
Abstract
The extracellular matrix (ECM) constitutes the scaffold of tissues and organs. It is a complex network of extracellular proteins, proteoglycans and glycoproteins, which form supramolecular aggregates, such as fibrils and sheet-like networks. In addition to its biochemical composition, including the covalent intermolecular cross-linkages, the ECM is also characterized by its biophysical parameters, such as topography, molecular density, stiffness/rigidity and tension. Taking these biochemical and biophysical parameters into consideration, the ECM is very versatile and undergoes constant remodeling. This review focusses on this remodeling of the ECM under the influence of a primary solid tumor mass. Within this tumor stroma, not only the cancer cells but also the resident fibroblasts, which differentiate into cancer-associated fibroblasts (CAFs), modify the ECM. Growth factors and chemokines, which are tethered to and released from the ECM, as well as metabolic changes of the cells within the tumor bulk, add to the tumor-supporting tumor microenvironment. Metastasizing cancer cells from a primary tumor mass infiltrate into the ECM, which variably may facilitate cancer cell migration or act as barrier, which has to be proteolytically breached by the infiltrating tumor cell. The biochemical and biophysical properties therefore determine the rates and routes of metastatic dissemination. Moreover, primed by soluble factors of the primary tumor, the ECM of distant organs may be remodeled in a way to facilitate the engraftment of metastasizing cancer cells. Such premetastatic niches are responsible for the organotropic preference of certain cancer entities to colonize at certain sites in distant organs and to establish a metastasis. Translational application of our knowledge about the cancer-primed ECM is sparse with respect to therapeutic approaches, whereas tumor-induced ECM alterations such as increased tissue stiffness and desmoplasia, as well as breaching the basement membrane are hallmark of malignancy and diagnostically and histologically harnessed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metastasis is the prominent cause of death in cancer. Uncontrolled growth of cells within a tissue, neoplasia, leads to solid tumors at the site of origin. Unless the neoplastic growth does not demand too much space and unless it obstructs a vessel or a nerve thereby indirectly causing ischemia, analgesia and paralysis, the tumor is considered benign and, in most cases, removable by surgery. In contrast, metastasis occurs when tumor cells invasively infiltrate normal tissues neighboring the primary tumor mass. Moreover, they spread throughout the body in a malignant way, a hallmark of cancer [1, 2]. This spreading requires the motility of malignant cells [3]. They move through the connective tissue with its meshwork of ECM proteins [4]. Moreover, metastatic cancer cells have to penetrate layers of tightly connected epithelial and endothelial cells and have to breach barriers of several ECM structures, such as the sheet-like basement membrane (BM) [5]. In addition, cancer cells must stay alive even in suspension during lymphatic and hematogenous dissemination. Attachment and detachment as well as movement require contacts of cancer cells with different ECM proteins and distinct supramolecular arrangements of the ECM. Moreover, the ECM also determines where cancer cells settle and establish a metastasis [6]. Recent years have highlighted that even in the primary tumor mass the ECM is qualitatively and quantitatively altered in a tumor-permissive way, thereby promoting tumor progression and influencing cancer cell invasion [7].
This review pinpoints the key role of ECM in the tumor environment and its interplay with tumor cells. It is of prime medical interest to decipher how the ECM is modulated by tumor cells and how the ECM affects metastasis in diverse aspects, acting as a mechanical support for migration and metastatic engraftment or as a barrier for cancer infiltration.
ECM, more than an intercellular filling material
The highly ordered meshwork of ECM-molecules
The distinction between an organ-specific parenchyma, such as the secreting epithelial cells of a gland, and the stromal compartment of the organ, which fills the space between the parenchyma and allows access of blood vessels and nerves, was a histologic description of an organ. This gave rise to the name ‘connective tissue’ for the stromal compartment. However, the stroma is not just an intercellular filling but an active element in the development, homeostasis, and functionality of the entire organ [4, 8]. This is based on the complex interplays between the cells of different lineages, such as epithelial cells and mesenchymal fibroblasts, and between the cells and the ECM.
The ECM, typically found in the large intercellular spaces of the connective tissue, consists of a meshwork of fibrous proteins, proteoglycans with their properties-determining carbohydrate conjugates, amorphous materials, minerals, and water [4, 9, 10]. The ECM also contains growth factors and other cytokines at orders of magnitude lower concentrations that orchestrate developmental processes in morphogenesis, regeneration and maintenance [8].
The molecular architecture of connective tissue underlines one function of the ECM, i.e., to shape and maintain the form of tissues and organs as a three-dimensional scaffold [4, 8,9,10]. In addition, the ECM also forms as a thin sheet-like BM that delimits the stroma from other tissues, such as the epithelial and endothelial cell layers, neurons and muscle cells and fibers, or adipocytes. This BM serves as mechanical substratum for cell adhesion [5, 11,12,13,14,15]. Forming a structural scaffold, fibrous ECM proteins can bear mechanical tensile forces generated by cells and transferred to bone and cartilage to move the body. Also, the tissue pressure caused by the swelling capacity of charged polysaccharides of proteoglycans sets the fibrillar network under tension [14, 16,17,18].
The molecular and supramolecular structures of its components enable the ECM to fulfil the scaffolding and mechanical force-bearing functions (Table 1). The most abundant ECM proteins are the collagens, of which 28 types have been described so far [4, 10, 19,20,21]. Their characteristic feature is the collagenous triple helix and its propensity to assemble into higher-order supramolecular structures, such as fibrils and networks [4, 10, 19]. The triple helix consists of three collagenous peptide strands with a typical Gly-X–Y motif, with X and Y predominantly being proline and 4-hydroxyproline. In a parallel direction, they wind around each other in a staggered manner, as their glycine residues line up along the helix axis [22, 23]. Unless the triple helix is unwound by high temperatures, it is robust against proteolytic cleavage. Several triple helical collagen molecules align laterally and in a staggered manner to form fibrils [22, 24]. From the core to the perimeter of collagenous fibrils, the different collagen types, I, V, and XI, vary in their relative abundance to contribute best to the overall function of the fibril in tissue [25]. In network-forming collagens, additional intermolecular contacts such as head-to-head and tail-to-tail interactions, including some more recently discovered redox-based methionine-derived crosslinks, stabilize the chicken wire-like sheets of type IV collagen. Together with type XV and XVIII collagens, it is the shape-determining components of BMs [12, 26,27,28,29]. Additional collagen types, such as fibrillar collagens with interrupted triple helix (FACITs) or type VII collagen [30, 31], interlink the collagenous fibrils and networks to the surrounding ECM proteins. Once assembled in the extracellular space, the collagen molecules within the scaffold can be covalently crosslinked by extracellular enzymes, family members of lysyl oxidase (LOX and lysyl oxidase-like, LOXL) and transglutaminase [32,33,34]. These enzymes also covalently crosslink proteins of the elastin network, comprising elastin, fibrillins, and other associating ECM proteins [35, 36].
Another ubiquitously found ECM protein is fibronectin, which consists of two disulfide-linked chains. 20 different isoforms due to alternative splicing are known (Table 1) [37,38,39]. Fibronectin molecules not only bind to the collagen scaffold, but also align into filaments which can aggregate into networks with the help of cellular contacts via the fibronectin receptor, integrin α5β1 [40]. As a multidomain and multifunctional protein, fibronectin also binds several growth factors and proteoglycans [41].
The tensile forces that collagen and other fibrillar networks absorb counteract the tissue pressure created by the swelling power of the highly charged glycosaminoglycan (GAG) chains of proteoglycans [16, 17, 42]. This large family of ECM proteins comprises the large size hyaluronic acid-binding hyalectans (Table 1), viz. aggrecan, neurocan, brevican and versican, and the 18 different small leucine-rich proteoglycans (SLRPs), such as decorin and biglycan [17, 18, 43]. Among the long list of potential functions, two features of proteoglycans are noteworthy in this context. With their GAG chain(s) they tether growth factors and cytokines, thereby enabling the ECM to serve as storage and present growth factors in a temporal and spatial order needed for tissue development and regeneration [13]. Moreover, by interacting with other ECM proteins, proteoglycans may regulate their assembly, such as the collagenous fibrils, and their function, such as perlecan, which is characteristically found in BMs and contributes to its negative charge and its filtration properties [13, 44, 45].
Laminins are other typical components of BMs (Table 1) [10, 12, 46]. All 16 different laminins consists of 3 chains, α, β, and γ, which characteristically are wound around each other in an α-helical coiled-coil domain [24]. This forms the long arm of these cruciform molecules, the three short arms are shaped by the N-terminal domains of the three individual chains. The C-terminal end of the long arm is flanked by a large globular G-domain, which is divided into five homologous LG domains [47]. The first three assemble into a clover leaf-like structure, while the last two LG4-LG5 domains, which bind heparan sulfate GAG chains of proteoglycans, might be proteolytically cleaved off from some laminin isoforms in a physiologically regulated manner [48]. The supramolecular assembly of laminins is essential for BMs [49].
Being abundant around cells, matricellular proteins (Table 1) affect cell contacts to the ECM [50,51,52]. They have characteristic domains with which they interact with ECM scaffold proteins, such as the multifunctional SLRPs, decorin and versican [18, 50, 53]. Other matricellular proteins (reviewed in [50]) from various protein families are CCN proteins, tenascins [54,55,56], SIBLINGs [57], galectins [58], SPARC [59], thrombospondins [60], and periostin [61] (Table 1). They undergo characteristic changes during tumor progression [62]. Moreover, matricellular proteins, such as thrombospondins, can alter redox signaling in cancer cells, thereby affecting the hypoxia-influenced redox status within a tumor mass [60, 63].
Cellular contacts with the ECM
Cells are mechanically anchored in the ECM perceiving information about the environment. Epithelial and endothelial cells are aligned along the BM, which provides the substratum and cues for cell survival, and proliferation, and other biochemical and physiological functions of the cells [11, 14]. Entirely embedded within the ECM, fibroblasts also intimately interact with the ECM by producing and assisting assembly of ECM molecules and by exerting mechanical forces onto the fibrillar network within the ECM, thereby contributing to ECM tension, an important biophysical parameter for the embedded cells [64, 65].
At the molecular level, interactions of cells with the ECM are mediated by various adhesion receptors, among which integrins and about 13 transmembrane proteoglycans take a principal role (Table 1) [17, 66, 67]. 18 members constitute the family of integrins, all of which consist of two subunits, α and β [66, 67]. The N-terminal domains of both subunits jointly form the head domain, which harbors the ECM ligand binding site. From the head, the two chains individually form two stalks, which can take different conformations representing different activation states. Via short membrane-spanning α-helices, both subunits possess short cytoplasmic domains, except for the large cytoplasmic domain of the integrin β4 chain [67]. Integrins lack, unlike growth factor receptors, any signaling domains. By recruiting adaptor molecules and signal transducing molecules, such as focal adhesion kinase (FAK) and members of the Src family, they serve as intracellular docking sites for the cytoskeleton and relay signals between the ECM and the cell interior [68]. This supramolecular protein array, which is recruited to the ligand-bound and clustered integrins, can be considered as an adhesion-dependent cell organelle, which was termed adhesome [69,70,71,72]. Depending on its stage of formation or on its function to support stable adhesion or cell migration, different types of adhesomes, such as focal contacts and focal adhesions, can be distinguished [72, 73]. However, due to experimental accessibility, most studies on these adhesion complexes have been carried out with cells adhering to and migrating on ECM-coated surfaces. These adhesome structures differ from the ones formed by cells in the three-dimensional system of the ECM. Nevertheless, integrins as transmembrane connectors between the ECM and the cytoskeleton allow transmission of forces, which the cells generate with their actomyosin system, onto the ECM [70, 72].
The four syndecan isoforms belong to the transmembrane glycoprotein, which also mediate cellular interaction with the ECM [4, 17]. Moreover, via their heparan and chondroitin sulfate GAG chains, syndecans and other cell surface proteoglycans are able to regulate integrin-ligand interactions, such as between fibronectin and laminins and their corresponding receptors (Table 1) [40, 74, 75]. Thus, they may assist integrins in recruiting signaling molecules to the adhesomes [40, 75,76,77].
Tumor-induced ECM remodeling
After injury, tissue healing begins by forming the fibrin network of a hemostatic blood thrombus. Platelets, enclosed into the thrombotic plug, release growth factors and attract fibroblasts, which replace the provisional fibrin matrix for a collagenous matrix. The immigrating and resident fibroblasts differentiate into myofibroblasts [78, 79]. Marker proteins for myofibroblast differentiation are α-smooth muscle actin (αSMA), indicative of a strengthened actomyosin activity, and a bunch of secreted and deposited ECM-proteins, such as fibrillar collagens and fibronectin. The strengthened actomyosin machinery results in increased force exertion, which brings the newly deposited ECM under tension and causes wound contraction. Resembling chronic wounds, neoplastic tumors have been compared to a ‘wound that never heals’ [80, 81]. In fact, resident fibroblasts within a solid tumor mass differentiate into myofibroblast-like, cancer-associated fibroblasts (CAFs) under the influence of neighboring cancer cells [7, 79, 82, 83]. Transforming growth factor-β (TGF-β) is a key regulator in myofibroblast differentiation during wound healing and in CAF differentiation during tumor progression [7, 84]. Within the ECM meshwork, TGF-β is complexed by latent TGFβ-binding protein (LTBP) and the latency-associated protein (LAP) and tethered to the fibronectin network, which is connected to the fibrillar meshwork of the ECM [38]. Notably, partial proteolysis of LAP and mechanical tension along the ECM fibrils cause the release of TGF-β from its LTBP-cage and hence its activation. This promotes the differentiation of fibroblasts into more contractile CAFs, which further increase tension and release of TGF-β in a self-perpetuating circle [38, 79, 82, 85]. In addition, CAFs express a repertoire of diverse growth factors, such as vascular endothelial growth factor-A (VEGF-A), hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), and several chemokines, thereby attracting other cells, such as endothelial and immune cells to the tumor mass, and orchestrating them to join the tumor-supportive environment [7, 86].
By secreting ECM components and ECM-modifying enzymes, cancer cells modify their environment [87]. But even more, CAFs, driven by cancer cells, produce and deposit substantial amounts of ECM components, thus altering the ECM of the tumor stroma qualitatively and quantitatively [88, 89]. Therefore, the ECM at the primary site of the tumor dramatically changes in its composition and relative abundance, respectively. In addition, the ECM components are more frequently crosslinked by members of the LOX and transglutaminase family, especially LOX-1, LOXL-2 and transglutaminase-2, which are upregulated in neoplastic tumor tissue [32, 33, 90, 91]. Other posttranslational modifications of the ECM scaffold may also occur, as the non-enzymatic glycation of ECM proteins during hyperglycemia may explain the increased risk to develop certain tumors in diabetes patients [92]. Caused by these biochemical alterations, the ECM also changes in its biophysical parameters, such as topography, stiffness/rigidity, and tension (Fig. 1) [65, 93, 94], as increased deposition of fibrillar collagen and cross-linkage inter alia stiffens the tissue and causes desmoplasia, the tumor-typical fibrotic deposition of ECM. Fibrillar collagens abundantly expressed in desmoplastic tumor tissue and other ectopically expressed ECM proteins can serve as diagnostic tumor stroma markers (Table 1) [89, 95]. Among these tumor stroma-typical ECM proteins are tenascin-W, laminin-332 [96,97,98,99], and the splice variant of fibronectin expressing the extra domains A and B (ED-A and -B).

