
Abstract
Understanding the population structure and evolutionary history of the eastern Pacific seahorse Hippocampus ingens is critical for the effective management of this threatened species. Life history characteristics of H. ingens (site fidelity and brooding of young) may limit gene flow and lead to population differentiation. A recent study analyzing conserved fragments of the mitochondrial cyt b and control region found no population structure. We re-assess this conclusion with a phylogeographic analysis of relationships among 115 individuals of H. ingens over a broader geographic range (San Diego Bay in California, Gulf of California, Central America, Ecuador, and Peru) based on a more variable 428 base pair fragment of the control region. This expanded analysis affirms low overall nucleotide diversity relative to other seahorses (θπ = 0.004), and shows evidence of a recent bottleneck and population expansion since the middle Pleistocene. AMOVA analysis shows moderate overall population structure (ΦST = 0.10, P val = 0.00), and pairwise ΦST estimates indicate structure between the Gulf of California and all Pacific coast localities. Knowledge of population structure in H. ingens may improve conservation efforts by identifying evolutionarily important management units, and could determine source regions in the continuing trade of seahorses for traditional Chinese medicine. The level of genetic divergence observed between the Gulf of California and all other localities sampled may distinguish the Gulf as a separate management unit. Additional phylogeographic research with more quickly evolving genetic markers and targeted sampling at the mouth of the Gulf of California is warranted to inform strategies for conservation of this threatened seahorse.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding the process by which marine populations diverge or remain homogenous presents a major challenge in evolutionary biology. The resulting patterns of genetic variation can have conservation implications by informing us about the past and present evolutionary dynamics of a species. Population genetic statistics are useful in testing hypotheses about the distribution of alleles within and between populations, and can therefore be used to interpret movement of individuals (Bohonak 1999). An understanding of dispersal patterns can identify evolutionary source populations of high conservation priority, while maintenance of overall intraspecific genetic variation is important for supporting adaptive response to global change (Moritz 1994). Measures of population structure can uncover these patterns and identify demographically independent populations that should be managed as separate units (Moritz 1994; Palsbøll et al. 2007). Population structure develops when genetic drift and local adaptation are strong enough forces to counteract gene flow. In the marine environment, the development of population structure is influenced by barriers to dispersal such as ocean currents, historical vicariance, geographic distance coupled with differences in dispersal ability, and habitat discontinuity (Dawson et al. 2002; Doherty et al. 1995; Hellberg 1996; Johnson and Black 1995).
The focal taxon of this study, Hippocampus ingens, lives exclusively in sub-tidal habitat of the eastern Pacific. The common ancestor of both H. ingens and its central Pacific sister species, H. fisheri, diverged from Atlantic H. reidi at the rise of the Isthmus of Panama during the early Pliocene (4–5 mya) (Teske et al. 2007; Jacobs et al. 2004). The later divergence of H. ingens and H. fisheri 2.5–3 mya (Teske et al. 2007) left H. ingens the only seahorse found in the eastern Pacific. H. ingens’ contemporary distribution includes a putatively transient population in San Diego, California (Hubbs and Hilton 1963; Jones et al. 1988), and otherwise extends from a northern limit of lower Pacific Baja to a southern limit of Peru (Lourie et al. 2004). The only known oceanic island population is found in the Galapagos Islands (Lourie et al. 2004). H. ingens’ lives in a diversity of tropical and subtropical coastal habitats found within the Eastern Pacific; mangrove forests, seagrass beds, rocky reefs, and coral reefs (Lourie et al. 2004). Analysis of the overall taxonomic configuration of this region yields five major biogeographic provinces; California (coastline north of the Gulf of California), the Gulf of California (GOC), the Panamic province, the oceanic islands province, and the Peruvian province (Muss et al. 2001; Robertson and Cramer 2009; Spalding et al. 2007). There has been disagreement in the way in which the Pacific coastline south of the GOC should be divided. Some have argued that there are significant differences north and south of the Central American Gap, a 1,200 km long span of sandy and muddy coastal habitat, such that the Panamic province which we use in this study should be divided into the Mexican Province north of the gap, and the Panamic province south of the gap (Robertson and Cramer 2009; Hastings 2000). Thus, the regional definitions used for this study are not always agreed upon, but are strongly supported by recent studies (Robertson and Cramer 2009).
The majority of breaks in species boundaries which define these provinces reflect differences in physical and ecological conditions rather than hard barriers to dispersal (Jacobs et al. 2004; Robertson and Cramer 2009). This is because dynamic oceanographic processes facilitate sporadic dispersal (Jacobs et al. 2004). This pattern is evidenced by the lack of genetic differentiation in wide-ranging reef fish species across thousands of kilometers of the mainland coast (Muss et al. 2001; Craig et al. 2006). In contrast to this pattern of genetic connectivity in wide ranging species along the mainland coast, genetic differentiation has been found between the mainland coast and oceanic islands east of Central America (Muss et al. 2001). H. ingens is not found on these oceanic islands, so is unaffected by this barrier. The Gulf represents a putative dispersal barrier which remains unresolved. H. ingens is found frequently within the GOC (Thomson et al. 2000), making the investigation of this barrier an important aspect in characterizing demographic and phylogeographic patterns in H. ingens.
