, Ruzzante DE, Loeschcke V (2005) Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Mol Ecol 14:3219–3234
Kenchington E, Heino M, Nielsen EE (2003) Managing marine genetic diversity: time for action? ICES J Mar Sci 60:1172–1176
Lopez A, Vera M, Planas M, Bouza C (2015) Conservation genetics of threatened Hippocampus guttulatus in vulnerable habitats in NW Spain: temporal and spatial stability of wild populations with flexible polygamous mating system in captivity. PLoS One 10:e0117538
Lourie SA, Foster SJ, Cooper EWT, Vincent ACJ (2004) A guide to the identification of seahorses, Washington D.C.
Luzzatto DC, Estalles ML, Diaz de Astarloa JM (2013) Rafting seahorses: the presence of the seahorse Hippocampus patagonicus in floating debris. J Fish Biol 83:677–681
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J, Olsen J, Perez KE, Stam W, Vainola R, Viard F, Wares J (2008) Evaluating valuating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89:S108–S122
Magoulas A, Castilho R, Caetano S, Marcato S, Patarnello T (2006) Mitochondrial DNA reveals a mosaic pattern of phylogeographical structure in Atlantic and Mediterranean populations of anchovy (Engraulis encrasicolus). Mol Phylogenet Evol 39:734–746
Merji R, Lo Butto S, Ben Hassine OK, Arculeo M (2009) A study on Pomatoschistus tortonesei Miller 1968 (Perciformes, Gobiidae) reveals the Siculo-Tunisian Strait (STS) as a breakpoint for gene flow in the Mediterranean basin. Mol Phylogenet Evol 53:596–601
Merjri R, Arculeo M, Ben Hassine OK, Brutto SL (2011) Genetic architecture of the marbled goby Pomatoschistus marmoratus (Perciformes, Gobiidae) in the Mediterranean Sea. Mol Phylogenet Evol 58:395–403
Neiva J, Pearson GA, Valero M, Serrao EA (2012) Fine-scale genetic breaks driven by historical range dynamics and ongoing density-barrier effects in the estuarine seaweed Fucus ceranoides L. BMC Evol Biol 12:78
Nicastro KR, Zardi GI, McQuaid CD, Teske PR, Barker NP (2008) Coastal topography drives genetic structure in marine mussels. Mar Ecol Prog Ser 368:189–195
Nolasco R, Dubert J, Dominges CP, Cordeiro Piers A, Queiroga H (2013) Model-derived connectivity patterns along the western Iberian Peninsula: asymmetrical larval flow and source-sink cell. Mar Ecol Prog Ser 485:123–142
Palsboll PJ, Berube M, Allendorf FW (2006) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Palumbi SR (2003) Population genetics, demographic connectivity and the design of marine reserves. Ecol Appl 13:S146–S158
Pardo BG, López A, Martínez P, Bouza C (2006) Novel microsatellite loci in the threatened European long-snouted seahorse (Hippocampus guttulatus) for genetic diversity and parentage analysis. Conserv Genet 8:1243–1245
Pascoal S, Creer S, Taylor MI, Queiroga H, Carvalho G, Mendo S (2009) Development and application of microsatellites in Carcinus maenas: genetic differentiation between northern and central Portuguese populations. PLoS One 4:e7268
Patarnello T, Volckaert FA, Castilho R (2007) Pillars of Hercules: is the Atlantic-Mediterranean transition a phylogeographical break? Mol Ecol 16:4426–4444
Perante NC, Pajaro MG, Meeuwig JJ, Vincent ACJ (2002) Biology of a seahorse species, Hippocampus comes in the central Philippines. J Fish Biol 60:821–837
Perez-Losada M, Guerra A, Carvalho GR, Sanjuan A, Shaw PW (2002) Extensive population subdivision of the cuttlefish Sepia officinalis (Mollusca: Cephalopoda) around the Iberian Peninsula indicated by microsatellite DNA variation. Heredity 89:417–424
Pérez-Ruzafa A, Marcos C, García-Charton JA, Salas F (2008) European marine protected areas (MPAs) as tools for fisheries management and conservation. J Nat Conserv 16:187–192
Piñeira J, Quesada H, Rolán-Alvarez E, Caballero A (2008) Genetic discontinuity associated with an environmentally induced barrier to gene exchange in the marine snail Littorina saxatilis. Mar Ecol Prog Ser 357:175–184
Planes S, Jones GP, Thorrold SR (2009) Larval dispersal connects fish populations in a network of marine protected areas. Proc Natl Acad Sci USA 106:5693–5697
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure from multilocus genotype data. Genetics 155:945–959
Quesada H, Gallagher C, Skibinski DAG, Skibinski DOF (1998) Patterns of polymorphism and gene flow of gender- associated mitochondrial DNA lineages in European mussel populations. Mol Ecol 7:1041–1051
Quinteiro J, Rodriguez-Castro J, Rey-Mendez M (2007) Population genetic structure of the stalked barnacle Pollicipes pollicipes (Gmelin, 1789) in the North Eastern Atlantic: influence of coastal currents and mesoscale hydrographic structures. Mar Biol 153:47–60
Riccioni G, Landi M, Ferrara G, Milano I, Cariani A, Zane L, Sella M, Barbujan G, Tinti F (2010) Spatio-temporal population structuring and genetic diversity retention in depleted Atlantic Bluefin tuna of the Mediterranean Sea. Proc Natl Acad Sci USA 107:2102–2107
Rousset F (2008) GENEPOP’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Notes 8:103–106
Ryman N, Palm S (2006) POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol 6:600–602