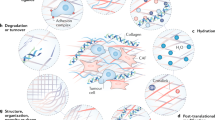
Potential roles of the ECM in tumor progression and metastasis. The ECM embeds tumor cells (TC), resident fibroblasts and their derivatives, the cancer-associated fibroblasts (CAFs). Tumor-induced CAF differentiation is a hallmark of tumor progression, as CAFs contribute to the tumor-permissive and supportive tumor microenvironment (TME), as CAFs remodel the ECM by synthesis, modification, and cross-linkage of ECM components, especially under the influence of transforming growth factor-β (TGF-β). ECM composition, supramolecular aggregates, such as collagenous fibrils, and the swelling potential of proteoglycans are biochemical parameters of the tumor stroma ECM. In addition, TGF-β and other growth factors are stored within the ECM and released in a tension-dependent manner. The increased ECM deposition and desmoplasia, and the biochemical cross-linkage increases the stiffness/rigidity. In addition, the topography of the ECM components and the tension on the ECM are biophysical parameters, which reinforce CAF differentiation and tumor progression. Both the swelling potential of proteoglycans and the high contractility of CAFs, the latter in a self-perpetuating manner, are responsible for the mechanical tension within the tumor stroma ECM. The biochemical and biophysical parameters of the tumor stroma ECM, together with the metabolic changes within the TME, attract endothelial and specific subsets of immune cells, which consequently contribute to tumor progression. Disseminating from the tumor mass, cancer cells undergo epithelial mesenchymal transition (EMT) and migrate either along tracks and channels within the ECM scaffold or are impaired by dense ECM obstacles, such as desmoplastic capsules of fibrillar collagen of the basement membrane. In the latter case, ECM-degrading enzymes clear a pathway and release ECM fragments, which as matrikines can stimulate cancer cell proliferation and invasion
Fibronectin is strongly expressed in the tumor stroma, in particular its splice variants ED-A and ED-B [37, 38, 100,101,102,103,104,105]. The two extra domains are partially redundant, as either of them can compensate for the loss of the other in the corresponding knockout mice. Nevertheless, the two extra domains contain additional binding sites for integrins, α9β1 and α4β1, in addition to the fibronectin-binding αv subunit-containing integrins and the classical fibronectin receptor, α5β1 integrin [101, 103, 104]. Presumably due to its enhanced ability to contact cells via integrins, the ED-A-containing FN variant releases TGF-β1 from its LTBP-LAP complex especially in a mechanical force-dependent manner, without any need of partial proteolysis [38, 84]. Thus, mechanical tension, based on integrin-transmitted forces, promotes CAF differentiation [85, 103]. Moreover, the fibronectin meshwork also tethers other growth factors and may deliver them to proliferating tumor cells [41]. With respect to the increased deposition of cell-adhesive ECM proteins in the tumor stroma, it is intriguing that the tenascin isoforms, tenascin-W and -C, are highly upregulated among the tenascin family in a TGF-β1 dependent manner [106, 107]. They interact with fibronectin and attenuate cell adhesion [50, 54, 56]. Tenascin-W may even serve as specific molecular marker for the tumor stroma (Table 1) [108,109,110].
In normal tissue, laminins are characteristically found in BMs. The ectopic expression of laminin-332 within the tumor stroma, especially at the invading front of tumor cells, is striking as other BM proteins are scarcely, if at all, expressed in this compartment (Table 1) [111,112,113]. However, some tumor entities apparently do not express laminin-332. For others, the expression of single laminin-332 chains, especially the laminin γ2 and β3 chains are reported. They correlate with poor prognosis for the patient (Table 1) [114,115,116,117]. Laminin-332 is also exceptional in terms of its diversity of partial proteolytic processing and the resulting altered cell migration [48]. Cleavage within the linker sequence between LG3 and LG4 of the laminin α3 chain may affect interaction sites for several integrins and the heparan sulfate chain of syndecan-4 and thus cell motility [48, 112, 118, 119]. Different proteases, also expressed by cancer cells, are involved in the proteolytic cleavage within the laminin chains, β3 and γ2, with different effects on tumor progression and cancer cell migration [48, 120, 121]. The ectopically expressed laminin-332 in tumor stroma promotes CAF differentiation and sustains the differentiated phenotype of CAFs [122].
The alterations in composition and cross-linkage of ECM, dictated by the cancer cells or mostly carried out by CAFs, result not only in biochemical properties and storage of growth factors [123], but also change biophysical parameters, such as stiffness and tension of the tumor stroma [65, 93, 124]. Although the ECM undergoes constant turnover, the life-time of these alterations limits plasticity of the tumor microenvironment, which can be considered as the ‘memory’ of the tissue or as a cancer-related ‘ECM signature’ [125,126,127]. This environmental niche provided by the biochemical and biophysical characteristics of the ECM is not only permissive to survival and proliferation of the cancer cells, but also it initiates and promotes oncogenic transformation and is able to influence somatic mutation rates [64, 65, 128]. This cancer growth-permissive tumor microenvironment (TME) also encompasses metabolic changes such as high concentration of lactic acid, acidosis, low support of oxygen and an altered redox status. These metabolic changes are caused by metabolic rewiring of cancer cells to use glycolysis as the primary source of energy, known as Warburg effect, their addiction to glutamine and their demand of oxygen [129, 130] [129, 130]. Other metabolic parameters of the TME are the altered metabolism of reactive oxygen species and of electrolytes, such as Ca2+-ions, which have been reviewed elsewhere [63, 131, 132]. CAFs also contribute to these metabolic changes and are driven by cancer cells to support their metabolic needs [97, 133]. The desmoplastic changes of the ECM and the metabolic reprogramming of both cancer cells and CAFs influence the susceptibility of tumors to the most common therapeutic strategies, chemo- and radiotherapy [130, 134,135,136,137].
The tumor stroma ECM influences endothelial and immune cells within the tumor mass
CAFs are derived from fibroblasts and other mesenchymal cells, such as stellate cells, preadipoctyes, bone marrow-derived cells, endothelial cells and pericytes [138]. During tumor-induced angiogenesis, endothelial cells are attracted to the tumor bulk by members of the VEGF family, which are expressed by cells within the hypoxic tumor mass [139, 140]. Among the VEGF family members, VEGF-A165 is the predominant angiogenic cytokine. In addition, CAF-produced cytokines, such as TGF-β and PDGF, stimulate angiogenic sprouting of vascular cells [7, 141]. These cytokines diffuse from the tumor mass and, by being tethered to GAG-chains of the ECM, form a stable gradient along which endothelial cells sprout into the tumor mass [142, 143]. Moreover, stiffness and other biophysical properties of the ECM influence angiogenetic sprouting, as endothelial cells form new capillaries especially at sites of high scaffold tension [144,145,146]. The outgrowing endothelial cells align longitudinally to form a lumenized tube that anastomoses and forms a closed circuit for blood flow [129, 147, 148]. The endothelial cell tubes are ensheathed by a newly formed BM. In contrast to vessels of normal tissue, tumor vessels are often tortuous, sometimes blunt-ended and with different calibers, which impairs blood flow through the tumor mass [147, 149]. Moreover, when reaching the tumor bulk, endothelial cells at the tip of the sprouts get in direct contact with the tumor stroma ECM and the tumor cells. Tumor cells not only coopt existing blood vessels, but also can integrate into the monolayer of endothelial cells or even replace the endothelial cells lining entirely, forming mosaic vessels or vasculogenic mimicry (VM) vessels [129, 147, 150,151,152]. Among other factors, ECM stiffness within the tumor mass favors the upregulation of the matricellular protein CCN1, which in turn induces N-cadherin expression in endothelial cells and thus allows direct intercellular contacts with tumor cells [153]. VM vessels are histologically recognized by their lack of the endothelial cell marker CD31 and by the presence of a sleeve of BM glycoproteins in periodic acid Schiff (PAS) staining [152, 154]. The presence of such VM vessels correlates, probably due to the direct access of tumor cells to the blood stream, with a poor prognosis [155]. Recent studies revealed that ECM proteins, their fragments and their supramolecular aggregates, foster the formation of such VM vessels. This has been reported for the matricellular protein CCN2, produced by CAFs, the proteolytically cleaved γ2 chain of laminin-332, and a dense three-dimensional collagen-network, respectively [156,157,158,159].
The new vascular tubes grown into the tumor mass provide nutrients and oxygen and remove waste products from the tumor cells, thereby supporting further tumor progression. Another advantage for the cancer cells is that ingrowing blood vessels facilitate their spread through the blood stream [160]. For this hematogenous metastasis, tumor cells gain access to the blood stream by penetrating the endothelial cell layer and its subjacent BM. Also, lymph vessels are subverted by cancer cells to spread throughout the body. This lymphogenic metastasis explains why metastases are frequently formed in the lymph nodes into which the tumor tissue-draining lymph vessels converge [161,162,163].
During vessel formation, endothelial cells are assisted by diverse immune cells [164,165,166]. Also on their route of immunological surveillance, immune cells encounter the tumor mass [167]. However, the TME keeps most cells of the innate and adaptive immune system in an immunosuppressive state [168]. The contact of immune cells with the desmoplastic ECM deposition plays a yet not fully understood role in this immunosuppression, which further fosters tumor progression [169]. Within the tumor stroma, monocyte-derived macrophages preferentially acquire the M2 phenotype. These tumor-associated macrophages (TAMs) secrete various cytokines and interleukins (ILs), especially the immune reaction-attenuating IL-10, as well as TGF-β [168, 170]. TGF-β attracts the subclass of regulatory T-cells (Tregs) and other adaptive immune cells to the tumor mass [84, 141]. Along with the myeloid suppressor cells, they suppress the attack of CD8+-T-cells and of natural killer (NK) cells to cancer cells [168, 171,172,173]. This is mediated by direct cellular contacts via membrane-bound receptors and counterreceptors, among them the immune checkpoint receptor PD-1 and its ligand, PD-L1 [164, 174,175,176]. Tregs also secrete TGF-β and help in activating ECM-tethered TGF-β, thereby reinforcing the tumor-supportive effects of this growth factor [173]. TGF-β was shown to cause expression of a set of ECM genes, which contribute to the immunosuppression of the TME [177]. Moreover, the selective recruitment of specific immune cells to the tumor stroma reinforces the desmoplastic process while the immune cell-secreted interleukins add an additional immunosuppressive tendency to the TME and contribute to the immunotolerance against cancer cells [168, 170, 178]. While reprogramming of Tregs via immune checkpoint inhibitors is already an approved therapy to reactivate immune response against cancer cells [174,175,176], selective immigration of subgroups of immune cells into the tumor stroma likely depends on the ECM of the tumor stroma and might become another strategy to curb tumor progression and metastasis [179].
Cancer cells on the move: along or across the ECM
In order to metastasize, cancer cells spread throughout the body along different routes in successive steps of the metastatic cascade (Fig. 2). Cancer cells disseminate from the primary tumor site, where they separate from their neighboring cancer cells. Then, they migrate through the ECM scaffold and have to overcome barriers made by the dense ECM meshwork. Sometimes, they are transported by the blood stream or by the lymph for long distances and thus get to other organs, where they settle down and form a metastasis. Along their routes, cancer cells encounter ECM proteins, and the ECM scaffold plays an ambivalent role in cancer cell migration and metastasis. On the one hand, ECM supports cancer cell dissemination as fibrillar supramolecular structures serve as stabilizing scaffold, tracks and channels, which enable cancer cells to move at considerable rates through the stromal tissue. On the other hand, the ECM meshwork may impair cell migration by barring the way of cancer cells and forming impermeable dense ECM barriers, such as at the BM [64]. Cells sense their ECM environment by forming very thin cell protrusions, the filopodia, in which molecular ECM receptors probe the ECM environment [68, 180]. In addition, cancer cells have molecular tools and strategies to deal with supportive or impairing types of ECM.

The metastatic cascade. After the oncogenically transformed epithelial cells have grown into a neoplastic tumor (carcinoma in situ), they reveal their malignancy by undergoing metastasis and colonizing distant organs. The metastatic cascade encompasses the following steps, in which cancer cells encounter ECM molecules. ➊ Breaching the basement membrane (BM) and infiltrating into the underlying connective tissue requires proteolytic activity of the cancer cells and is a hallmark of malignancy. ➋ Taking advantage of collagen fibrils, cancer cells migrate through the interstitial space along these fibrillar tracks or through channels between collagen fibrils. ➌ Approaching the blood vessels, cancer cells again breach the subendothelial BM, intravasate and reach the blood stream. ➍ Decorated with thrombocytes in a fibrin-dependent manner, blood-borne cancer cells reach distant organs via the circulation. ➎ After attaching to the vessel wall, cancer cells extravasate by breaching the subendothelial BM and access the interstitial stroma. ➏ Again by migrating along the fibrillar components of the ECM scaffold, cancer cells migrate towards the premetastatic niche. ➐ Reaching it, cancer cells engraft within the ECM of the metastatic niche and progress to a secondary tumor mass or temporally remain in a dormant state, until tumor progression resumes
Role of ECM in cancer cell dissemination
The vast majority of solid tumors are epithelial cell-derived carcinomas, which are characterized by their close cell–cell contacts. These intercellular connections are mediated via cadherins, a family of cell-membrane anchored immunoglobulin fold-containing proteins, which favor a homophilic, Ca2+-dependent trans-interaction between cells of the same tissue [181,182,183,184,185]. These cadherin-mediated contacts allow the selective formation of epithelial cell layers and enable cells to withstand intercellular forces thereby clustering epithelial cells. Such intercellular bonds are loosened when individual cancer cells or a group of cancer cells leave the carcinoma in situ to disseminate (Fig. 2, step ➊ [183]. Along with the loss of cell–cell contacts, these carcinoma cells undergo additional changes, such as changing their morphology and becoming motile. This step of epithelial-mesenchymal transition (EMT) depends on several factors, such as the ECM and growth factors [186, 187]. The process is conveyed by intracellular signaling pathways, receiving signals from the ECM via integrins and from several growth factors, such as TGF-β and hepatocyte growth factor (HGF) via the corresponding growth factor receptors [141, 186, 187]. In particular, TGF-β is of key importance not only to CAF differentiation and immunosuppression but also to EMT of carcinoma cells, initiating their metastatic movement [141]. Additional CAF-derived growth factors, such as SDF1/CXCL12, reinforce this process [188].
Cancer cells breach ECM barriers
Epithelial neoplasias progress but remain in their original site. However, a decisive hallmark of malignancy is that they breach the BM which separates the epithelium from the neighboring connective tissue [2]. The BM is impermeable to cells except immune cells and malignant tumor cells. To penetrate the dense ECM network of the BM, cancer cells secrete several proteases of different types [87, 189,190,191]. Among these proteases, the Zn2+ ion-dependent matrix metalloproteinases (MMPs) play a crucial role and hence have a high prognostic value [192,193,194,195]. Among the 28 members of the MMP family, most studies in the last few years have focused on the two collagenases, MMP-1 and membrane-type 1 MMP (MT1-MMP, MMP-14), as well as on the gelatinases, MMP-9 and MMP-2 (Table 1) [194, 196,197,198,199,200,201,202,203]. Whereas gelatinases cleave only denatured collagen chains, collagenases are able to partially unwind and cleave the otherwise proteolytically stable triple helix of collagen [204,205,206]. After collagenolytic cleavage by MMP1 and MT1-MMP, the triple helix is destabilized, unwinds and becomes a substrate to MMP-9 and MMP-2. Moreover, activation of MMP-2 also depends on the membrane-anchored MT1-MMP in a complex binding and cleaving mechanism [206,207,208]. Synthesis and secretion of MMPs by cancer cells depend on various factors of the TME, such as certain growth factors and the tumor-specific variants of fibronectin [209].