The GOC displays high levels of endemism (17% of continental shelf fish) and genetic differentiation in some species (Bernardi et al. 2003; Thomson et al. 2000; Walker 1960). Genetic studies assessing dispersal barriers separating the GOC have been limited. Most studies of in-shore fishes have focused on endemics and species with a range extending north of the GOC. These studies have revealed population structure in only some of the species investigated. Furthermore, only the minority of studies have sited barriers with potential to influence H. ingens, while most have attributed population structure to past vicariance only influential to species with more temperate distributions than H. ingens (Bernardi et al. 2003; Riginos 2005; Sandoval-Castillo et al. 2004), or to ecological barriers (Pfeiler et al. 2008) only influential to species with more specialized habitat needs than H. ingens. Thus, the only published barriers with potential to influence H. ingens include geographic distance coupled with low dispersal ability (Craig et al. 2006; Riginos and Nachman 2001), and contemporary oceanographic barriers (Bernardi et al. 2003; Selkoe et al. 2007; Terry et al. 2000). These potential oceanographic barriers are caused by the formation of two gyres in the upper and central GOC regions (Alvarez-Borrego 2002; Beier 1997; Lavin et al. 1997), a persistent oceanic front in the cape of Baja region, formed by the convergence of the California Current Water, Tropical Surface Water, and the GOC Water (Castro et al. 2006), and inflow of surface waters during summer months (Lavin and Marinone 2003). Despite the existence of these potential barriers, a study of gastropod distribution data shows that the majority of gastropods found in the region are unaffected by a barrier at the mouth of the GOC (Simison 2006). This leaves the question of a contemporary barrier to dispersal at the mouth of the GOC unresolved.
Here we present the phylogeography of a fish with a distribution extending both north and south from the GOC. Only a few phylogeographic studies that present molecular data have been conducted in fishes with a similar range, even though 73% of GOC fishes have this distribution. Furthermore, studies of wide ranging species have been conducted exclusively in taxa with high dispersal potential. They have revealed structure only between offshore islands and the mainland coastline (Craig et al. 2006; Muss et al. 2001). H. ingens is unique in that it presumably has much lower dispersal potential than species previously examined, which predicts a higher degree of population structure. Site fidelity and brooding of young suggest relatively restricted dispersal capabilities (Foster and Vincent 2004). Previous studies within Hippocampus spp. (Lourie et al. 2005; Teske et al. 2003) are largely consistent with this prediction of population structure. However, results from a previous study in H. ingens (Sanders et al. 2008) detected no population structure among 4 localities along the Pacific coast based on two mitochondrial gene fragments compared among 19 and 13 specimens separately (Sanders et al. 2008). The limited scale of the previous study merits reassessment of population structure with a more comprehensive sample. Here, we present mitochondrial phylogeographic analyses in H. ingens from a larger set of specimens more densely sampled across H. ingens’ geographic range.
H. ingens is not only a representative of a geographic distribution understudied within the framework of eastern Pacific phylogeographic patterns, but it is also a threatened fish of ecological, economic and cultural value. More than 20 million seahorses are sold each year for use in traditional Chinese medicine (TCM), for aquarium exhibits, and as curiosities (Foster and Vincent 2005). This high demand threatens the genus Hippocampus globally, which were placed on Appendix II of the Convention on International Trade in Endangered Species of Wild Fauna and Flora in 2004 (CITES 2004). During the process of listing this genus, H. ingens emerged as one of the six most impacted seahorse species (CITES 2002). Furthermore, Mexico and Peru are classified as major seahorse exporters with more than 1 dry ton sold annually (Baum and Vincent 2005). To manage and conserve seahorse populations, as required by CITES, resource managers need basic information such as species distributions, population size and structure, and genetic indices of geographic partitioning that could help define management units (Moritz 1994; Palsbøll et al.2007). Additionally, phylogeographic analyses may allow preliminary assessments of the origin of specimens confiscated in light of the trade regulations mandated by CITES in 2004 (Lourie et al.2004).
The objectives of the present study are to (i) examine demographic and phylogeographic patterns of H. ingens sampled from multiple localities within four biogeographic provinces (ii) interpret these demographic and phylogeographic findings considering likely variables of past and present geology, oceanography, and ecology and (iii) elucidate conservation implications. We hypothesize that geographic genetic structure in H. ingens will correspond with oceanographic barriers that have caused biogeographic and phylogeographic breaks in other eastern Pacific species (e.g. Bernardi et al. 2003; Selkoe et al. 2007; Terry et al. 2000; Jacobs et al. 2004; Muss et al. 2001; Robertson and Cramer 2009). We further hypothesize that the barriers most likely to have influenced H. ingens’ phylogeography include historical vicariance and oceanographic barriers between the GOC and the Pacific coast, and the combination of ocean currents and geographic distance between the extreme northern and southern limits of H. ingens’ range; San Diego, California and Peru.
Materials and methods
Localities sampled
A total of 115 H. ingens individuals were sampled from 13 localities: San Diego, California (n = 18); Loreto, Mexico (n = 1); Guaymas, Mexico (n = 9); Mazatlan, Mexico (n = 6); Salina Cruz, Mexico (n = 5); Buena Vista, Guatemala (n = 18); Puerto San Jose, Guatemala (n = 12); Guayaquil, Ecuador (n = 6); Manta, Ecuador (n = 14); Salango, Ecuador (n = 3); Sechura, Peru (n = 3); Chiclayo, Peru (n = 4); Pisco, Peru (n = 4). Another 12 samples of unknown port of entry are from within the political boundaries of Peru. DNA sequence data from 56 of these samples were donated without corresponding tissues via Dr. K. Koepfli, Dept. of Ecology and Evolutionary Biology, UCLA. Additional Genomic DNA extractions were obtained via Dr. Stephen Palumbi, Dept. of Biology, Stanford University, in collaboration with the Redpath Museum, McGill University, and Project Seahorse. The remaining 29 H. ingens samples were obtained as whole specimens in collaboration with the Birch Aquarium at Scripps Institution of Oceanography, the Ecuador Dept of Fisheries, and the U.S. Fish and Wildlife Service.