Saarman NP, Louie KD, Hamilton H (2010) Genetic differentiation across eastern Pacific oceanographic barriers in the threatened seahorse Hippocampus ingens. Conserv Genet 11:1989–2000
Sanna D, Biagi F, Alaya HB, Maltagliati F, Addis A, Romero A, De Juan J, Quignard J-P, Castelli A, Franzoi P, Torricelli P, Casu M, Carcupino M, Francalacci P (2013) Mitochondrial DNA variability of the pipefish Syngnathus abaster. J Fish Biol 82:856–876
Schunter C, Carreras-Carbonell J, Macpherson E, Tintore J, Vidal-Vijande E, Pascual A, Guidetti P, Pascual M (2011) Matching genetics with oceanography: directional gene flow in a Mediterranean fish species. Mol Ecol 20:5167–5181
Serra IA, Innocenti AM, Dimaida G, Calvo S, Migliaccio M, Zambianchi E, Pizzigalli C, Arnaud-Haond S, Duarte CM, Serrao EA, Procaccini G (2010) Genetic structure in the Mediterranean seagrass Posidonia oceanica: disentangling past vicariance events from contemporary patterns of gene flow. Mol Ecol 19:557–568
Sorokin YI (2002) The Black Sea: ecology and oceanography. Backhuis Publishing, Leiden
Swofford DL (2003) PAUP*. Phylogenetic analysis using parsimony (* and other methods). Sinauer Associates, Sunderland
Teske PR, Cherry MI, Matthee CA (2003) Population genetics of the endangered Knysna seahorse, Hippocampus capensis. Mol Ecol 12:1703–1715
Teske PR, Hamilton H, Matthee CA, Barker NP (2007) Signatures of seaway closures and founder dispersal in the phylogeny of a circumglobally distributed seahorse lineage. BMC Evol Biol 7:138
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Vandendriessche S, Messiaen M, Vincx M, Degraer S (2005) Juvenile Hippocampus guttulatus from a neuston tow at the French-Belgian border. Belg J Zool 135:101–102
Veitch L, Dulvy NK, Koldewey HJ, Lieberman S, Pauly D, Roberts CM, Rodgers AD, Baillie JEM (2012) Avoiding empty ocean commitments at Rio+ 20. Science 336:1383–1385
Vincent AC, Foster SJ, Koldewey HJ (2011) Conservation and management of seahorses and other Syngnathidae. J Fish Biol 78:1681–1724
Volkmann L, Martyn I, Moulton V, Spillner A, Mooers AO (2014) Prioritizing populations for conservation using phylogenetic networks. PLoS One 9:e88945
Ward RD, Woodmark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44:213–232
Wilson AB (2006) Genetic signature of recent glaciation on populations of a near-shore marine fish species (Syngnathus leptorhynchus). Mol Ecol 15:1857–1871
Wilson AB, Eigenmann Veraguth I (2010) The impact of Pleistocene glaciation across the range of a widespread European coastal species. Mol Ecol 19:4535–4553
Winnepenninckx B, Backeljau T, Dewachter R (1993) Extraction of high molecular-weight DNA from molluscs. Trends Genet 9:407
Woodall LC (2009) The population genetics and mating systems of European seahorses. PhD thesis, Royal Holloway-University of London, UK
Woodall LC (2012) Hippocampus guttulatus. In: IUCN 2013. IUCN Red List of Threatened Species, Version 2013.1 http://www.iucnredlist.org. Assessed 24 Nov 2013
Woodall LC, Koldewey HJ, Shaw PW (2011a) Serial monogamy in the European long-snouted seahorse Hippocampus guttulatus. Conserv Genet 12:1645–1649
Woodall LC, Koldewey HJ, Shaw PW (2011b) Historical and contemporary population genetic connectivity of the European short-snouted seahorse Hippocampus hippocampus and implications for management. J Fish Biol 78:1738–1756
Woodall LC, Jones R, Zimmerman B, Guillaume S, Stubbington T, Shaw P, Koldewey HJ (2012) Partial fin-clipping as an effective tool for tissue sampling seahorses, Hippocampus spp. J Mar Biol Assoc UK 92:1427–1432
Zarraonaindia I, Iriondo M, Albaina A, Pardo MP, Manzano C, Grant WS, Irigoien X, Estonba A (2012) Multiple SNP markers reveal fine-scale population and deep phylogeographic structure in European anchovy (Engraulis encrasicolus L.). PLoS One 7:e42201
Acknowledgments
This is a contribution from Project Seahorse. We gratefully acknowledge France: C. L. Milinaire, P. Moriniere, S. Auffret, X. De Montandouin, P. Louisy, J-B Senegas, P. LeLong, Spain: J.A. Rodriguez, Scubadoo, A.Martinez de Murguia, B. Moya, Greece: F. Vilanikis, Y. Issaris, M. Salomidi, Tethys dive club, Thalassa, Portugal, Parque Natural da Ria Formosa, M. Gaspar, F. Gil, Italy: A. Tavaglini, S. Repetto, L. Castellano, Bulgaria: A. Seaman, T. Gallati, Gallati Divecenter, Volunteers: J. Marcus, V. Santos, D. Mason, M. Naud, T-T. Ang and S. DeAmicis for support with field work and providing samples. We also express thanks to J. Curtis and N. McKeown for enlightening discussion and technical assistance. Finally we wish to thank reviewers for their useful comments.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
This project was funded by Chocolaterie Guylian and a Natural Environment Research Council Industrial Case studentship (NER/S/C/2005/13461) to LCW.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Woodall, L.C., Koldewey, H.J., Boehm, J.T. et al. Past and present drivers of population structure in a small coastal fish, the European long snouted seahorse Hippocampus guttulatus . Conserv Genet 16, 1139–1153 (2015). https://doi.org/10.1007/s10592-015-0728-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0728-y