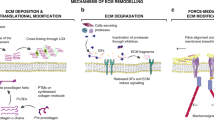
Invasion of cancer cells is hindered by the BM and capsules of fibrillar collagens surrounding desmoplastic tumors [2, 93]. Yet, invasive cancer cells can break through these dense collagen meshworks that are in their way with the aid of MT1-MMP-containing invadopodia, which take a central role in the proteolytic penetration of the ECM barrier [198, 201, 210, 211] (Fig. 3). These cellular protrusions are formed under the influence of the TME, especially TGF-β, HGF, and epidermal growth factor (EGF). Hypoxia also promotes invadopodia formation in cancer cells [198, 212,213,214]. The most appropriate stimulus is mechanical stiffness of the ECM, which is sensed via integrins in a Rho- and WASP/WAVE-dependent manner [68, 212, 215, 216]. This results in a Rac-, PAK1- and cortactin-dependent formation of the core structure of the invadopodia [198, 212, 217, 218]. This core structure consists of F-actin bundles, which stabilize the cell protrusion and extend it into the ECM barrier at the front of an invading cell [198, 212, 219]. During maturation, various signaling molecules, such as PAK-1 and -4, which prolong the half-life of invadopodia, are recruited [220]. In addition, collagenolytic MT1-MMP is recruited to the invadopodium, which enables local proteolysis to facilitate the penetration of barriers and to activate the soluble gelatinase MMP-2 [198, 220]. Due to the recruitment of Tks-4 and -5, invadopodia are stabilized for more than 60 min, in contrast to cellular protrusions, called podosomes, of endothelial cells during tumor-induced angiogenesis, which are also equipped with proteolytic MT1-MMP activity [221,222,223]. Structurally homologous to MT1-MMP, the membrane-anchored proteinases of the ADAMTS family (a disintegrin and metalloproteinase with thrombospondin-1 motif) have been identified to cleave proteoglycans if they are obstacles to the cancer cell movement (Table 1) [224]. To allow attachment to the ECM, integrins are located in a ring-like fashion at the base of the invadopodium [225]. Either lacking or possessing ECM-cleaving proteinases, adhesomes and invadopodia, respectively, are cellular organelles, which mediate cellular contacts with the ECM in a metastasis-relevant manner (Fig. 3).

Invadopodia and adhesomes are ECM-contacting cell organelles, which are relevant in metastasis. Cancer cells can produce special membrane protrusions, termed invadopodia, which in many ways resemble adhesomes but also show differences [71, 180, 212, 349, 350]. Like adhesomes, invadopodia are equipped with integrins as ECM adhesion molecules, and similar to endothelial cell podosomes, these form an adhesion ring during invadopodium maturation [225]. In addition to the adhesive capability of adhesomes, invadopodia have ECM-degrading abilities. Therefore, invadopodia are important promoters of the metastatic cascade. Invadopodia are remarkably long-lived compared to adhesomes. Preferably using the nucleus as a mechanical abutment, they possess a stiff actin core that, together with various adapter and signaling molecules, can propel the invadopodium far into the ECM like a molecular drilling rod. Essential components of invadopodia and adhesomes are listed for comparison
ECM barrier-penetrating proteinases also cleave protease-activated receptors (PARs) on cancer cells and CAFs, such as PAR-1 and PAR-2 [226]. These G-protein coupled receptors are cleaved canonically by thrombin and non-canonically by MMPs, such as MMP-1 and MMP-13, at their extracellular N-terminus [227, 228]. In the tumor stroma, activated monocytes/macrophages secrete thrombin. This is activated by cancer cell-derived tissue factor (TF) via an extrinsic coagulation cascade [226]. Likewise, MMPs are also activated at sites of ECM penetration. Proteolytic processing activates Gα12/13 of the heterotrimeric G-protein and thereby Rho signaling. This affects the actomyosin machinery with its motor protein myosin II and thus increases cell contractility and cell movement. This further promotes invasion through the ECM barrier. Moreover, Rho activation promotes EMT of cadherin-interlinked cancer cells [227, 228].
In addition, ECM barrier-penetrating proteinases release soluble and bioactive fragments, so-called matrikines (Table 1), from insoluble matrix proteins that indirectly affect the TME [229,230,231]. Thus, fragments of BM collagen types IV, XV, XVIII, and XIX are released by proteolytically active infiltrating tumor cells. In addition to affecting cancer cells [232], endostatin and other such fragments are angiostatic and prevent sprouting of endothelial cells into the tumor mass [143, 229, 233, 234]. Endostatin also reverses immunosuppression [235], and a versican-derived matrikine causes selective recruitment of a specific subset of dendritic cells to the tumor stroma [236]. Similarly, the perlecan fragment endorepellin impairs angiogenesis by interacting with integrin α2β1 on endothelial cells [237, 238]. Some fragments of matricellular proteins and of laminin-332 agonistically bind to the EGF receptor, thereby promoting cancer cell from motility [230, 239]. Likewise, matrikines can be cleaved off elastin [240,241,242].
ECM provides the molecular tracks for cancer cell dissemination
The molecular details of cell migration are described in other reviews [243,244,245,246,247,248]. Here, it may suffice to name the relevant molecules: (i) the force-generating actomyosin system, consisting of F-actin fibers and the interconnecting motor proteins myosin II [249,250,251,252] (ii) the actin cytoskeleton-organizing members of the Rho family, the WAVE/WASP family and Arf family [215, 216, 253,254,255], (iii) the motor protein-regulating kinases [256], and (iv) the adhesome proteins including integrins [251, 257] as well as adaptor proteins, such as vinculin, α-actinin, and paxillin. Vinculin and α-actinin change their conformation and thus unmask cryptic binding sites for other proteins upon force transmission [258,259,260,261,262,263]. Moreover, paxillin and vinculin are recruited after phosphorylation to the adhesome in a sustained and force-dependent manner [264, 265]. Therefore, they sense mechanical forces, including the stiffness/rigidity and the tension of the ECM, and serve as receptors for mechanotransduction. Moreover, linkage of the cytoskeleton with the nuclear membrane and force-dependent translocation of the transcription-factors, YAP and TAZ, results in a force-dependent gene expression along the Hippo pathway [266,267,268].
Cancer cell migration depends on the biochemical properties of the ECM, as cell adhesion receptors must interact with the appropriate strength to the scaffold protein to allow both attachment and detachment of the cell in an ordered manner [269] Collagen fibers are ideal tracks, along which tumor cells migrate (Fig. 2, steps ➋ and ➏ [270, 271]. In haptotactic migration, cells sense the density of cell attachment sites of the deposited ECM proteins [272, 273]. In addition, biophysical parameters of the ECM, such as fiber topography and geometry, stiffness/rigidity and tension determine cell migration (Fig. 1) [64, 65, 274]. Moreover, cancer cells perceive the mechanical conditions of their environment and regulate the migration rate in correlation to the ECM stiffness in durotactic migration [65].
Following the path of least resistance, infiltrating cells recognize and use open pores within the ECM. These pores must have an open cross-sectional area of at least 7 µm2, which is necessary for the rather rigid nucleus of a eukaryotic cell to be squeezed through [3, 275, 276]. Otherwise, protease-assisted invadopodia open the pore to the necessary size. But also disruption and subsequent repair of nuclei have been observed [277]. Usually, cancer cells move through channel-like tracks of the ECM which are 3–30 µm in diameter and 100–600 µm in length [3]. Once such a migration track through the ECM scaffold is found or opened by a path-finding cancer cell, several cancer cells follow this path [127, 278, 279]. If adhesion strength to the ECM proteins in these channels is too low, cancer cells move in an amoeboid fashion, which is driven by the contractile activity of the cortical actin network. Upon integrin-mediated firm adhesion, cells migrate faster with a spindle-like fibroblastic shape. In adhesomes, integrins are connected to the actin cytoskeleton. Together with F-actin stress fibers, which stretch through the entire cell soma, myosin motor proteins generate the forces, which move the cell relative to the ECM. There are other additional types of migration between amoeboid and fibroblast-like migration, such as lobopodial migration with characteristic bleb-like membrane protrusions [3, 278,279,280].
Depending of the degree of intercellular contacts between the cancer cells, not only single cells but also groups of interconnected cancer cells can invade the ECM [281, 282]. Collectively migrating cancer cells are linked by cadherins and gap junctions, making them slower but also more robust to antimetastatic factors [283, 284]. In contrast, single cells or cohorts of less interconnected cancer cells migrate faster [281]. The transition from single cell migration to collective cell migration is multifactorial [127, 278, 279, 285]. Among other factors, it depends on the biochemical composition of the ECM, since, for example, fibronectin supports single cell migration [286]. Moreover, cancer cells adapt their migration mode to the biophysical properties of the ECM [286], to the status of pericellular proteolysis [287] and to metabolic conditions, including the hypoxic status, of the tissue [288, 289].
Also during cancer cell infiltration and migration, CAFs are not mere bystanders but reportedly promote cancer cell infiltration into the surrounding normal tissue by taking the lead and forming ECM tracks in which tumor cells follow [290]. Integrin α3β1 on CAFs and its ligand, laminin-332, in the tumor stroma play key roles in this context and confer migratory and invasive properties on cancer cells [122].
Hematogenous metastasis also depends on the ECM
The fastest way for cancer cells to colonize distant organs is the transport by the blood stream. During intravasation and extravasation, cancer cells gain access to and exit from the blood stream by penetrating the dense ECM network of the BM (Fig. 2, steps ➌ and ➎). Blood-borne cancer cells recruit platelets (Fig. 2, step ➍). The latter support the cancer cells with growth factors and shelter them from immune attack [291]. The formation of such tumor cell-platelet aggregates is indispensably required for hematogenous metastasis [292]. Most of the multifactorial interactions between cancer cells and platelets are mediated via direct receptor-counterreceptor contacts. Nevertheless, fibrin is also an essential bridging molecule, which is recognized by both tumor cells and platelets via the integrins, αvβ3 and αIIbβ3, respectively [291]. Tissue factor (TF) expressing tumor cells activate the extrinsic pathway of the coagulation cascade and initiate the conversion of soluble fibrinogen to insoluble fibrin [293]. Like the scaffolding proteins of the ECM, fibrin molecules form highly ordered fibrin bundles, which bridge tumor cells and platelets, and thus contribute to hematogenous metastasis [226]. Von Willebrand factor (vWF), an ECM proteins usually expressed by endothelial cells, but also by cancer cells, may serve the same tumor cell-platelet-bridging function, and assist tumor cells to attach to the endothelial cells upon extravasation, along with cell adhesion molecules on tumor cells and platelets (Fig. 2, step ➎) [294, 295].
The metastatic niche, the site of settlement and colonization of cancer cells
If cancer cells find suitable conditions in a distant organ, they settle there and form a secondary, metastatic tumor (Fig. 2, step ➐). The question, whether the tumor cells are actively targeting a particular tissue or accidentally encountering an environment suitable for further cancer progression, has not yet been fully resolved. The first option is underlined by the fact that certain cancer entities show organotropism and form metastases preferentially in certain organs [296,297,298,299]. Another option was first phrased as ‘seed and soil’ theory by Stephen Paget in 1889, which states that metastasizing cancer cells like seeds engraft and grow only in tumor cell-permissive tissues (soil). In reality, both options are partly realized, as a primary tumor mass is able to influence secondary sites of the organism to prepare those distant sites for tumor cell engraftment, even before cancer cells take their metastatic routes. Hence, these primed secondary tissue sites were named premetastatic niches [300, 301].
Chemokines and matrikines, as well as exosomes, 30-100 nm-sized extracellular vesicles, are systemically released from the primary tumor [298, 302,303,304]. On their surface, exosomes bear integrins, which adhere to the ECM of the targeted tissue after being systemically distributed via blood and lymph throughout the body [298]. At the target sites, they fuse with normal cells and release their contents, among them translatable mRNA and regulatory microRNA (miRNA), along with proteins [303, 304]. Thus, these cells may change their phenotype, alter their metabolism, and secrete ECM proteins or ECM-modifying enzymes, such as LOXs [305]. In addition, systemically spread cytokines and growth factors are recognized by corresponding receptors in the target tissue [126, 306]. Among the latter, also TGF-β educates the designated sites of metastatic settlement by altering the ECM and resident cells in a biochemical and biophysical manner similar to the ones in the primary tumor mass [306,307,308]. For example, tenascin-W is a component of the (pre)metastatic niche in bones [106]. LOX family members, secreted from a primary tumor and distributed systemically via the blood circulation, can interfere with bone homeostasis by promoting osteoclastogenesis, leading to the formation of premetastatic lesions that can be colonized by circulating cancer cells [309]. LOX also promotes tissue stiffening and induces premetastatic niche and metastasis in breast cancer [310, 311]. Also, immune cells immigrate into the chemokine- and exosome-responsive tissue, further preparing the properties of the (pre)metastatic niche [312]. Upon engraftment of metastasizing cancer cells, the premetastatic niche becomes a metastatic niche, which fosters its own progression, thus closing the metastatic cycle, or its dormancy [299, 313, 314].
Translational aspects and perspectives
The knowledge about the biophysical alterations of tumor tissue has been used diagnostically for a long time. For example, palpation examination of the breast detects tissue regions of stiffer ECM and desmoplasia, which are suspicious and prompt further examination of the patient. Biochemical alteration of the tumor stroma is analyzed histologically in tissue biopsies. Fibronectin splice variants containing the domains ED-A and ED-B, laminin-332, periostin and tenascin-W are such markers typical of tumor stroma [7, 61, 98, 109, 114, 315, 316]. Microscopic and other imaging techniques have been improved to diagnostically analyze more accurately tumor-associated alterations of the ECM [97, 271]. Therapeutic approaches to use these markers as antigenic targets to direct antitumor agents to the tumor site have been only experimental so far [96, 99]. Some of these tumor stroma-typical proteins and fragments thereof are diagnostically detected in blood samples, such as the laminin γ2 chains [117]. They may develop into more robust and easily accessible tumor markers.
Therapeutics which directly target ECM components are still awaited. Several attempts, also in clinical trials, have been made to pharmacologically target ECM-modifying enzymes such MMPs and LOXs [32, 189, 191, 202]. Inhibition of MMPs might support the endogenous ECM barrier. Therefore, such inhibitors might prevent cancer cells from breaching the BM and curb or even prevent metastasis [5, 317]. Moreover, inhibitors of MMP-activated PARs, which enhance the metastatic invasion of cancer cells, have been used in clinical trial [227].