DNA extraction, amplification, and sequencing
DNA was extracted from a 3 × 3 mm piece of skin and muscle tissue using the Qiagen DNAeasy™ tissue extraction protocol and quantified using a Nanodrop nucleic acid calibrator. A 428 basepair (bp) fragment spanning the last 70 bp of mitochondrial tRNA-Pro and the first 359 bp of the control region was sequenced for all individuals. This region is designated as bp 15,580–16,008 in the annotated H. kuda mitochondrial genome available on Genbank (NC-010272.1, Kawahara et al. 2008). The target region was amplified in a 25 μl polymerase chain reaction (PCR) using Taq polymerase (New England Biolabs Inc.) at the recommended final concentration of 25 units per ml. A PTC-100 Thermocycler was used under the following PCR conditions: 94°C for 2 min 30 s; 35 cycles of 94°C for 30 s, 50°C for 45 s, and 72°C for 1 min 15 s; and then a final extension step of 72°C for 5 min. We used the following primers: forward HCBF1009 5′-TGAATTGGGGGAATACCTGT-3′ and reverse HCRLrv 5′-AAGCGTCGATGAAAGTGTG-3′ designed for this study. PCR products were cleaned using shrimp alkaline phosphatase and exonucleotide I (USB Corp., Cleveland, OH, USA). Cycle sequencing was performed in both directions with BigDye Terminator™ (vers. 3.0, Applied Biosystems Inc.) following the manufacturer’s protocol scaled to a final volume of 10 μl, using the following reaction mix: 5.3 μl of H2O, 1.5 μl of BigDye Terminator™ buffer (5X), 0.2 μl of BigDye Terminator™ (3:1 dilution), 0.2 μl primer (10 uM), 5.0 μl PCR product under the following PCR conditions (PTC-100 Thermocycler): 24 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min. Sequencing reactions were precipitated in Sephadex gel filtration spin columns (Sigma-Aldrich, Inc.), dried in a speed-vacuum for 30 min on medium heat, re-suspended in 10 μl formamide, and loaded on an ABI Prism 3730 DNA sequencer (Applied Biosystems Inc.). Both strands were sequenced in all samples. Sequences were assembled and edited using Sequencher (Gene Codes Corp.) and aligned using the Clustal-W algorithm in BioEdit (Hall 1999). Sequences are available on Genbank, accession numbers GQ386655–GQ386769.
Sequence variation
The number of unique haplotypes, segregating sites, pairwise average nucleotide diversity (θπ) (Nei and Li 1979), segregating sites diversity (θs) (Watterson 1975), and haplotype diversity (Hd) were calculated using DnaSP (vers. 4.1, Rozas et al. 2003). Deviation from neutrality expectations was tested with Tajima’s D (Tajima 1989) and Fu’s Fs statistic (Fu 1997), calculated using DnaSP (vers. 4.1, Rozas et al. 2003) and Arlequin (vers. 3.0, Excoffier et al. 2005). Tajima’s test is the most conservative test of neutrality, while Fu’s Fs is the most powerful test for population growth (Ramos-Onsins and Rozas 2002). Fu and Li’s D* and F* statistics (Fu and Li 1993) were also calculated in DnaSP (vers. 4.1, Rozas et al. 2003) and compared with Fu’s Fs statistic (Fu 1997) to test for population expansion. In conjunction, these statistics are able to distinguish between demographic effects and selection which are the two major causes of deviation from mutation/drift equilibrium.
A signal of demographic non-equilibrium was further tested using the coalescence-based method implemented in FLUCTUATE (vers. 1.4, Kuhner et al. 1998) to jointly estimate regionally specific exponential growth rate (g) and nucleotide diversity (θ). All runs were completed including only the in-group sequences, for the total data set and for each regional definition with the following model parameters; random starting trees, empirical nucleotide frequencies, initial g value of 0, starting θ value estimated with Watterson’s (1975) approach, overall transition-transversion bias ti/tv = 2.7545 determined in MrModeltest (vers. 2.3, Nylander et al. 2004) under the best fit model, HKY+I+G. The search strategy implemented is as follows; 10 short chains of 4,000 steps and 4 long chains of 400,000 steps, trees sampled every 20 steps. Analyses were repeated five times per data partition to ensure stability of the estimates; the mean values are reported. Significance was assessed using a two-tailed chi-square likelihood test.
For regions with significant results from the coalescence based test of growth, we estimated the time of coalescence as the time at which effective population size was 1% of the present day effective population size, following Wares and Cunningham (2001). The time at which the population was 1% of its current size is given as ln(0.01)/μg, a calculation derived from the equation Nt = θe −(gμ)t, where Nt is the population size at time t, θ is the nucleotide diversity (=2 Nμ), μ is the mutation rate per site for mtDNA, and g is the estimated exponential growth rate (Wares and Cunningham 2001). Mutation rate μ was estimated at 6.3 × 10−9 per site based on a generation time of approximately 1 year (Curtis and Vincent 2006), and time since divergence from outgroup H. kuda of 5 mya as calibrated in Teske et al. (2007).
Phylogenetic analysis
The minimum spanning haplotype network was built using a parsimony based median-joining method in Network (vers. 4.5, Bandelt et al. 1999). This method combines multiple equally parsimonious minimum-spanning trees into a single minimum-spanning network with the addition of a few consensus sequences (median vectors) of three mutually close sequences to improve visualization (Bandelt et al. 1999).
Phylogenetic relationships among sequences were assessed using the maximum parsimony method as implemented in PAUP* (vers. 4.0, Swofford 2002), and Bayesian methods as implemented in MrBayes (vers. 3.1, Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). The HKY+I+G model of molecular evolution was applied in Bayesian analyses, which was the best fit model selected by a hierarchical likelihood ratio test as implemented in MrModeltest (vers. 2.3, Nylander et al. 2004). A consensus tree was constructed from 15 million generations sampled every 1,000 trees after stationarity was observed. A burn in of 12,000 generations was excluded from the consensus tree. The corresponding control-region sequence from the H. kuda mitochondrial genome (Genbank NC-010272.1) was used as the outgroup (Kawahara et al. 2008; Teske et al. 2004).