While the use of MMP inhibitors as anti-cancer drugs was initially difficult due to lack of specificity and side effects, several new MMP inhibitors with improved properties have been developed and are currently under investigation ([318] and references therein). Neovastat (Benefin/AE-941) inhibits MMPs-2, -9, and -12, which is well tolerated by cancer patients in phase I/II clinical trials [319, 320]. Yet, in a phase III trial, no efficacy was found in patients with non-small cell lung carcinoma [321]. The broad-spectrum MMP inhibitor BMS-275291 was well tolerated in a phase I study and showed little musculoskeletal toxicity [322], but in a phase III study, it did not improve patient survival in advanced non-small cell lung carcinoma [323]. Also, another phase III study with tanomastat (BAY 12-9566), a non-peptidomimetic MMP inhibitor directed against MMPs-2, -3, and -9, did not show any efficacy in slowing or halting cancer progression [324]. Similarly, prinomastat (AG-3340), which inhibits MMPs-2, -3, 9, -13, and -14, caused arthralgia and myalgia in a phase I study and failed in a phase III trial of efficacy in non-small cell lung cancer [325, 326]. Yet, several monoclonal antibodies against MMP-9 [327, 328] and MMP-14 [329, 330] appear promising in preclinical models. With regard to MMP-directed tumor therapy, microRNA-mediated post-transcriptional MMP regulation is also of interest [331,332,333].
The other group of collagen-modifying enzymes, the copper-dependent LOX family members are also tangible targets for tumor therapy [32]. In a phase II study on copper depletion using tetrathiomolybdate, triple-negative breast cancer patients showed a marked decrease in the LOX activity involved in the formation of a premetastatic niche [334]. In other phase II trials, however, the LOXL2 function-blocking antibody simtuzumab did not improve clinical outcomes in patients with KRAS mutant colorectal or pancreatic adenocarcinoma [335, 336], although it is well tolerated in patients with solid tumors and inhibits the desmoplastic reaction in vitro [337, 338].
When inhibitors of ECM-modifying enzymes are used, in addition to limited specificity, undesirable side effects pose a considerable problem. For example, the broad-spectrum MMP inhibitor marimastat, although better bioavailable than its analog batimastat [339], proved inappropriate in phase III trials because it caused musculoskeletal pain and inflammation [340]. Similarly, the development of the low molecular weight inhibitor CGS 27023A/MMI270 directed against MMPs-2, -8 and -9 was discontinued because of poorly tolerated joint and muscular pains in phase II studies in early stage non-small cell lung carcinoma patients [341].
The potential to harness ECM molecules as antimetastatic therapeutics or to deliver anticancer compounds to the tumor have recently been evaluated in detail in a series of excellent reviews [16, 20, 39, 42, 43, 52, 237, 318, 342, 343]. Also, the ECM-receptors might be relevant pharmacological targets in blocking cancer cell progression and migration [243, 344, 345]. However, attempts to “normalize” the tumor stroma and its ECM into a non-tumor supporting environment [346] or to prevent the tumor-induced formation of premetastatic niches [347, 348] are desirable goals for the future.
Abbreviations
- ADAMTS:
-
A disintegrin and metalloproteinase with thrombospondin motifs
- CAF:
-
Cancer-associated fibroblast
- CCN:
-
CTGF, Cyr61, and NOV
- CTGF:
-
Connective tissue growth
- CXCL12 = SDF.1:
-
C-X-X chemokine 12 = stroma cell-derived factor-1
- Cyr61:
-
Cysteine-rich angiogenic protein 61
- DDR:
-
Discoidin domain receptor
- ECM:
-
Extracellular matrix
- ED-A, -B:
-
Extra domain-A, -B
- EGF-L:
-
Epidermal growth factor-like
- EGFR:
-
Epidermal growth factor receptor
- Ela-2:
-
Neutrophil elastase
- ERC:
-
Elastin receptor complex
- Endo180:
-
Endocytic receptor 180 = C-type mannose receptor 2
- FNFr:
-
Fibronectin fragment
- GAG:
-
Glycosaminoglycan
- GPVI:
-
Glycoprotein VI
- HGF:
-
Hepatocyte growth factor
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- IL:
-
Interleukin
- KRAS:
-
Kirsten rat sarcoma oncogene
- LAIR-1:
-
Leukocyte-associated immunoglobulin-like receptor 1
- LG3, 4:
-
Laminin globular domain 3, 4
- LOX/LOXL:
-
Lysyl oxidase/lysyl oxidase-like
- LRP6:
-
Low-density lipoprotein receptor-related protein 6
- LY75:
-
Lymphocyte antigen 75 (CD205, DEC-205)
- MET:
-
Mesenchymal-epithelial transition factor proto-oncogene, hepatocyte growth factor receptor, HGFR
- MMP:
-
Matrix metalloproteinase
- MR:
-
Mannose receptor
- MuSK:
-
Muscle-specific kinase
- NC1:
-
Non-collagenous domain 1
- NCAM-L1:
-
Neural cell adhesion molecule L1
- NOV:
-
Nephroblastoma overexpressed gene
- NrCAM:
-
Neuron-glial related cell adhesion molecule
- PAR:
-
Protease-activated receptor
- PD-1/PD-L1:
-
Programmed death-1/programmed death ligand-1
- PDGF:
-
Platelet-derived growth factor
- PLA2R:
-
Phospholipase-A2-receptor
- Sema3F:
-
Semaphorin 3F
- SIBLING:
-
Small integrin-binding ligand-N-linked glycoprotein
- SLRP:
-
Small leucine-rich proteoglycan
- SPARC:
-
Secreted protein, acid and rich in cysteine
- TAM:
-
Tumor-associated macrophage
- TF:
-
Tissue factor
- TLR-2, -4:
-
Toll-like receptor-2, -4
- TME:
-
Tumor microenvironment
- TGF-β:
-
Transforming growth factor-β
- Treg:
-
Regulatory T cell
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- VM:
-
Vasculogenic mimicry
References
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Paul CD, Mistriotis P, Konstantopoulos K (2017) Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer 17(2):131–140. https://doi.org/10.1038/nrc.2016.123
Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK (2016) Extracellular matrix structure. Adv Drug Deliv Rev 97:4–27. https://doi.org/10.1016/j.addr.2015.11.001
Chang TT, Thakar D, Weaver VM (2017) Force-dependent breaching of the basement membrane. Matrix Biol 57–58:178–189. https://doi.org/10.1016/j.matbio.2016.12.005
Walker C, Mojares E, Del Rio Hernandez A (2018) Role of extracellular matrix in development and cancer progression. Int J Mol Sci 19(10):3028. https://doi.org/10.3390/ijms19103028
Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16(9):582–598. https://doi.org/10.1038/nrc.2016.73
Rozario T, DeSimone DW (2010) The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol 341(1):126–140. https://doi.org/10.1016/j.ydbio.2009.10.026
Halper J, Kjaer M (2014) Basic components of connective tissues and extracellular matrix: elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv Exp Med Biol 802:31–47. https://doi.org/10.1007/978-94-007-7893-1_3
Singh B, Fleury C, Jalalvand F, Riesbeck K (2012) Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol Rev 36(6):1122–1180. https://doi.org/10.1111/j.1574-6976.2012.00340.x
Halfter W, Oertle P, Monnier CA, Camenzind L, Reyes-Lua M, Hu H, Candiello J, Labilloy A, Balasubramani M, Henrich PB, Plodinec M (2015) New concepts in basement membrane biology. FEBS J 282(23):4466–4479. https://doi.org/10.1111/febs.13495
Mak KM, Mei R (2017) Basement membrane type IV collagen and laminin: an overview of their biology and value as fibrosis biomarkers of liver disease. Anat Rec (Hoboken) 300(8):1371–1390. https://doi.org/10.1002/ar.23567
McCarthy KJ (2015) The basement membrane proteoglycans perlecan and agrin: something old, something new. Curr Top Membr 76:255–303. https://doi.org/10.1016/bs.ctm.2015.09.001
Miller RT (2017) Mechanical properties of basement membrane in health and disease. Matrix Biol 57–58:366–373. https://doi.org/10.1016/j.matbio.2016.07.001
Randles MJ, Humphries MJ, Lennon R (2017) Proteomic definitions of basement membrane composition in health and disease. Matrix Biol 57–58:12–28. https://doi.org/10.1016/j.matbio.2016.08.006
Liang J, Jiang D, Noble PW (2016) Hyaluronan as a therapeutic target in human diseases. Adv Drug Deliv Rev 97:186–203. https://doi.org/10.1016/j.addr.2015.10.017
Iozzo RV, Schaefer L (2015) Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol 42:11–55. https://doi.org/10.1016/j.matbio.2015.02.003
Schaefer L, Tredup C, Gubbiotti MA, Iozzo RV (2017) Proteoglycan neofunctions: regulation of inflammation and autophagy in cancer biology. FEBS J 284(1):10–26. https://doi.org/10.1111/febs.13963
Ricard-Blum S (2011) The collagen family. Cold Spring Harb Perspect Biol 3(1):a004978. https://doi.org/10.1101/cshperspect.a004978
An B, Lin YS, Brodsky B (2016) Collagen interactions: drug design and delivery. Adv Drug Deliv Rev 97:69–84. https://doi.org/10.1016/j.addr.2015.11.013
Mao M, Alavi MV, Labelle-Dumais C, Gould DB (2015) Type IV collagens and basement membrane diseases: cell biology and pathogenic mechanisms. Curr Top Membr 76:61–116. https://doi.org/10.1016/bs.ctm.2015.09.002
Bhattacharjee A, Bansal M (2005) Collagen structure: the Madras triple helix and the current scenario. IUBMB Life 57(3):161–172. https://doi.org/10.1080/15216540500090710
Brodsky B, Persikov AV (2005) Molecular structure of the collagen triple helix. Adv Protein Chem 70:301–339. https://doi.org/10.1016/S0065-3233(05)70009-7
Beck K, Brodsky B (1998) Supercoiled protein motifs: the collagen triple-helix and the α-helical coiled coil. J Struct Biol 122(1–2):17–29. https://doi.org/10.1006/jsbi.1998.3965
Provenzano PP, Vanderby R Jr (2006) Collagen fibril morphology and organization: implications for force transmission in ligament and tendon. Matrix Biol 25(2):71–84. https://doi.org/10.1016/j.matbio.2005.09.005
Boudko SP, Danylevych N, Hudson BG, Pedchenko VK (2018) Basement membrane collagen IV: isolation of functional domains. Methods Cell Biol 143:171–185. https://doi.org/10.1016/bs.mcb.2017.08.010
Cummings CF, Pedchenko V, Brown KL, Colon S, Rafi M, Jones-Paris C, Pokydeshava E, Liu M, Pastor-Pareja JC, Stothers C, Ero-Tolliver IA, McCall AS, Vanacore R, Bhave G, Santoro S, Blackwell TS, Zent R, Pozzi A, Hudson BG (2016) Extracellular chloride signals collagen IV network assembly during basement membrane formation. J Cell Biol 213(4):479–494. https://doi.org/10.1083/jcb.201510065
Vanacore RM, Friedman DB, Ham AJ, Sundaramoorthy M, Hudson BG (2005) Identification of S-hydroxylysyl-methionine as the covalent cross-link of the noncollagenous (NC1) hexamer of the α1α1α2 collagen IV network: a role for the post-translational modification of lysine 211 to hydroxylysine 211 in hexamer assembly. J Biol Chem 280(32):29300–29310. https://doi.org/10.1074/jbc.M502752200
Heljasvaara R, Aikio M, Ruotsalainen H, Pihlajaniemi T (2017) Collagen XVIII in tissue homeostasis and dysregulation—lessons learned from model organisms and human patients. Matrix Biol 57–58:55–75. https://doi.org/10.1016/j.matbio.2016.10.002
Shaw LM, Olsen BR (1991) FACIT collagens: diverse molecular bridges in extracellular matrices. Trends Biochem Sci 16(5):191–194
Has C, Bruckner-Tuderman L (2006) Molecular and diagnostic aspects of genetic skin fragility. J Dermatol Sci 44(3):129–144. https://doi.org/10.1016/j.jdermsci.2006.08.003
Barker HE, Cox TR, Erler JT (2012) The rationale for targeting the LOX family in cancer. Nat Rev Cancer 12(8):540–552. https://doi.org/10.1038/nrc3319
Eckert RL, Fisher ML, Grun D, Adhikary G, Xu W, Kerr C (2015) Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol Carcinog 54(10):947–958. https://doi.org/10.1002/mc.22375
Li B, Cerione RA, Antonyak M (2011) Tissue transglutaminase and its role in human cancer progression. Adv Enzymol Relat Areas Mol Biol 78:247–293
Saitow CB, Wise SG, Weiss AS, Castellot JJ, Kaplan DL (2013) Elastin biology and tissue engineering with adult cells. Biomol Concepts 4(2):173–185. https://doi.org/10.1515/bmc-2012-0040
Kim YM, Kim EC, Kim Y (2011) The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol Biol Rep 38(1):145–149. https://doi.org/10.1007/s11033-010-0088-0
Mezzenga R, Mitsi M (2018) The molecular dance of fibronectin: conformational flexibility leads to functional versatility. Biomacromol. https://doi.org/10.1021/acs.biomac.8b01258
White ES, Muro AF (2011) Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 63(7):538–546. https://doi.org/10.1002/iub.493
Kumra H, Reinhardt DP (2016) Fibronectin-targeted drug delivery in cancer. Adv Drug Deliv Rev 97:101–110. https://doi.org/10.1016/j.addr.2015.11.014
Woods A, Longley RL, Tumova S, Couchman JR (2000) Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch Biochem Biophys 374(1):66–72. https://doi.org/10.1006/abbi.1999.1607
Prasad A, Clark RA (2018) Fibronectin interaction with growth factors in the context of general ways extracellular matrix molecules regulate growth factor signaling. G Ital Dermatol Venereol 153(3):361–374. https://doi.org/10.23736/S0392-0488.18.05952-7
Dosio F, Arpicco S, Stella B, Fattal E (2016) Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv Drug Deliv Rev 97:204–236. https://doi.org/10.1016/j.addr.2015.11.011
Neill T, Schaefer L, Iozzo RV (2016) Decorin as a multivalent therapeutic agent against cancer. Adv Drug Deliv Rev 97:174–185. https://doi.org/10.1016/j.addr.2015.10.016
Gubbiotti MA, Neill T, Iozzo RV (2017) A current view of perlecan in physiology and pathology: a mosaic of functions. Matrix Biol 57–58:285–298. https://doi.org/10.1016/j.matbio.2016.09.003
Harvey SJ, Miner JH (2008) Revisiting the glomerular charge barrier in the molecular era. Curr Opin Nephrol Hypertens 17(4):393–398. https://doi.org/10.1097/MNH.0b013e32830464de
Aumailley M (2013) The laminin family. Cell Adhes Migr 7(1):48–55. https://doi.org/10.4161/cam.22826
Hohenester E, Engel J (2002) Domain structure and organisation in extracellular matrix proteins. Matrix Biol 21(2):115–128
Rousselle P, Beck K (2013) Laminin 332 processing impacts cellular behavior. Cell Adhes Migr 7(1):122–134. https://doi.org/10.4161/cam.23132
Hohenester E, Yurchenco PD (2013) Laminins in basement membrane assembly. Cell Adhes Migr 7(1):56–63. https://doi.org/10.4161/cam.21831
Thakur R, Mishra DP (2016) Matrix reloaded: CCN, tenascin and SIBLING group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol Ther 168:61–74. https://doi.org/10.1016/j.pharmthera.2016.09.002
Viloria K, Hill NJ (2016) Embracing the complexity of matricellular proteins: the functional and clinical significance of splice variation. Biomol Concepts 7(2):117–132. https://doi.org/10.1515/bmc-2016-0004
Sawyer AJ, Kyriakides TR (2016) Matricellular proteins in drug delivery: therapeutic targets, active agents, and therapeutic localization. Adv Drug Deliv Rev 97:56–68. https://doi.org/10.1016/j.addr.2015.12.016
Sofeu Feugaing DD, Gotte M, Viola M (2013) More than matrix: the multifaceted role of decorin in cancer. Eur J Cell Biol 92(1):1–11. https://doi.org/10.1016/j.ejcb.2012.08.004
Yoshida T, Akatsuka T, Imanaka-Yoshida K (2015) Tenascin-C and integrins in cancer. Cell Adhes Migr 9(1–2):96–104. https://doi.org/10.1080/19336918.2015.1008332
Brellier F, Martina E, Degen M, Heuze-Vourc’h N, Petit A, Kryza T, Courty Y, Terracciano L, Ruiz C, Chiquet-Ehrismann R (2012) Tenascin-W is a better cancer biomarker than tenascin-C for most human solid tumors. BMC Clin Pathol 12:14. https://doi.org/10.1186/1472-6890-12-14
Chiquet-Ehrismann R, Hagios C, Matsumoto K (1994) The tenascin gene family. Perspect Dev Neurobiol 2(1):3–7
Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS (2008) Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer 8(3):212–226. https://doi.org/10.1038/nrc2345
Kamili NA, Arthur CM, Gerner-Smidt C, Tafesse E, Blenda A, Dias-Baruffi M, Stowell SR (2016) Key regulators of galectin-glycan interactions. Proteomics 16(24):3111–3125. https://doi.org/10.1002/pmic.201600116
Naschberger E, Liebl A, Schellerer VS, Schutz M, Britzen-Laurent N, Kolbel P, Schaal U, Haep L, Regensburger D, Wittmann T, Klein-Hitpass L, Rau TT, Dietel B, Meniel VS, Clarke AR, Merkel S, Croner RS, Hohenberger W, Sturzl M (2016) Matricellular protein SPARCL1 regulates tumor microenvironment-dependent endothelial cell heterogeneity in colorectal carcinoma. J Clin Invest 126(11):4187–4204. https://doi.org/10.1172/JCI78260
Roberts DD, Kaur S, Isenberg JS (2017) Regulation of cellular redox signaling by matricellular proteins in vascular biology, immunology, and cancer. Antioxid Redox Signal 27(12):874–911. https://doi.org/10.1089/ars.2017.7140
Gonzalez-Gonzalez L, Alonso J (2018) Periostin: a matricellular protein with multiple functions in cancer development and progression. Front Oncol 8:225. https://doi.org/10.3389/fonc.2018.00225
Vincent KM, Postovit LM (2018) Matricellular proteins in cancer: a focus on secreted Frizzled-related proteins. J Cell Commun Signal 12(1):103–112. https://doi.org/10.1007/s12079-017-0398-2
Grosche J, Meissner J, Eble JA (2018) More than a syllable in fib-ROS-is: the role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol Aspects Med 63:30–46. https://doi.org/10.1016/j.mam.2018.03.005
Holle AW, Young JL, Van Vliet KJ, Kamm RD, Discher D, Janmey P, Spatz JP, Saif T (2018) Cell-extracellular matrix mechanobiology: forceful tools and emerging needs for basic and translational research. Nano Lett 18(1):1–8. https://doi.org/10.1021/acs.nanolett.7b04982
Stroka KM, Konstantopoulos K (2014) Physical biology in cancer. 4. Physical cues guide tumor cell adhesion and migration. Am J Physiol Cell Physiol 306(2):C98–C109. https://doi.org/10.1152/ajpcell.00289.2013
Barczyk M, Carracedo S, Gullberg D (2010) Integrins. Cell Tissue Res 339(1):269–280. https://doi.org/10.1007/s00441-009-0834-6
Campbell ID, Humphries MJ (2011) Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol 3(3):a004994. https://doi.org/10.1101/cshperspect.a004994
Humphries JD, Chastney MR, Askari JA, Humphries MJ (2018) Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol 56:14–21. https://doi.org/10.1016/j.ceb.2018.08.004
Horton ER, Humphries JD, James J, Jones MC, Askari JA, Humphries MJ (2016) The integrin adhesome network at a glance. J Cell Sci 129(22):4159–4163. https://doi.org/10.1242/jcs.192054
Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM (2010) Nanoscale architecture of integrin-based cell adhesions. Nature 468(7323):580–584. https://doi.org/10.1038/nature09621
Winograd-Katz SE, Fassler R, Geiger B, Legate KR (2014) The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol 15(4):273–288. https://doi.org/10.1038/nrm3769
Geiger B, Yamada KM (2011) Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol 3(5):a005033. https://doi.org/10.1101/cshperspect.a005033
Zaidel-Bar R, Geiger B (2010) The switchable integrin adhesome. J Cell Sci 123(Pt 9):1385–1388. https://doi.org/10.1242/jcs.066183
Carulli S, Beck K, Dayan G, Boulesteix S, Lortat-Jacob H, Rousselle P (2012) Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin α3 LG45 protein domain. J Biol Chem 287(15):12204–12216. https://doi.org/10.1074/jbc.M111.300061
Bachy S, Letourneur F, Rousselle P (2008) Syndecan-1 interaction with the LG4/5 domain in laminin-332 is essential for keratinocyte migration. J Cell Physiol 214(1):238–249. https://doi.org/10.1002/jcp.21184
Soares MA, Teixeira FC, Fontes M, Areas AL, Leal MG, Pavao MS, Stelling MP (2015) Heparan sulfate proteoglycans may promote or inhibit cancer progression by interacting with integrins and affecting cell migration. Biomed Res Int 2015:453801. https://doi.org/10.1155/2015/453801
Pantazaka E, Papadimitriou E (2014) Chondroitin sulfate-cell membrane effectors as regulators of growth factor-mediated vascular and cancer cell migration. Biochim Biophys Acta 1840(8):2643–2650. https://doi.org/10.1016/j.bbagen.2014.01.009
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180(4):1340–1355. https://doi.org/10.1016/j.ajpath.2012.02.004
Otranto M, Sarrazy V, Bonte F, Hinz B, Gabbiani G, Desmouliere A (2012) The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr 6(3):203–219. https://doi.org/10.4161/cam.20377
Dvorak HF (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315(26):1650–1659. https://doi.org/10.1056/nejm198612253152606
Dvorak HF (2015) Tumors: wounds that do not heal-redux. Cancer Immunol Res 3(1):1–11. https://doi.org/10.1158/2326-6066.Cir-14-0209
Kuzet SE, Gaggioli C (2016) Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res 365(3):607–619. https://doi.org/10.1007/s00441-016-2467-x
Desmouliere A, Guyot C, Gabbiani G (2004) The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol 48(5–6):509–517. https://doi.org/10.1387/ijdb.041802ad
Caja L, Dituri F, Mancarella S, Caballero-Diaz D, Moustakas A, Giannelli G, Fabregat I (2018) TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mol Sci 19(5):1294. https://doi.org/10.3390/ijms19051294
Khan Z, Marshall JF (2016) The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res 365(3):657–673. https://doi.org/10.1007/s00441-016-2474-y
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C (2017) Role of tumor microenvironment in tumorigenesis. J Cancer 8(5):761–773. https://doi.org/10.7150/jca.17648
Crotti S, Piccoli M, Rizzolio F, Giordano A, Nitti D, Agostini M (2017) Extracellular matrix and colorectal cancer: how surrounding microenvironment affects cancer cell behavior? J Cell Physiol 232(5):967–975. https://doi.org/10.1002/jcp.25658
Erdogan B, Webb DJ (2017) Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans 45(1):229–236. https://doi.org/10.1042/BST20160387
Cunderlikova B (2016) Clinical significance of immunohistochemically detected extracellular matrix proteins and their spatial distribution in primary cancer. Crit Rev Oncol Hematol 105:127–144. https://doi.org/10.1016/j.critrevonc.2016.04.017
Rizzacasa B, Morini E, Pucci S, Murdocca M, Novelli G, Amati F (2017) LOX-1 and its splice variants: a new challenge for atherosclerosis and cancer-targeted therapies. Int J Mol Sci 18(2):290. https://doi.org/10.3390/ijms18020290
Huang L, Xu AM, Liu W (2015) Transglutaminase 2 in cancer. Am J Cancer Res 5(9):2756–2776
Rojas A, Anazco C, Gonzalez I, Araya P (2018) Extracellular matrix glycation and receptor for advanced glycation end-products activation: a missing piece in the puzzle of the association between diabetes and cancer. Carcinogenesis 39(4):515–521. https://doi.org/10.1093/carcin/bgy012
Malik R, Lelkes PI, Cukierman E (2015) Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol 33(4):230–236. https://doi.org/10.1016/j.tibtech.2015.01.004
Malik R, Luong T, Cao X, Han B, Shah N, Franco-Barraza J, Han L, Shenoy VB, Lelkes PI, Cukierman E (2018) Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2. Matrix Biol 1:2. https://doi.org/10.1016/j.matbio.2018.11.001
Tolle RC, Gaggioli C, Dengjel J (2018) Three-dimensional cell culture conditions affect the proteome of cancer-associated fibroblasts. J Proteome Res 17(8):2780–2789. https://doi.org/10.1021/acs.jproteome.8b00237
Ronca R, Sozzani S, Presta M, Alessi P (2009) Delivering cytokines at tumor site: the immunocytokine-conjugated anti-EDB-fibronectin antibody case. Immunobiology 214(9–10):800–810. https://doi.org/10.1016/j.imbio.2009.06.005
Ramamonjisoa N, Ackerstaff E (2017) Characterization of the tumor microenvironment and tumor-stroma interaction by non-invasive preclinical imaging. Front Oncol 7:3. https://doi.org/10.3389/fonc.2017.00003
Micke P, Ostman A (2004) Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 45(Suppl 2):S163–S175. https://doi.org/10.1016/j.lungcan.2004.07.977
Hirata E, Sahai E (2017) Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med 7(7):a026781. https://doi.org/10.1101/cshperspect.a026781
Topalovski M, Brekken RA (2016) Matrix control of pancreatic cancer: new insights into fibronectin signaling. Cancer Lett 381(1):252–258. https://doi.org/10.1016/j.canlet.2015.12.027
Zollinger AJ, Smith ML (2017) Fibronectin, the extracellular glue. Matrix Biol 60–61:27–37. https://doi.org/10.1016/j.matbio.2016.07.011
Han Z, Lu ZR (2017) Targeting fibronectin for cancer imaging and therapy. J Mater Chem B 5(4):639–654. https://doi.org/10.1039/C6TB02008A
Wang K, Seo BR, Fischbach C, Gourdon D (2016) Fibronectin mechanobiology regulates tumorigenesis. Cell Mol Bioeng 9:1–11. https://doi.org/10.1007/s12195-015-0417-4
Bachman H, Nicosia J, Dysart M, Barker TH (2015) Utilizing fibronectin integrin-binding specificity to control cellular responses. Adv Wound Care (New Rochelle) 4(8):501–511. https://doi.org/10.1089/wound.2014.0621
Kanchanawong P, Waterman CM (2012) Advances in light-based imaging of three-dimensional cellular ultrastructure. Curr Opin Cell Biol 24(1):125–133. https://doi.org/10.1016/j.ceb.2011.11.010
Chiovaro F, Martina E, Bottos A, Scherberich A, Hynes NE, Chiquet-Ehrismann R (2015) Transcriptional regulation of tenascin-W by TGF-β signaling in the bone metastatic niche of breast cancer cells. Int J Cancer 137(8):1842–1854. https://doi.org/10.1002/ijc.29565
Adams JC, Chiquet-Ehrismann R, Tucker RP (2015) The evolution of tenascins and fibronectin. Cell Adhes Migr 9(1–2):22–33. https://doi.org/10.4161/19336918.2014.970030
Martina E, Chiquet-Ehrismann R, Brellier F (2010) Tenascin-W: an extracellular matrix protein associated with osteogenesis and cancer. Int J Biochem Cell Biol 42(9):1412–1415. https://doi.org/10.1016/j.biocel.2010.06.004
Degen M, Brellier F, Schenk S, Driscoll R, Zaman K, Stupp R, Tornillo L, Terracciano L, Chiquet-Ehrismann R, Ruegg C, Seelentag W (2008) Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int J Cancer 122(11):2454–2461. https://doi.org/10.1002/ijc.23417
Degen M, Brellier F, Kain R, Ruiz C, Terracciano L, Orend G, Chiquet-Ehrismann R (2007) Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res 67(19):9169–9179. https://doi.org/10.1158/0008-5472.CAN-07-0666
Kim BG, An HJ, Kang S, Choi YP, Gao MQ, Park H, Cho NH (2011) Laminin-332-rich tumor microenvironment for tumor invasion in the interface zone of breast cancer. Am J Pathol 178(1):373–381. https://doi.org/10.1016/j.ajpath.2010.11.028
Tsuruta D, Kobayashi H, Imanishi H, Sugawara K, Ishii M, Jones JC (2008) Laminin-332-integrin interaction: a target for cancer therapy? Curr Med Chem 15(20):1968–1975
Marinkovich MP (2007) Tumour microenvironment: laminin 332 in squamous-cell carcinoma. Nat Rev Cancer 7(5):370–380. https://doi.org/10.1038/nrc2089
Guess CM, Lafleur BJ, Weidow BL, Quaranta V (2009) A decreased ratio of laminin-332 β3 to gamma2 subunit mRNA is associated with poor prognosis in colon cancer. Cancer Epidemiol Biomark Prev 18(5):1584–1590. https://doi.org/10.1158/1055-9965.EPI-08-1027
Guess CM, Quaranta V (2009) Defining the role of laminin-332 in carcinoma. Matrix Biol 28(8):445–455. https://doi.org/10.1016/j.matbio.2009.07.008
Chen J, Wang W, Wei J, Zhou D, Zhao X, Song W, Sun Q, Huang P, Zheng S (2015) Overexpression of β3 chains of laminin-332 is associated with clinicopathologic features and decreased survival in patients with pancreatic adenocarcinoma. Appl Immunohistochem Mol Morphol 23(7):516–521. https://doi.org/10.1097/PAI.0000000000000115
Katayama M, Funakoshi A, Sumii T, Sanzen N, Sekiguchi K (2005) Laminin gamma2-chain fragment circulating level increases in patients with metastatic pancreatic ductal cell adenocarcinomas. Cancer Lett 225(1):167–176. https://doi.org/10.1016/j.canlet.2004.11.052
Ramovs V, Te Molder L, Sonnenberg A (2017) The opposing roles of laminin-binding integrins in cancer. Matrix Biol 57–58:213–243. https://doi.org/10.1016/j.matbio.2016.08.007
Yamada M, Sekiguchi K (2015) Molecular basis of laminin-integrin interactions. Curr Top Membr 76:197–229. https://doi.org/10.1016/bs.ctm.2015.07.002