Population subdivision
Biogeographic provinces define regions that have been influenced by a unique set of evolutionary and ecological forces. However, they do not necessarily correlate well with intraspecific phylogeographic boundaries in eastern Pacific marine taxa (Jacobs et al. 2004). For this reason, we conducted analyses at multiple scales with attention to evolutionary processes that have operated in other species of similar contemporary and historical range. Population-level geographic definitions were used in descriptive statistics, neutrality statistics, Analysis of Molecular Variance (AMOVA), estimates of ΦST (analogous to FST), and a Mantel test for ‘isolation by distance’ (IBD). Regional-level geographic definitions were used in AMOVA, descriptive statistics, neutrality statistics, estimates of ΦST, growth rate (g), and time of coalescence (Tc). For regional analyses we pooled samples into five groupings (California, GOC, Central America, Ecuador, and Peru) on the basis of recognized biogeographic provinces (Hastings 2000; Robertson and Cramer 2009). Both the Central American and Ecuador localities fall into the same extensive Panamic biogeographic province defined by Robertson and Cramer (2009). However, we maintain separate regional definitions to improve geographic resolution because there has been discussion regarding whether this region should be considered two provinces divided north and south of the Central American Gap (e.g. Hastings 2000; Robertson and Cramer 2009).
To test for population structure among localities, an AMOVA was performed using Arlequin (vers. 3.0, Excoffier et al. 2005). Overall ΦST and pairwise ΦST values at both the population and regional scales were calculated. The FST analog ΦST was used to estimate population structure in this study to take nucleotide differences between haplotypes of sequence data into account (Excoffier et al. 1992). Significance of these values was tested using a non-parametric permutation approach described in Excoffier et al. (1992).
A pattern of IBD was tested with a Mantel test using GENEPOP (vers. 3.0, Raymond and Rousset 1995). For this test, four populations were pooled to increase sample size; Loreto and Guaymas, Mexico (separated by 183 km), Buena Vista and Puerto San Jose, Guatemala (separated by 19 km); Manta and Salango, Ecuador (separated by 57 km); and Sechura and Chiclayo, Peru (separated by 131 km). Matrices were calculated for both geographic and genetic distances. The logarithmic distance matrix was based on geographic distance in km measured in Google Earth (vers. 4.3, Google 2008) using the ruler function with shortest line defined by eye, avoiding land. The genetic distance was defined by pairwise ΦST generated in Arlequin (vers. 3.0, Excoffier et al. 2005). The analysis was repeated excluding the GOC to compare with the overall results.
Results
Sequence variation and estimates of demographic history
The 115 H. ingens mitochondrial sequences, 428 bp in length, contained 23 polymorphic sites defining 28 unique H. ingens haplotypes (Table 1), with 1–9 base differences between sequences. Nucleotide diversity θπ, and segregating site θs diversity estimates were about half that found in several other species of seahorses (e.g. θπ = 0.016 in H. kuda; θπ = 0.013 in H. spinosissimus, Lourie et al. 2005) (Table 1), and consistent with estimates of diversity in cyt b found in this species by Sanders et al. (2008). A single insertion was detected at basepair number 207 of this dataset which falls within the control region at basepair 15,787 in the annotated H. kuda mitochondrial genome available on Genbank (NC-010272.1, Kawahara et al. 2008). This insertion defines one unique haplotype (named HapA207) with a count of just four.
Tajima’s D (Tajima 1989) was significantly different from neutral expectations for the total data set (Table 1). Fu and Li’s D* and F* statistics (Fu and Li 1993) did not reject neutral expectations while Fu’s Fs (Fu 1997) was significantly negative and rejected neutral expectations in four population definitions; Buena Vista and Puerto San Jose, Manta and Salango, Guayaquil, and unknown Peru, and in three regional definitions; Central America, Ecuador, and Peru, and in the overall dataset (Table 1).
Results from the coalescent based analysis using FLUCTUATE (vers. 1.4, Kuhner et al. 1998) suggest a high growth rate for the entire geographic range sampled (Table 2). At a regional scale, non-significant growth estimates were observed in California, and the GOC, and high estimates of growth with significant probability values were found in Central America, Ecuador and Peru (Table 2). Time since coalesence approximates the lower limit in timing of population growth. All four analyses that produced significant results (Central America, Ecuador, Peru, and the overall dataset) indicated a coalescent time during the middle Pleistocene (0.25–0.54 mya) (Table 2).
Phylogenetic analysis
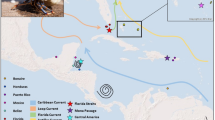
The minimum spanning network demonstrates that the most common haplotype (A) was found in 47 sequences representing all geographic regions (Fig. 1). Median vectors were calculated and are shown in this network as solid squares at the intersection of two separate network branches. Most haplotypes were separated by a single mutation, with no clear assortment of haplotypes based on geographic locality. The two most distant haplotypes were found in the GOC, separated from one another by six polymorphic sites, and a minimum of three polymorphic sites from closest haplotype C. There were four common haplotypes with total counts ranging from 9 to 47. All San Diego, California individuals sampled (n = 18) shared three haplotypes; A, A207 and B (Fig. 1).

Minimum spanning network with haplotype name, sample number of each haplotype, and indication of proportion from each region in shades that correspond to adjacent map. Median vectors indicated by solid squares at branch intersections. The map on the right shows sample localities with sample number in parentheses. Shaded areas indicate which region each locality was assigned
Both parsimony and Bayesian phylogenetic methods recovered a star phylogeny with minimal topology. Bayesian analyses produced trees with an average likelihood score of −ln L = 895.40. The resulting consensus tree was not included as a figure in this manuscript because of the simple topology and the low posterior probability node support values recovered. However, it is worth noting that two haplotypes sampled exclusively from within the GOC (F and H) formed a sub-clade with posterior probability node support of 0.88 nested within the poorly supported clade (posterior probability 0.67) comprised of haplotypes C, P, F, and H.