Tripathi M, Nandana S, Yamashita H, Ganesan R, Kirchhofer D, Quaranta V (2008) Laminin-332 is a substrate for hepsin, a protease associated with prostate cancer progression. J Biol Chem 283(45):30576–30584. https://doi.org/10.1074/jbc.M802312200
Tripathi M, Potdar AA, Yamashita H, Weidow B, Cummings PT, Kirchhofer D, Quaranta V (2011) Laminin-332 cleavage by matriptase alters motility parameters of prostate cancer cells. Prostate 71(2):184–196. https://doi.org/10.1002/pros.21233
Cavaco ACM, Rezaei M, Caliandro MF, Lima AM, Stehling M, Dhayat SA, Haier J, Brakebusch C, Eble JA (2018) The interaction between laminin-332 and α3β1 integrin determines differentiation and maintenance of CAFs, and supports invasion of pancreatic duct adenocarcinoma cells. Cancers (Basel) 11(1):14. https://doi.org/10.3390/cancers11010014
Luo Z, Wang Q, Lau WB, Lau B, Xu L, Zhao L, Yang H, Feng M, Xuan Y, Yang Y, Lei L, Wang C, Yi T, Zhao X, Wei Y, Zhou S (2016) Tumor microenvironment: the culprit for ovarian cancer metastasis? Cancer Lett 377(2):174–182. https://doi.org/10.1016/j.canlet.2016.04.038
Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM (2005) Tensional homeostasis and the malignant phenotype. Cancer Cell 8(3):241–254. https://doi.org/10.1016/j.ccr.2005.08.010
Giussani M, Merlino G, Cappelletti V, Tagliabue E, Daidone MG (2015) Tumor-extracellular matrix interactions: identification of tools associated with breast cancer progression. Semin Cancer Biol 35:3–10. https://doi.org/10.1016/j.semcancer.2015.09.012
Sundquist E, Renko O, Salo S, Magga J, Cervigne NK, Nyberg P, Risteli J, Sormunen R, Vuolteenaho O, Zandonadi F, Paes Leme AF, Coletta RD, Ruskoaho H, Salo T (2016) Neoplastic extracellular matrix environment promotes cancer invasion in vitro. Exp Cell Res 344(2):229–240. https://doi.org/10.1016/j.yexcr.2016.04.003
Te Boekhorst V, Friedl P (2016) plasticity of cancer cell invasion-mechanisms and implications for therapy. Adv Cancer Res 132:209–264. https://doi.org/10.1016/bs.acr.2016.07.005
Bhowmick NA, Neilson EG, Moses HL (2004) Stromal fibroblasts in cancer initiation and progression. Nature 432(7015):332–337. https://doi.org/10.1038/nature03096
Cavaco A, Rezaei M, Niland S, Eble JA (2017) Collateral damage intended: cancer-associated fibroblasts and vasculature are potential targets in cancer therapy. Int J Mol Sci 18(11):2355. https://doi.org/10.3390/ijms18112355
Marchiq I, Pouyssegur J (2016) Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl) 94(2):155–171. https://doi.org/10.1007/s00109-015-1307-x
Nielsen N, Lindemann O, Schwab A (2014) TRP channels and STIM/ORAI proteins: sensors and effectors of cancer and stroma cell migration. Br J Pharmacol 171(24):5524–5540. https://doi.org/10.1111/bph.12721
Tochhawng L, Deng S, Pervaiz S, Yap CT (2013) Redox regulation of cancer cell migration and invasion. Mitochondrion 13(3):246–253. https://doi.org/10.1016/j.mito.2012.08.002
Xing Y, Zhao S, Zhou BP, Mi J (2015) Metabolic reprogramming of the tumour microenvironment. FEBS J 282(20):3892–3898. https://doi.org/10.1111/febs.13402
Baluna RG, Eng TY, Thomas CR (2006) Adhesion molecules in radiotherapy. Radiat Res 166(6):819–831. https://doi.org/10.1667/RR0380.1
Borek C (2004) Dietary antioxidants and human cancer. Integr Cancer Ther 3(4):333–341. https://doi.org/10.1177/1534735404270578
Westbury CB, Yarnold JR (2012) Radiation fibrosis–current clinical and therapeutic perspectives. Clin Oncol (R Coll Radiol) 24(10):657–672. https://doi.org/10.1016/j.clon.2012.04.001
Yahyapour R, Motevaseli E, Rezaeyan A, Abdollahi H, Farhood B, Cheki M, Rezapoor S, Shabeeb D, Musa AE, Najafi M, Villa V (2018) Reduction-oxidation (redox) system in radiation-induced normal tissue injury: molecular mechanisms and implications in radiation therapeutics. Clin Transl Oncol 20(8):975–988. https://doi.org/10.1007/s12094-017-1828-6
Polanska UM, Orimo A (2013) Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. J Cell Physiol 228(8):1651–1657. https://doi.org/10.1002/jcp.24347
Koch S, Claesson-Welsh L (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2(7):a006502. https://doi.org/10.1101/cshperspect.a006502
Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M, Moldovan IM, Roman AL, Mihu CM (2018) Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol 59(2):455–467
Miyazono K, Katsuno Y, Koinuma D, Ehata S, Morikawa M (2018) Intracellular and extracellular TGF-β signaling in cancer: some recent topics. Front Med 12(4):387–411. https://doi.org/10.1007/s11684-018-0646-8
Crafts TD, Jensen AR, Blocher-Smith EC, Markel TA (2015) Vascular endothelial growth factor: therapeutic possibilities and challenges for the treatment of ischemia. Cytokine 71(2):385–393. https://doi.org/10.1016/j.cyto.2014.08.005
Iozzo RV, Sanderson RD (2011) Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med 15(5):1013–1031. https://doi.org/10.1111/j.1582-4934.2010.01236.x
Zanotelli MR, Reinhart-King CA (2018) Mechanical forces in tumor angiogenesis. Adv Exp Med Biol 1092:91–112. https://doi.org/10.1007/978-3-319-95294-9_6
Edgar LT, Maas SA, Guilkey JE, Weiss JA (2015) A coupled model of neovessel growth and matrix mechanics describes and predicts angiogenesis in vitro. Biomech Model Mechanobiol 14(4):767–782. https://doi.org/10.1007/s10237-014-0635-z
Sund M, Xie L, Kalluri R (2004) The contribution of vascular basement membranes and extracellular matrix to the mechanics of tumor angiogenesis. APMIS 112(7–8):450–462. https://doi.org/10.1111/j.1600-0463.2004.t01-1-apm11207-0806.x
Knopik-Skrocka A, Kręplewska P, Jarmołowska-Jurczyszyn D (2017) Tumor blood vessels and vasculogenic mimicry – Current knowledge and searching for new cellular/molecular targets of anti-angiogenic therapy. Adv Cell Biol 5(1):50–71. https://doi.org/10.1515/acb-2017-0005
Zuazo-Gaztelu I, Casanovas O (2018) Unraveling the role of angiogenesis in cancer ecosystems. Front Oncol 8:248. https://doi.org/10.3389/fonc.2018.00248
Dudley AC (2012) Tumor endothelial cells. Cold Spring Harb Perspect Med 2(3):a006536. https://doi.org/10.1101/cshperspect.a006536
Angara K, Borin TF, Arbab AS (2017) Vascular mimicry: a novel neovascularization mechanism driving anti-angiogenic therapy (AAT) resistance in glioblastoma. Transl Oncol 10(4):650–660. https://doi.org/10.1016/j.tranon.2017.04.007
Dunleavey JM, Dudley AC (2012) Vascular mimicry: concepts and implications for anti-angiogenic therapy. Curr Angiogenes 1(2):133–138. https://doi.org/10.2174/2211552811201020133
Seftor RE, Hess AR, Seftor EA, Kirschmann DA, Hardy KM, Margaryan NV, Hendrix MJ (2012) Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. Am J Pathol 181(4):1115–1125. https://doi.org/10.1016/j.ajpath.2012.07.013
Reid SE, Kay EJ, Neilson LJ, Henze AT, Serneels J, McGhee EJ, Dhayade S, Nixon C, Mackey JB, Santi A, Swaminathan K, Athineos D, Papalazarou V, Patella F, Roman-Fernandez A, ElMaghloob Y, Hernandez-Fernaud JR, Adams RH, Ismail S, Bryant DM, Salmeron-Sanchez M, Machesky LM, Carlin LM, Blyth K, Mazzone M, Zanivan S (2017) Tumor matrix stiffness promotes metastatic cancer cell interaction with the endothelium. EMBO J 36(16):2373–2389. https://doi.org/10.15252/embj.201694912
Folberg R, Maniotis AJ (2004) Vasculogenic mimicry. APMIS 112(7–8):508–525. https://doi.org/10.1111/j.1600-0463.2004.apm11207-0810.x
Cao Z, Bao M, Miele L, Sarkar FH, Wang Z, Zhou Q (2013) Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: a systemic review and meta-analysis. Eur J Cancer 49(18):3914–3923. https://doi.org/10.1016/j.ejca.2013.07.148
Hutchenreuther J, Vincent K, Norley C, Racanelli M, Gruber SB, Johnson TM, Fullen DR, Raskin L, Perbal B, Holdsworth DW, Postovit LM, Leask A (2018) Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. https://doi.org/10.1016/j.matbio.2018.06.003
Seftor RE, Seftor EA, Kirschmann DA, Hendrix MJ (2002) Targeting the tumor microenvironment with chemically modified tetracyclines: inhibition of laminin 5 gamma2 chain promigratory fragments and vasculogenic mimicry. Mol Cancer Ther 1(13):1173–1179
Qiao L, Liang N, Zhang J, Xie J, Liu F, Xu D, Yu X, Tian Y (2015) Advanced research on vasculogenic mimicry in cancer. J Cell Mol Med 19(2):315–326. https://doi.org/10.1111/jcmm.12496
Velez DO, Tsui B, Goshia T, Chute CL, Han A, Carter H, Fraley SI (2017) 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat Commun 8(1):1651. https://doi.org/10.1038/s41467-017-01556-7
Labelle M, Hynes RO (2012) The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov 2(12):1091–1099. https://doi.org/10.1158/2159-8290.CD-12-0329
Sleeman JP, Cady B, Pantel K (2012) The connectivity of lymphogenous and hematogenous tumor cell dissemination: biological insights and clinical implications. Clin Exp Metastasis 29(7):737–746. https://doi.org/10.1007/s10585-012-9489-x
Zhu T, Hu X, Wei P, Shan G (2018) Molecular background of the regional lymph node metastasis of gastric cancer. Oncol Lett 15(3):3409–3414. https://doi.org/10.3892/ol.2018.7813
Jones D, Pereira ER, Padera TP (2018) Growth and immune evasion of lymph node metastasis. Front Oncol 8:36. https://doi.org/10.3389/fonc.2018.00036
Pircher A, Wolf D, Heidenreich A, Hilbe W, Pichler R, Heidegger I (2017) Synergies of targeting tumor angiogenesis and immune checkpoints in non-small cell lung cancer and renal cell cancer: from basic concepts to clinical reality. Int J Mol Sci 18(11):2291. https://doi.org/10.3390/ijms18112291
Schito L (2018) Bridging angiogenesis and immune evasion in the hypoxic tumor microenvironment. Am J Physiol Regul Integr Comp Physiol 1:4. https://doi.org/10.1152/ajpregu.00209.2018
Cimpean AM, Tamma R, Ruggieri S, Nico B, Toma A, Ribatti D (2017) Mast cells in breast cancer angiogenesis. Crit Rev Oncol Hematol 115:23–26. https://doi.org/10.1016/j.critrevonc.2017.04.009
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022. https://doi.org/10.1038/ni.2703
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437. https://doi.org/10.1038/nm.3394
Sangaletti S, Chiodoni C, Tripodo C, Colombo MP (2017) The good and bad of targeting cancer-associated extracellular matrix. Curr Opin Pharmacol 35:75–82. https://doi.org/10.1016/j.coph.2017.06.003
Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM (2012) Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012:948098. https://doi.org/10.1155/2012/948098
Shevach EM (2009) Mechanisms of foxp3 + T regulatory cell-mediated suppression. Immunity 30(5):636–645. https://doi.org/10.1016/j.immuni.2009.04.010
Sica A, Massarotti M (2017) Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun 85:117–125. https://doi.org/10.1016/j.jaut.2017.07.010
Zhao H, Liao X, Kang Y (2017) Tregs: where we are and what comes next? Front Immunol 8:1578. https://doi.org/10.3389/fimmu.2017.01578
Dermani FK, Samadi P, Rahmani G, Kohlan AK, Najafi R (2019) PD-1/PD-L1 immune checkpoint: potential target for cancer therapy. J Cell Physiol 234(2):1313–1325. https://doi.org/10.1002/jcp.27172
Hahn AW, Gill DM, Pal SK, Agarwal N (2017) The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy 9(8):681–692. https://doi.org/10.2217/imt-2017-0024
Hatae R, Chamoto K (2016) Immune checkpoint inhibitors targeting programmed cell death-1 (PD-1) in cancer therapy. Rinsho Ketsueki 57(10):2224–2231. https://doi.org/10.11406/rinketsu.57.2224
Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD (2018) TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 9(1):4692. https://doi.org/10.1038/s41467-018-06654-8
Rhee I (2016) Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res 39(11):1588–1596. https://doi.org/10.1007/s12272-016-0820-y
Torcellan T, Stolp J, Chtanova T (2017) In vivo imaging sheds light on immune cell migration and function in cancer. Front Immunol 8:309. https://doi.org/10.3389/fimmu.2017.00309
Jacquemet G, Hamidi H, Ivaska J (2015) Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol 36:23–31. https://doi.org/10.1016/j.ceb.2015.06.007
Angst BD, Marcozzi C, Magee AI (2001) The cadherin superfamily. J Cell Sci 114(Pt 4):625–626
Bertocchi C, Wang Y, Ravasio A, Hara Y, Wu Y, Sailov T, Baird MA, Davidson MW, Zaidel-Bar R, Toyama Y, Ladoux B, Mege RM, Kanchanawong P (2017) Nanoscale architecture of cadherin-based cell adhesions. Nat Cell Biol 19(1):28–37. https://doi.org/10.1038/ncb3456
Gloushankova NA, Rubtsova SN, Zhitnyak IY (2017) Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 5(3):e1356900. https://doi.org/10.1080/21688370.2017.1356900
Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, Troyanovsky RB, Ben-Shaul A, Frank J, Troyanovsky SM, Shapiro L, Honig B (2011) The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19(2):244–256. https://doi.org/10.1016/j.str.2010.11.016
Leckband DE, de Rooij J (2014) Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol 30:291–315. https://doi.org/10.1146/annurev-cellbio-100913-013212
Cichon MA, Radisky DC (2014) Extracellular matrix as a contextual determinant of transforming growth factor-β signaling in epithelial-mesenchymal transition and in cancer. Cell Adhes Migr 8(6):588–594. https://doi.org/10.4161/19336918.2014.972788
Pietila M, Ivaska J, Mani SA (2016) Whom to blame for metastasis, the epithelial-mesenchymal transition or the tumor microenvironment? Cancer Lett 380(1):359–368. https://doi.org/10.1016/j.canlet.2015.12.033
Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM (2014) Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer 110(3):724–732. https://doi.org/10.1038/bjc.2013.768
Eatemadi A, Aiyelabegan HT, Negahdari B, Mazlomi MA, Daraee H, Daraee N, Eatemadi R, Sadroddiny E (2017) Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed Pharmacother 86:221–231. https://doi.org/10.1016/j.biopha.2016.12.021
Wolf K, Friedl P (2011) Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol 21(12):736–744. https://doi.org/10.1016/j.tcb.2011.09.006