Population subdivision
The overall ΦST value 0.109 revealed statistically significant (P value = 0.00) moderate genetic differentiation between geographic regions (Table 3). An AMOVA analysis excluding the GOC revealed low structure; ΦST was calculated at 0.023 with a probability value of 0.03 (Table 3). Almost 90% of the control region genetic variation resided within populations rather than among the five regions (Table 3). The Central American regional definition spans the largest distance (2,195 km), from Mazatlan to Panama (Table 4). This regional grouping is justified by ΦST calculations that indicated much greater differentiation north of Mazatlan than south of Mazatlan. ΦST between Mazatlan and the GOC was moderate (Table 4), though marginally significant (probability value = 0.05, value not shown in table), while the overall ΦST among the 3 Central American populations was non-significant (Table 3). Pairwise estimates of genetic structure produced high and significant ΦST values, ranging from 0.21 to 0.45 between the GOC and all other regions (Table 5). San Diego, California had statistically significant but low pairwise ΦST values of 0.10 and 0.04 when compared to Ecuador and Central America, respectively. We found no evidence of population structure south of the GOC.
The Mantel test performed in GENEPOP (vers. 3.0, Raymond and Rousset 1995) with the genetic and geographic distances displayed in Table 4 show a slight but non-significant pattern of IBD, with a one-tailed probability value of 0.91 (Slatkin 1993). When analysis was repeated excluding the GOC, there was no pattern of IBD, with a one-tailed probability value of 0.74.
Discussion
Demographic history
Our results are consistent with a dynamic demographic history for the H. ingens lineage. Evidence of a recent population bottleneck in H. ingens include deviation from neutrality expectations, FLUCTUATE estimates of positive population growth, and a star phylogeny. Significant deviation from neutrality expectations in Tajima’s and Fu’s statistics rejected a neutral model with constant population size. Negative values of these statistics are signatures of population expansion or purifying selection (Tajima 1989), specifically in the southern portion of H. ingens’ range (Table 1). Non-significant Fu and Li’s D* and F* statistics point to demographic expansion rather than selection (Fu 1997). Inferences drawn from these neutrality statistics were consistent with FLUCTUATE results and provide evidence of population expansion within the last 0.25–0.54 mya (Table 2). We hypothesize that H. ingens experienced a severe bottleneck (where effective population size dropped below 1% of its current size) subsequent to its divergence from H. fisheri during the middle Pleistocene. This bottleneck may have been associated with a number of dynamic processes that occurred in the eastern Pacific during the Pleistocene. This epoch is characterized by the re-establishment of an upwelling regime, significant sea level flux, increasingly rocky shorelines, multiple glaciation events, and finally a general increase in sea level at the end of the epoch (Jacobs et al. 2004). A more detailed reconstruction of historical population dynamics awaits increased sampling in the northern portion of H. ingens’ range.
Standard diversity statistics revealed interesting dynamics at the northern end of H. ingens’ range. Comparison of regional sequence variation shows that San Diego, California had extremely low relative diversity (Table 1) suggesting a recent founder event. This evidence supports the hypothesis that H. ingens populations are mainly transient north of the Tropical Eastern Pacific (TEP). Warming of coastal water during El Niño events (Robertson and Cramer 2009) can temporarily extend the range of tropical endemics, and may play a role in the periodic H. ingens colonization of San Diego Bay (Hubbs and Hilton 1963; Jones et al. 1988). A single H. ingens specimen, housed in the Harvard Museum of Comparative Zoology (specimen number MCZ 53292), was collected as far north as San Francisco Bay in 1960 following the El Niño event of 1957–1959 (Hubbs and Hilton 1963). Collection records (Jones et al. 1988) and recent field work done for this study suggest that there has been a breeding population of H. ingens in San Diego Bay since the mid 1980s, putatively following the major El Niño event of 1982–1983 (Jones et al. 1988). The low haplotype diversity observed in San Diego, California relative to all other localities sampled (Table 1) implies that the founding population was small. Non-significant FLUCTUATE results from this region leave this interpretation a tentative hypothesis.
Phylogeographic patterns and potential barriers to gene flow
Population structure develops when gene flow between two populations is low enough to allow genetic drift and local adaptation to create genetic divergence. Discrete genetic breaks can be caused by both incomplete and complete barriers that operate across multiple scales in both time and space. Examples include persistent oceanographic barriers and geological events. Even when these barriers are lacking, simple geographic distance coupled with limited dispersal ability can be enough to prevent gene flow between separate populations.
In contrast to previous analysis of H. ingens phylogeography (Sanders et al. 2008), our study indicates moderate and statistically significant overall population structure (Table 3). Our results are consistent with vicariance by persistent oceanographic processes. Vicariance yields a pattern of high ΦST values between isolated populations, and low ΦST among populations on either side of the barrier. We observed high and significant ΦST values between the GOC and Pacific coast populations from California to Peru, and low ΦST values among Pacific coast populations. Spatial resolution of sampling both north and south of the GOC was not sufficient to fully evaluate this genetic break in H. ingens. Nonetheless, lack of structure along the Pacific coast is exemplified by non-significant pairwise ΦST estimates between the two most geographically distant regions, San Diego, California and Peru, and between all localities and regions south of the GOC (Tables 4 and 5). The exception to this trend is found in minimal but still significant pairwise ΦST estimates between San Diego, California and Central America and Ecuador. This signal is difficult to interpret in light of the low haplotype diversity found in San Diego, California and the unresolved transient status of this population.
We hypothesize that two main processes have contributed to the pattern of a single distinct genetic break; the life history of H. ingens, and oceanographic conditions of both the eastern Pacific and the GOC. Seahorses are thought to display site fidelity as adults, but dispersal ability in juveniles is largely unknown. There is genetic evidence for limited dispersal in all Hippocampus species that have been previously analyzed in a phylogeographic context (Lourie et al. 2005; Teske et al. 2003). Nonetheless, after male pregnancy, fully formed juveniles are delivered into the water column and the few that survive seem to rise in the water column to surface waters and float with little mobility in currents for unknown duration (Lourie et al. 2004). Hitch-hiking on macro algae floats may be one mode of dispersal that could increase connectivity across large geographic distances dependent on oceanographic processes.
The eastern Pacific coast is characterized by stochastic processes which facilitate sporadic dispersal. Siegel et al. (2008) demonstrate through modeling that alongshore larval connectivity is inherently a stochastic process due to chaotic coastal currents. This local process, coupled with extensive mixing due to a seasonal shift of the Inter-tropical Convergence Zone and periodic El Niño events, facilitate sporadic dispersal between regions of the eastern Pacific (Muss et al. 2001; Pisias and Mix 1997). The geographic scale of the resulting pattern of genetic connectivity depends on dispersal potential, which in turn depends on larval characteristics such as duration and mobility. A species with low dispersal potential would experience reduced likelihood of gene flow with greater distance, which may be reflected in statistically significant ΦST estimates and population structure exhibiting IBD. On the other hand, a species with high dispersal potential would display a pattern of minimal or non-significant ΦST estimates and no IBD (e.g. Epinephelus labriformis, Craig et al. 2006; Ophioblennius steindachneri, Muss et al. 2001). H. ingens exhibited moderate ΦST estimates driven by structure across one discrete barrier, but no pattern of IBD. Although there remains the possibility that population structure exists on the Pacific coast at scales not captured in this dataset, the most likely explanation is that dynamic oceanographic conditions facilitate connectivity along the Pacific coast, while unique oceanographic conditions of the GOC facilitate population structure.
We hypothesize that population structure developed between the GOC and Pacific coast localities because of restricted genetic exchange due to site fidelity in adults and contemporary oceanographic barriers to juvenile dispersal between regions. Site fidelity is typical in adult seahorses (Foster and Vincent 2004). Potential juvenile dispersal barriers include the formation of two gyres in the upper and central GOC regions (Alvarez-Borrego 2002; Beier 1997; Lavin et al. 1997), a persistent oceanic front where the California Current Water, Tropical Surface Water, and the Gulf of California Waters converge (Castro et al. 2006), and inflow of surface waters during summer months (Lavin and Marinone 2003). The patterns of gyre formation, positioning of the oceanic front, and patterns of inflow and outflow are heavily influence by inter-annual variability caused by El Niño (Lavin and Marinone 2003; Castro et al. 2000). Despite this variability, conditions within the GOC may increase self-recruitment of juvenile H. ingens. High diversity and the presence of endemic haplotypes in the GOC are congruent with this hypothesis. Additionally, the oceanic inflow of surface waters during the summer may coincide with H. ingens’ peak in reproductive output, pending confirmation that H. ingens follows the trend in breeding season timing and duration reported in other seahorses (Foster and Vincent 2004). Coincidence of breeding season with surface water inflow would facilitate a decrease in juvenile migration out of the GOC while allowing genetic influx from the outer coast. Detailed studies of breeding habits in H. ingens and finer scaled sampling within the GOC and along the adjacent outer coasts would be necessary to further resolve this question.
Conservation implications
Understanding broad patterns of gene flow is an important advancement in creating a successful international management plan for H. ingens. Moderate overall geographic structure with only one distinct genetic break suggests that H. ingens populations comprise a large management unit of the entire Pacific coast, and a possible second management unit within the GOC. Significant ΦST values (Tables 4 and 5) between the GOC and all other localities sampled correlate with migration levels of less than two individuals per generation under the Wright-Fisher island population model. This implies demographic independence (Palsbøll et al. 2007). Evidence of demographic independence and high relative nucleotide diversity in the GOC suggest that the GOC depends heavily on self-recruitment and may be more vulnerable to over-fishing than populations with greater connectivity such as those along the Central American coast.
These results have important implications for management of seahorse populations in Mexican waters. Mexico is considered a major exporter of dried seahorses (Baum and Vincent 2005). The majority of these seahorses are caught as bycatch by shrimp trawlers operating along the western coast of Mexico (Baum and Vincent 2005, Sarah Foster pers obs. 2010). Furthermore, the effort of trawlers measured in number of boats operating are much greater in the GOC than elsewhere, evidenced by the fact that the states of Sonora, Sinaloa and Nayarit produce the most shrimp in Mexico (SAGARPA 2008). The combination of higher bycatch of seahorses in the region surrounding the GOC, and low connectivity of the GOC with the outer coast populations leaves GOC populations particularly vulnerable to overharvest. Current regulations of shrimp trawl bycatch within the states of Sonora, Sinaloa and Nayarit and regulation of seahorse exports from northern Mexico may need to be strengthened in order to meet CITES requirements.
Although results of this study have increased our understanding of the geographic genetic relationships among H. ingens populations, several pertinent questions remain unanswered. The limited size of some sampled populations and the relatively low diversity of the mitochondrial gene fragments sequenced do not allow identification of TCM trade source populations derived from Pacific coast populations. Furthermore, these data provide insufficient power to determine migration rates with coalescent based methods, and specific demographic dynamics in the GOC and San Diego, California could not be resolved. This study highlights the need for further genetic characterization with more rapidly evolving markers and fine-scaled geographic sampling focused on the GOC. Field work that can yield improved estimates of life history characteristics such as adult movement, generation time, and breeding season are further steps to ensure the persistence of this valuable and threatened species.