Stefanidakis M, Koivunen E (2006) Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood 108(5):1441–1450. https://doi.org/10.1182/blood-2006-02-005363
Nagase H, Visse R, Murphy G (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69(3):562–573. https://doi.org/10.1016/j.cardiores.2005.12.002
Jacob A, Prekeris R (2015) The regulation of MMP targeting to invadopodia during cancer metastasis. Front Cell Dev Biol 3:4. https://doi.org/10.3389/fcell.2015.00004
Ren F, Tang R, Zhang X, Madushi WM, Luo D, Dang Y, Li Z, Wei K, Chen G (2015) Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: a systematic review and meta-analysis. PLoS ONE 10(8):e0135544. https://doi.org/10.1371/journal.pone.0135544
Vandooren J, Van den Steen PE, Opdenakker G (2013) Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol 48(3):222–272. https://doi.org/10.3109/10409238.2013.770819
Huang H (2018) Matrix metalloproteinase-9 (MMP-9) as a cancer biomarker and MMP-9 biosensors: recent advances. Sensors (Basel) 18(10):3249. https://doi.org/10.3390/s18103249
Zhang X, Huang S, Guo J, Zhou L, You L, Zhang T, Zhao Y (2016) Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics (review). Int J Oncol 48(5):1783–1793. https://doi.org/10.3892/ijo.2016.3400
Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rosse C, Chavrier P (2016) Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu Rev Cell Dev Biol 32:555–576. https://doi.org/10.1146/annurev-cellbio-111315-125227
Pahwa S, Stawikowski MJ, Fields GB (2014) Monitoring and inhibiting MT1-MMP during cancer initiation and progression. Cancers (Basel) 6(1):416–435. https://doi.org/10.3390/cancers6010416
Alcantara MB, Dass CR (2013) Regulation of MT1-MMP and MMP-2 by the serpin PEDF: a promising new target for metastatic cancer. Cell Physiol Biochem 31(4–5):487–494. https://doi.org/10.1159/000350069
Poincloux R, Lizarraga F, Chavrier P (2009) Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci 122(Pt 17):3015–3024. https://doi.org/10.1242/jcs.034561
Radisky ES, Radisky DC (2015) Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front Biosci 20:1144–1163. https://doi.org/10.2741/4364
Parvanescu V, Georgescu M, Georgescu I, Surlin V, Patrascu S, Picleanu AM, Georgescu E (2015) The role of matrix metalloproteinase-9 (MMP-9) as a prognostic factor in epithelial and lymphatic neoplasia. Chirurgia (Bucur) 110(6):506–510
Tam EM, Moore TR, Butler GS, Overall CM (2004) Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J Biol Chem 279(41):43336–43344. https://doi.org/10.1074/jbc.M407186200
Farina AR, Mackay AR (2014) Gelatinase B/MMP-9 in tumour pathogenesis and progression. Cancers (Basel) 6(1):240–296. https://doi.org/10.3390/cancers6010240
Overall CM (2001) Matrix metalloproteinase substrate binding domains, modules and exosites. Overview and experimental strategies. Methods Mol Biol 151:79–120
Thakur V, Bedogni B (2016) The membrane tethered matrix metalloproteinase MT1-MMP at the forefront of melanoma cell invasion and metastasis. Pharmacol Res 111:17–22. https://doi.org/10.1016/j.phrs.2016.05.019
Sato H, Takino T (2010) Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci 101(4):843–847. https://doi.org/10.1111/j.1349-7006.2010.01498.x
Saad S, Gottlieb DJ, Bradstock KF, Overall CM, Bendall LJ (2002) Cancer cell-associated fibronectin induces release of matrix metalloproteinase-2 from normal fibroblasts. Cancer Res 62(1):283–289
Itoh Y (2006) MT1-MMP: a key regulator of cell migration in tissue. IUBMB Life 58(10):589–596. https://doi.org/10.1080/15216540600962818
Itoh Y, Seiki M (2006) MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol 206(1):1–8. https://doi.org/10.1002/jcp.20431
Eddy RJ, Weidmann MD, Sharma VP, Condeelis JS (2017) Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol 27(8):595–607. https://doi.org/10.1016/j.tcb.2017.03.003
Revach OY, Geiger B (2014) The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adhes Migr 8(3):215–225
Bagnato A, Rosano L (2018) Endothelin-1 receptor drives invadopodia: exploiting how β-arrestin-1 guides the way. Small GTPases 9(5):394–398. https://doi.org/10.1080/21541248.2016.1235526
Alekhina O, Burstein E, Billadeau DD (2017) Cellular functions of WASP family proteins at a glance. J Cell Sci 130(14):2235–2241. https://doi.org/10.1242/jcs.199570
Frugtniet B, Jiang WG, Martin TA (2015) Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer (Dove Med Press) 7:99–109. https://doi.org/10.2147/BCTT.S59006
Parekh A, Weaver AM (2016) Regulation of invadopodia by mechanical signaling. Exp Cell Res 343(1):89–95. https://doi.org/10.1016/j.yexcr.2015.10.038
Jeannot P, Besson A (2017) Cortactin function in invadopodia. Small GTPases. https://doi.org/10.1080/21541248.2017.1405773
Linder S (2007) The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol 17(3):107–117. https://doi.org/10.1016/j.tcb.2007.01.002
Nicholas NS, Pipili A, Lesjak MS, Wells CM (2017) Differential role for PAK1 and PAK4 during the invadopodia lifecycle. Small GTPases. https://doi.org/10.1080/21541248.2017.1295830
Seano G, Primo L (2015) Podosomes and invadopodia: tools to breach vascular basement membrane. Cell Cycle 14(9):1370–1374. https://doi.org/10.1080/15384101.2015.1026523
Deryugina EI, Quigley JP (2015) Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol 44–46:94–112. https://doi.org/10.1016/j.matbio.2015.04.004
Genis L, Galvez BG, Gonzalo P, Arroyo AG (2006) MT1-MMP: universal or particular player in angiogenesis? Cancer Metastasis Rev 25(1):77–86. https://doi.org/10.1007/s10555-006-7891-z
Binder MJ, McCoombe S, Williams ED, McCulloch DR, Ward AC (2017) The extracellular matrix in cancer progression: role of hyalectan proteoglycans and ADAMTS enzymes. Cancer Lett 385:55–64. https://doi.org/10.1016/j.canlet.2016.11.001
Branch KM, Hoshino D, Weaver AM (2012) Adhesion rings surround invadopodia and promote maturation. Biol Open 1(8):711–722. https://doi.org/10.1242/bio.20121867
Zara M, Canobbio I, Visconte C, Canino J, Torti M, Guidetti GF (2018) Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell Signal 48:45–53. https://doi.org/10.1016/j.cellsig.2018.04.008
Covic L, Kuliopulos A (2018) Protease-activated receptor 1 as therapeutic target in breast, lung, and ovarian cancer: pepducin approach. Int J Mol Sci 19(8):2237. https://doi.org/10.3390/ijms19082237
Liu X, Yu J, Song S, Yue X, Li Q (2017) Protease-activated receptor-1 (PAR-1): a promising molecular target for cancer. Oncotarget 8(63):107334–107345. https://doi.org/10.18632/oncotarget.21015
Monboisse JC, Oudart JB, Ramont L, Brassart-Pasco S (1840) Maquart FX (2014) Matrikines from basement membrane collagens: a new anti-cancer strategy. Biochim Biophys Acta 8:2589–2598. https://doi.org/10.1016/j.bbagen.2013.12.029
Tran KT, Lamb P, Deng JS (2005) Matrikines and matricryptins: implications for cutaneous cancers and skin repair. J Dermatol Sci 40(1):11–20. https://doi.org/10.1016/j.jdermsci.2005.05.001
Hornebeck W, Maquart FX (2003) Proteolyzed matrix as a template for the regulation of tumor progression. Biomed Pharmacother 57(5–6):223–230
Ramont L, Brassart-Pasco S, Thevenard J, Deshorgue A, Venteo L, Laronze JY, Pluot M, Monboisse JC, Maquart FX (2007) The NC1 domain of type XIX collagen inhibits in vivo melanoma growth. Mol Cancer Ther 6(2):506–514. https://doi.org/10.1158/1535-7163.MCT-06-0207
Brassart-Pasco S, Senechal K, Thevenard J, Ramont L, Devy J, Di Stefano L, Dupont-Deshorgue A, Brezillon S, Feru J, Jazeron JF, Diebold MD, Ricard-Blum S, Maquart FX, Monboisse JC (2012) Tetrastatin, the NC1 domain of the α4(IV) collagen chain: a novel potent anti-tumor matrikine. PLoS ONE 7(4):e29587. https://doi.org/10.1371/journal.pone.0029587
Folkman J (2006) Antiangiogenesis in cancer therapy–endostatin and its mechanisms of action. Exp Cell Res 312(5):594–607. https://doi.org/10.1016/j.yexcr.2005.11.015
Liu X, Nie W, Xie Q, Chen G, Li X, Jia Y, Yin B, Qu X, Li Y, Liang J (2018) Endostatin reverses immunosuppression of the tumor microenvironment in lung carcinoma. Oncol Lett 15(2):1874–1880. https://doi.org/10.3892/ol.2017.7455
Hope C, Emmerich PB, Papadas A, Pagenkopf A, Matkowskyj KA, Van De Hey DR, Payne SN, Clipson L, Callander NS, Hematti P, Miyamoto S, Johnson MG, Deming DA, Asimakopoulos F (2017) Versican-derived matrikines regulate Batf3-dendritic cell differentiation and promote T cell infiltration in colorectal cancer. J Immunol 199(5):1933–1941. https://doi.org/10.4049/jimmunol.1700529
Poluzzi C, Iozzo RV, Schaefer L (2016) Endostatin and endorepellin: a common route of action for similar angiostatic cancer avengers. Adv Drug Deliv Rev 97:156–173. https://doi.org/10.1016/j.addr.2015.10.012
Woodall BP, Nystrom A, Iozzo RA, Eble JA, Niland S, Krieg T, Eckes B, Pozzi A, Iozzo RV (2008) Integrin α2β1 is the required receptor for endorepellin angiostatic activity. J Biol Chem 283(4):2335–2343. https://doi.org/10.1074/jbc.M708364200
Grahovac J, Wells A (2014) Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion. Lab Invest 94(1):31–40. https://doi.org/10.1038/labinvest.2013.132
Da Silva J, Lameiras P, Beljebbar A, Berquand A, Villemin M, Ramont L, Dukic S, Nuzillard JM, Molinari M, Gautier M, Brassart-Pasco S, Brassart B (2018) Structural characterization and in vivo pro-tumor properties of a highly conserved matrikine. Oncotarget 9(25):17839–17857. https://doi.org/10.18632/oncotarget.24894
Scandolera A, Odoul L, Salesse S, Guillot A, Blaise S, Kawecki C, Maurice P, El Btaouri H, Romier-Crouzet B, Martiny L, Debelle L, Duca L (2016) The elastin receptor complex: a unique matricellular receptor with high anti-tumoral potential. Front Pharmacol 7:32. https://doi.org/10.3389/fphar.2016.00032
Duca L, Floquet N, Alix AJ, Haye B, Debelle L (2004) Elastin as a matrikine. Crit Rev Oncol Hematol 49(3):235–244. https://doi.org/10.1016/j.critrevonc.2003.09.007
Alexander S, Friedl P (2012) Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med 18(1):13–26. https://doi.org/10.1016/j.molmed.2011.11.003
Devreotes P, Horwitz AR (2015) Signaling networks that regulate cell migration. Cold Spring Harb Perspect Biol 7(8):a005959. https://doi.org/10.1101/cshperspect.a005959
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR (2003) Cell migration: integrating signals from front to back. Science 302(5651):1704–1709. https://doi.org/10.1126/science.1092053
Schwartz MA, Horwitz AR (2006) Integrating adhesion, protrusion, and contraction during cell migration. Cell 125(7):1223–1225. https://doi.org/10.1016/j.cell.2006.06.015
Vicente-Manzanares M, Horwitz AR (2011) Cell migration: an overview. Methods Mol Biol 769:1–24. https://doi.org/10.1007/978-1-61779-207-6_1
Vicente-Manzanares M, Webb DJ, Horwitz AR (2005) Cell migration at a glance. J Cell Sci 118(Pt 21):4917–4919. https://doi.org/10.1242/jcs.02662
Aguilar-Cuenca R, Juanes-Garcia A, Vicente-Manzanares M (2014) Myosin II in mechanotransduction: master and commander of cell migration, morphogenesis, and cancer. Cell Mol Life Sci 71(3):479–492. https://doi.org/10.1007/s00018-013-1439-5
Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR (2009) Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10(11):778–790. https://doi.org/10.1038/nrm2786
Vicente-Manzanares M, Choi CK, Horwitz AR (2009) Integrins in cell migration–the actin connection. J Cell Sci 122(Pt 2):199–206. https://doi.org/10.1242/jcs.018564
Yamaguchi H, Condeelis J (2007) Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 1773(5):642–652. https://doi.org/10.1016/j.bbamcr.2006.07.001
Ungefroren H, Witte D, Lehnert H (2018) The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev Dyn 247(3):451–461. https://doi.org/10.1002/dvdy.24505
Lawson CD, Ridley AJ (2018) Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol 217(2):447–457. https://doi.org/10.1083/jcb.201612069
Casalou C, Faustino A, Barral DC (2016) Arf proteins in cancer cell migration. Small GTPases 7(4):270–282. https://doi.org/10.1080/21541248.2016.1228792
Kale VP, Hengst JA, Desai DH, Amin SG, Yun JK (2015) The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett 361(2):185–196. https://doi.org/10.1016/j.canlet.2015.03.017
Huttenlocher A, Horwitz AR (2011) Integrins in cell migration. Cold Spring Harb Perspect Biol 3(9):a005074. https://doi.org/10.1101/cshperspect.a005074
Bays JL, DeMali KA (2017) Vinculin in cell-cell and cell-matrix adhesions. Cell Mol Life Sci 74(16):2999–3009. https://doi.org/10.1007/s00018-017-2511-3
Goldmann WH (2016) Role of vinculin in cellular mechanotransduction. Cell Biol Int 40(3):241–256. https://doi.org/10.1002/cbin.10563
Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA (2010) Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466(7303):263–266. https://doi.org/10.1038/nature09198
Ehrlicher AJ, Krishnan R, Guo M, Bidan CM, Weitz DA, Pollak MR (2015) Α-actinin binding kinetics modulate cellular dynamics and force generation. Proc Natl Acad Sci U S A 112(21):6619–6624. https://doi.org/10.1073/pnas.1505652112
Roca-Cusachs P, del Rio A, Puklin-Faucher E, Gauthier NC, Biais N, Sheetz MP (2013) Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc Natl Acad Sci USA 110(15):E1361–E1370. https://doi.org/10.1073/pnas.1220723110
Carisey A, Tsang R, Greiner AM, Nijenhuis N, Heath N, Nazgiewicz A, Kemkemer R, Derby B, Spatz J, Ballestrem C (2013) Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr Biol 23(4):271–281. https://doi.org/10.1016/j.cub.2013.01.009
Zaidel-Bar R, Milo R, Kam Z, Geiger B (2007) A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci 120(Pt 1):137–148. https://doi.org/10.1242/jcs.03314
Bays JL, Peng X, Tolbert CE, Guilluy C, Angell AE, Pan Y, Superfine R, Burridge K, DeMali KA (2014) Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J Cell Biol 205(2):251–263. https://doi.org/10.1083/jcb.201309092