References
Alvarez-Borrego S (2002) Physical oceanography. In: Case TJ, Cody M, Ezcura E (eds) A New Island Biogeography of the Sea of Cortés. Oxford University Press, New York, pp 41–59
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Baum JK, Vincent ACJ (2005) Magnitude and inferred impacts of the seahorse trade in Latin America. Environ Conserv 32:305–319
Beier E (1997) A numerical investigation of the annual variability in the Gulf of California. J Phys Oceanogr 27:615–632
Bernardi G, Findley L, Rocha-Olivares A (2003) Vicariance and dispersal across Baja California in disjunct marine fish populations. Evolution 57:1599–1609
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74(1):21–45
Castro R, Mascarenhas AS, Durazo R, Collins CA (2000) Seasonal variation of the temperature and salinity at the entrance to the Gulf of California, Mexico. Ciencias Marinas 26:561–583
Castro R, Durazo R, Mascarenhas A, Collins CA, Trasvina A (2006) Thermohaline variability and geostrophic circulation in the southern portion of the Gulf of California. Deep-Sea Res I-Oceanogr Res Pap 53(1):188–200
CITES (2002) Consideration of proposals for amendment of Appendices I and II. http://www.cites.org/eng/cop/12/prop/E12-P37.pdf. Accessed 15 May 2009
CITES (2004) Notification to the parties-trade in seahorses-implementation of decision 12.54. http://www.cites.org/eng/notif/2004/033.pdf. Accessed 15 May 2009
Craig MT, Hastings PA, Pondella DJ, Robertson DR, Rosales-Casian JA (2006) Phylogeography of the flag cabrilla Epinephelus labriformis (Serranidae): implications for the biogeography of the tropical eastern Pacific and the early stages of speciation in a marine shore fish. J Biogeogr 33:969–979
Curtis JR, Vincent AJ (2006) Life history of an unusual marine fish: survival, growth and movement patterns of the European long-snouted seahorse, Hippocampus guttulatus (Cuvier 1829). J Fish Biol 68:1–27
Dawson MN, Louie KD, Barlow M, Jacobs DK, Swift CC (2002) Comparative phylogeography of sympatric sister species, Clevelandia ios and Eucyclogobius newberryi (Teleostei, Gobiidae), across the California transition zone. Mol Ecol 11:1065–1075
Doherty PJ, Planes S, Mather P (1995) Gene flow and larval duration in 7 species of fish from the Great-Barrier-Reef. Ecology 76:2373–2391
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial-DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetic data analysis. Evol Bioinform 1:47–50
Foster SJ, Vincent ACJ (2004) Life history and ecology of seahorses: implications for conservation and management. J Fish Biol 65:1–61
Foster SJ, Vincent ACJ (2005) Enhancing sustainability of the international trade in seahorses with a single minimum size limit. Conserv Biol 19:1044–1050
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Google (2008) Google Earth vers. 4.3. http://earth.google.com. Accessed 15 May 2009
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hastings PA (2000) Biogoegraphy of the tropical eastern Pacific: distribution and phylogeny of chaenopsid fishes. Zool J Linn Soc 128:319–335
Hellberg ME (1996) Dependence of gene flow on geographic distance in two solitary corals with different larval dispersal capabilities. Evolution 50:1167–1175
Hubbs CL, Hilton SD (1963) The giant seahorse returns. Pac Discov 16(5):12–15
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jacobs DK, Haney TA, Louie KD (2004) Genes, diversity, and geologic process on the Pacific coast. Annu Rev Earth Planet Sci 32:601–652
Johnson MS, Black R (1995) Neighborhood size and the importance of barriers to gene flow in an intertidal snail. Heredity 75:142–154
Jones AT, Dutton P, Snodgrass RE (1988) Reoccurrence of the Pacific seahorse, Hippocampus ingens, in San Diego Bay. Calif Fish Game 74(4):236–248
Kawahara R, Miya M, Mabuchi K et al (2008) Interrelationships of the 11 gasterosteiform families (sticklebacks, pipefishes, and their relatives): a new perspective based on whole mitogenome sequences from 75 higher teleosts. Mol Phylogenet Evol 46:224–236
Kuhner MK, Yamato J, Felsenstein J (1998) Maximum likelihood estimation of population growth rates based on the coalescent. Genetics 149(1):429–434
Lavin MF, Durazo R, Palacios E, Argote ML, Carrillo L (1997) Lagrangian observations of the circulation in the Northern Gulf of California. J Phys Oceanogr 27:2298–2305
Lavin MF, Marinone SG (2003) An overview of the physical oceanography of the Gulf of California. In: Velasco Fuentes OU et al (eds) Nonlinear processes in geophysical fluid dynamics. Kluwer Academic Publishers, Netherlands, pp 173–204
Lourie SA, Foster SJ, Cooper EWT, Vincent AC (2004) A guide to the identification of seahorses. Project Seahorse and TRAFFIC North America. University of British Columbia and World Wildlife Fund, Washington, DC
Lourie SA, Green DM, Vincent AC (2005) Dispersal, habitat differences, and comparative phylogeography of Southeast Asia seahorses (Syngnathidae: Hippocampus). Mol Ecol 14:1073–1093
Moritz C (1994) Defining evolutionary-significant-units for conservation. Trends Ecol Evol 9:373–375
Muss A, Robertson DR, Stepien CA, Writz P, Bowen BW (2001) Phylogeography of Ophioblennius: the role of ocean currents and geography in reef fish evolution. Evolution 55:561–572
Nei M, Li WH (1979) Mathematical-model for studying genetic-variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Nylander JAA, Ronquist F, Huelsenbeck JP, Nieves-Aldrey JL (2004) Bayesian phylogenetic analysis of combined data. Syst Biol 53(1):47–67
Palsbøll PJ, Berube M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Pfeiler E, Watts T, Pugh J, van der Heiden AM (2008) Speciation and demographic history of the Cortez bonefish, Albula sp A (Albuliformes: Albulidae), in the Gulf of California inferred from mitochondrial DNA. J Fish Biol 73:382–394
Pisias NG, Mix A (1997) Spatial and temporal oceanographic variability of the eastern equatorial Pacific during the late Pleistocene, evidence from radiolarian microfossils. Paleoceanography 12:381–393
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Raymond M, Rousset F (1995) GENEPOP (version-1.2)—population-genetics software for exact tests and ecumenicism. J Hered 86(3):248–249
Riginos C (2005) Cryptic vicariance in Gulf of California fishes parallels vicariant patterns found in Baja California mammals and reptiles. Evolution 59:2678–2690
Riginos C, Nachman MW (2001) Population subdivision in marine environments: the contributions of biogeography, geographical distance and discontinuous habitat to genetic differentiation in a blennioid fish, Axoclinus nigricaudus. Mol Ecol 10:1439–1453
Robertson DR, Cramer KL (2009) Shore fishes and biogeographic subdivisions of the tropical eastern Pacific. Mar Ecol-Prog Ser 380:1–17
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
SAGARPA (2008) Anuario Estadý′stico de Acuacultura y Pesca, Mexico. Annual report 2008. http://www.conapesca.sagarpa.gob.mx/wb/cona/cona_anuario_estadistico_de_pesca. Accessed 11 Apr 2010
Sanders JG, Cribbs JE, Fienberg HG et al (2008) The tip of the tail: molecular identification of seahorses for sale in apothecary shops and curio stores in California. Conserv Genet 9:65–71
Sandoval-Castillo J, Rocha-Olivares A, Villavicencio-Garayzar C, Balart E (2004) Cryptic isolation of Gulf of California shovelnose guitarfish evidenced by mitochondrial DNA. Mar Biol 145:983–988
Selkoe KA, Vogel A, Gaines SD (2007) Effects of ephemeral circulation on recruitment and connectivity of nearshore fish populations spanning southern and Baja California. Mar Ecol-Prog Ser 351:209–220
Siegel DA, Mitarai S, Costello CJ, Gaines SD, Kendall BE, Warner RR, Winters KB (2008) The stochastic nature of larval connectivity among nearshore marine populations. Proc Natl Acad Sci 105(26):8974–8979
Simison WB (2006) Patterns of endemism for nearshore gastropod molluscs: a new view of the Gulf of California. J Molluscan Stud 72:53–63
Slatkin M (1993) Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47:264–279
Spalding MD, Fox HE, Halpern BS, McManus MA, Molnar J, Allen GR, Davidson N, Jorge ZA, Lombana AL, Lourie SA, Martin KD, McManus E, Recchia CA, Robertson J (2007) Marine ecoregions of the world: a bioregionalization of coastal and shelf areas. Bioscience 57:573–583
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinaur Associates, Massachussets
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Terry A, Bucciarelli G, Bernardi G (2000) Restricted gene flow and incipient speciation in disjunct Pacific Ocean and Sea of Cortez populations of a reef fish species, Girella nigricans. Evolution 54:652–659
Teske PR, Cherry MI, Matthee CA (2003) Population genetics of the endangered Knysna seahorse Hippocampus capensis. Mol Ecol 12:1703–1715
Teske PR, Cherry MI, Matthee CA (2004) The evolutionary history of seahorses (Syngnathidae: Hippocampus): molecular data suggest a west Pacific origin and two invasions of the Atlantic Ocean. Mol Phylogenet Evol 30:273–286
Teske PR, Hamilton H, Matthee CA, Barker NP (2007) Signatures of seaway closures and founder dispersal in the phylogeny of a circum globally distributed seahorse lineage. BMC Evol Biol 7:138
Thomson DA, Findley LT, Kerstitch AN (2000) Reef fishes of the Sea of Cortez. The rocky-shore fishes of the Gulf of California. Revised edition. Corrie Herring Hooks Series 44:i–xx, 1–353
Walker BW (1960) The distribution and affinities of the marine fish fauna of the Gulf of California. Syst Zool 9:123–133
Wares JP, Cunningham CW (2001) Phylogeography and historical ecology of the North Atlantic intertidal. Evolution 55:2455–2469
Watterson GA (1975) Number of segregating site in genetic models without recombination. Theor Popul Biol 7:256–276
Acknowledgments
We kindly thank Per Palsbøll from the department of Environmental Science, Policy and Management of UC Berkeley for his sponsorship of this project. We thank Anna Sellas for help and training in the lab. We are grateful to Grant Pogson, Giacomo Bernardi, Jon Sanders and Sara Lourie for inspiration and intellectual input. We greatly appreciate the resources provided by the Conservation Genetics lab of the College of Natural Resources, UC Berkeley, the Museum of Vertebrate Zoology of UC Berkeley, and the Center for Comparative Genomics of the California Academy of Sciences. This study would not have been possible without the donation of Hippocampus ingens samples from various individuals and institutions: Dr. K. Koepfli of the Dept. of Ecology and Evolutionary Biology, UCLA; Dr. Stephen Palumbi of the Dept. of Biology, Stanford University; the Redpath Museum at McGill University, Project Seahorse; Bob Burhans and Leslee Matsushige at the Birch Aquarium at Scripps Institution of Oceanography; the Ecuador Dept of Fisheries; and the U.S. Fish and Wildlife Service. This study was financially supported by the 2005 College of Natural Resources, UC Berkeley Undergraduate Research Opportunity Award and the Seahorse Research and Conservation Program of the California Academy of Sciences. We are grateful to three anonymous reviewers and Sarah Foster for their valuable comments on this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Saarman, N.P., Louie, K.D. & Hamilton, H. Genetic differentiation across eastern Pacific oceanographic barriers in the threatened seahorse Hippocampus ingens . Conserv Genet 11, 1989–2000 (2010). https://doi.org/10.1007/s10592-010-0092-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0092-x