Dobrokhotov O, Samsonov M, Sokabe M, Hirata H (2018) Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin Transl Med 7(1):23. https://doi.org/10.1186/s40169-018-0202-9
Noguchi S, Saito A, Nagase T (2018) YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int J Mol Sci 19(11):3674. https://doi.org/10.3390/ijms19113674
Zanconato F, Cordenonsi M, Piccolo S (2016) YAP/TAZ at the roots of cancer. Cancer Cell 29(6):783–803. https://doi.org/10.1016/j.ccell.2016.05.005
Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF (1997) Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385(6616):537–540. https://doi.org/10.1038/385537a0
Gritsenko PG, Ilina O, Friedl P (2012) Interstitial guidance of cancer invasion. J Pathol 226(2):185–199. https://doi.org/10.1002/path.3031
Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P (2009) Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol 20(8):931–941. https://doi.org/10.1016/j.semcdb.2009.08.005
Deng J, Zhao C, Spatz JP, Wei Q (2017) Nanopatterned adhesive, stretchable hydrogel to control ligand spacing and regulate cell spreading and migration. ACS Nano 11(8):8282–8291. https://doi.org/10.1021/acsnano.7b03449
Arnold M, Hirschfeld-Warneken VC, Lohmuller T, Heil P, Blummel J, Cavalcanti-Adam EA, Lopez-Garcia M, Walther P, Kessler H, Geiger B, Spatz JP (2008) Induction of cell polarization and migration by a gradient of nanoscale variations in adhesive ligand spacing. Nano Lett 8(7):2063–2069. https://doi.org/10.1021/nl801483w
Alfano M, Nebuloni M, Allevi R, Zerbi P, Longhi E, Luciano R, Locatelli I, Pecoraro A, Indrieri M, Speziali C, Doglioni C, Milani P, Montorsi F, Salonia A (2016) Linearized texture of three-dimensional extracellular matrix is mandatory for bladder cancer cell invasion. Sci Rep 6:36128. https://doi.org/10.1038/srep36128
Krause M, Wolf K (2015) Cancer cell migration in 3D tissue: negotiating space by proteolysis and nuclear deformability. Cell Adh Migr 9(5):357–366. https://doi.org/10.1080/19336918.2015.1061173
Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P (2013) Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201(7):1069–1084. https://doi.org/10.1083/jcb.201210152
Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352(6283):353–358. https://doi.org/10.1126/science.aad7297
Petrie RJ, Yamada KM (2016) Multiple mechanisms of 3D migration: the origins of plasticity. Curr Opin Cell Biol 42:7–12. https://doi.org/10.1016/j.ceb.2016.03.025
Doyle AD, Petrie RJ, Kutys ML, Yamada KM (2013) Dimensions in cell migration. Curr Opin Cell Biol 25(5):642–649. https://doi.org/10.1016/j.ceb.2013.06.004
Petrie RJ, Harlin HM, Korsak LI, Yamada KM (2017) Activating the nuclear piston mechanism of 3D migration in tumor cells. J Cell Biol 216(1):93–100. https://doi.org/10.1083/jcb.201605097
Friedl P, Locker J, Sahai E, Segall JE (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14(8):777–783. https://doi.org/10.1038/ncb2548
Friedl P, Gilmour D (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10(7):445–457. https://doi.org/10.1038/nrm2720
Khalil AA, Ilina O, Gritsenko PG, Bult P, Span PN, Friedl P (2017) Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin Exp Metastasis 34(6–7):421–429. https://doi.org/10.1007/s10585-017-9858-6
Das T, Spatz JP (2016) Getting a grip on collective cell migration. Nat Cell Biol 18(12):1265–1267. https://doi.org/10.1038/ncb3447
Park JA, Atia L, Mitchel JA, Fredberg JJ, Butler JP (2016) Collective migration and cell jamming in asthma, cancer and development. J Cell Sci 129(18):3375–3383. https://doi.org/10.1242/jcs.187922
Ramos Gde O, Bernardi L, Lauxen I, Sant’Ana Filho M, Horwitz AR, Lamers ML (2016) Fibronectin modulates cell adhesion and signaling to promote single cell migration of highly invasive oral squamous cell carcinoma. PLoS ONE 11(3):e0151338. https://doi.org/10.1371/journal.pone.0151338
Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P (2007) Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9(8):893–904. https://doi.org/10.1038/ncb1616
Han T, Kang D, Ji D, Wang X, Zhan W, Fu M, Xin HB, Wang JB (2013) How does cancer cell metabolism affect tumor migration and invasion? Cell Adh Migr 7(5):395–403. https://doi.org/10.4161/cam.26345
Lehmann S, Te Boekhorst V, Odenthal J, Bianchi R, van Helvert S, Ikenberg K, Ilina O, Stoma S, Xandry J, Jiang L, Grenman R, Rudin M, Friedl P (2017) Hypoxia induces a HIF-1-dependent transition from collective-to-amoeboid dissemination in epithelial cancer cells. Curr Biol 27(3):392–400. https://doi.org/10.1016/j.cub.2016.11.057
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E (2007) Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9(12):1392–1400. https://doi.org/10.1038/ncb1658
Erpenbeck L, Schön MP (2010) Deadly allies: the fatal interplay between platelets and metastizing cancer cells. Blood 115(17):3427–3436
Camerer E (2004) Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 104:397–401
Weisel JW, Litvinov RI (2017) Fibrin formation, structure and properties. Subcell Biochem 82:405–456. https://doi.org/10.1007/978-3-319-49674-0_13
O’Sullivan JM, Preston RJS, Robson T, O’Donnell JS (2018) Emerging Roles for von Willebrand Factor in Cancer Cell Biology. Semin Thromb Hemost 44(2):159–166. https://doi.org/10.1055/s-0037-1607352
Bauer AT, Suckau J, Frank K, Desch A, Goertz L, Wagner AH, Hecker M, Goerge T, Umansky L, Beckhove P, Utikal J, Gorzelanny C, Diaz-Valdes N, Umansky V, Schneider SW (2015) von Willebrand factor fibers promote cancer-associated platelet aggregation in malignant melanoma of mice and humans. Blood 125(20):3153–3163. https://doi.org/10.1182/blood-2014-08-595686
Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, Bissell MJ, Cox TR, Giaccia AJ, Erler JT, Hiratsuka S, Ghajar CM, Lyden D (2017) Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17(5):302–317. https://doi.org/10.1038/nrc.2017.6
Psaila B, Lyden D (2009) The metastatic niche: adapting the foreign soil. Nat Rev Cancer 9(4):285–293. https://doi.org/10.1038/nrc2621
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527(7578):329–335. https://doi.org/10.1038/nature15756
Geraud C, Koch PS, Damm F, Schledzewski K, Goerdt S (2014) The metastatic cycle: metastatic niches and cancer cell dissemination. J Dtsch Dermatol Ges 12(11):1012–1019. https://doi.org/10.1111/ddg.12451
Aguado BA, Bushnell GG, Rao SS, Jeruss JS, Shea LD (2017) Engineering the pre-metastatic niche. Nat Biomed Eng 1. https://doi.org/10.1038/s41551-017-0077
Liu Y, Cao X (2016) Characteristics and significance of the pre-metastatic niche. Cancer Cell 30(5):668–681. https://doi.org/10.1016/j.ccell.2016.09.011
Rezaeeyan H, Shirzad R, McKee TD, Saki N (2018) Role of chemokines in metastatic niche: new insights along with a diagnostic and prognostic approach. APMIS 126(5):359–370. https://doi.org/10.1111/apm.12818
Nogues L, Benito-Martin A, Hergueta-Redondo M, Peinado H (2018) The influence of tumour-derived extracellular vesicles on local and distal metastatic dissemination. Mol Aspects Med 60:15–26. https://doi.org/10.1016/j.mam.2017.11.012
Lobb RJ, Lima LG, Moller A (2017) Exosomes: key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol 67:3–10. https://doi.org/10.1016/j.semcdb.2017.01.004
Gartland A, Erler JT, Cox TR (2016) The role of lysyl oxidase, the extracellular matrix and the pre-metastatic niche in bone metastasis. J Bone Oncol 5(3):100–103. https://doi.org/10.1016/j.jbo.2016.04.001
Peinado H, Lavotshkin S, Lyden D (2011) The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 21(2):139–146. https://doi.org/10.1016/j.semcancer.2011.01.002
Hoye AM, Erler JT (2016) Structural ECM components in the premetastatic and metastatic niche. Am J Physiol Cell Physiol 310(11):C955–C967. https://doi.org/10.1152/ajpcell.00326.2015
Descot A, Oskarsson T (2013) The molecular composition of the metastatic niche. Exp Cell Res 319(11):1679–1686. https://doi.org/10.1016/j.yexcr.2013.04.017
Cox TR, Rumney RMH, Schoof EM, Perryman L, Hoye AM, Agrawal A, Bird D, Latif NA, Forrest H, Evans HR, Huggins ID, Lang G, Linding R, Gartland A, Erler JT (2015) The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522(7554):106–110. https://doi.org/10.1038/nature14492
El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, Csizmadia E, Mariani O, Zhu C, Campagne A, Toner M, Bhatia SN, Irimia D, Vincent-Salomon A, Karnoub AE (2012) Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci USA 109(43):17460–17465. https://doi.org/10.1073/pnas.1206653109
Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, Aakre M, Weaver VM, Moses HL (2013) Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-β-deficient mouse mammary carcinomas. Cancer Res 73(17):5336–5346. https://doi.org/10.1158/0008-5472.CAN-13-0012
Jablonska J, Lang S, Sionov RV, Granot Z (2017) The regulation of pre-metastatic niche formation by neutrophils. Oncotarget 8(67):112132–112144. https://doi.org/10.18632/oncotarget.22792
Yumoto K, Eber MR, Berry JE, Taichman RS, Shiozawa Y (2014) Molecular pathways: niches in metastatic dormancy. Clin Cancer Res 20(13):3384–3389. https://doi.org/10.1158/1078-0432.CCR-13-0897
Oskarsson T, Batlle E, Massague J (2014) Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14(3):306–321. https://doi.org/10.1016/j.stem.2014.02.002
Leprini A, Querze G, Zardi L (1994) Tenascin isoforms: possible targets for diagnosis and therapy of cancer and mechanisms regulating their expression. Perspect Dev Neurobiol 2(1):117–123
Nicolo G, Salvi S, Oliveri G, Borsi L, Castellani P, Zardi L (1990) Expression of tenascin and of the ED-B containing oncofetal fibronectin isoform in human cancer. Cell Differ Dev 32(3):401–408
Vandooren J, Opdenakker G, Loadman PM, Edwards DR (2016) Proteases in cancer drug delivery. Adv Drug Deliv Rev 97:144–155. https://doi.org/10.1016/j.addr.2015.12.020
Piperigkou Z, Manou D, Karamanou K, Theocharis AD (2018) Strategies to target matrix metalloproteinases as therapeutic approach in cancer. Methods Mol Biol 1731:325–348. https://doi.org/10.1007/978-1-4939-7595-2_27
Gingras D, Batist G, Beliveau R (2001) AE-941 (Neovastat): a novel multifunctional antiangiogenic compound. Expert Rev Anticancer Ther 1(3):341–347. https://doi.org/10.1586/14737140.1.3.341
Gingras D, Boivin D, Deckers C, Gendron S, Barthomeuf C, Beliveau R (2003) Neovastat–a novel antiangiogenic drug for cancer therapy. Anticancer Drugs 14(2):91–96. https://doi.org/10.1097/01.cad.0000054520.85618.3f
Lu C, Lee JJ, Komaki R, Herbst RS, Feng L, Evans WK, Choy H, Desjardins P, Esparaz BT, Truong MT, Saxman S, Kelaghan J, Bleyer A, Fisch MJ (2010) Chemoradiotherapy with or without AE-941 in stage III non-small cell lung cancer: a randomized phase III trial. J Natl Cancer Inst 102(12):859–865. https://doi.org/10.1093/jnci/djq179
Rizvi NA, Humphrey JS, Ness EA, Johnson MD, Gupta E, Williams K, Daly DJ, Sonnichsen D, Conway D, Marshall J, Hurwitz H (2004) A phase I study of oral BMS-275291, a novel nonhydroxamate sheddase-sparing matrix metalloproteinase inhibitor, in patients with advanced or metastatic cancer. Clin Cancer Res 10(6):1963–1970
Leighl NB, Paz-Ares L, Douillard JY, Peschel C, Arnold A, Depierre A, Santoro A, Betticher DC, Gatzemeier U, Jassem J, Crawford J, Tu D, Bezjak A, Humphrey JS, Voi M, Galbraith S, Hann K, Seymour L, Shepherd FA (2005) Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J Clin Oncol 23(12):2831–2839. https://doi.org/10.1200/JCO.2005.04.044
Hirte H, Vergote IB, Jeffrey JR, Grimshaw RN, Coppieters S, Schwartz B, Tu D, Sadura A, Brundage M, Seymour L (2006) A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol 102(2):300–308. https://doi.org/10.1016/j.ygyno.2005.12.020
Hande KR, Collier M, Paradiso L, Stuart-Smith J, Dixon M, Clendeninn N, Yeun G, Alberti D, Binger K, Wilding G (2004) Phase I and pharmacokinetic study of prinomastat, a matrix metalloprotease inhibitor. Clin Cancer Res 10(3):909–915
Bissett D, O’Byrne KJ, von Pawel J, Gatzemeier U, Price A, Nicolson M, Mercier R, Mazabel E, Penning C, Zhang MH, Collier MA, Shepherd FA (2005) Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol 23(4):842–849. https://doi.org/10.1200/JCO.2005.03.170
Paemen L, Martens E, Masure S, Opdenakker G (1995) Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur J Biochem 234(3):759–765
Martens E, Leyssen A, Van Aelst I, Fiten P, Piccard H, Hu J, Descamps FJ, Van den Steen PE, Proost P, Van Damme J, Liuzzi GM, Riccio P, Polverini E, Opdenakker G (2007) A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim Biophys Acta 1770(2):178–186. https://doi.org/10.1016/j.bbagen.2006.10.012
Devy L, Huang L, Naa L, Yanamandra N, Pieters H, Frans N, Chang E, Tao Q, Vanhove M, Lejeune A, van Gool R, Sexton DJ, Kuang G, Rank D, Hogan S, Pazmany C, Ma YL, Schoonbroodt S, Nixon AE, Ladner RC, Hoet R, Henderikx P, Tenhoor C, Rabbani SA, Valentino ML, Wood CR, Dransfield DT (2009) Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res 69(4):1517–1526. https://doi.org/10.1158/0008-5472.CAN-08-3255
Lemaitre V, D’Armiento J (2006) Matrix metalloproteinases in development and disease. Birth Defects Res C 78(1):1–10. https://doi.org/10.1002/bdrc.20065
Li L, Li H (2013) Role of microRNA-mediated MMP regulation in the treatment and diagnosis of malignant tumors. Cancer Biol Ther 14(9):796–805. https://doi.org/10.4161/cbt.25936
Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, Krichevsky AM (2008) MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol 28(17):5369–5380. https://doi.org/10.1128/MCB.00479-08
Costa PM, Cardoso AL, Custodia C, Cunha P, Pereira de Almeida L, Pedroso de Lima MC (2015) MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: a new multimodal gene therapy approach for glioblastoma. J Control Release 207:31–39. https://doi.org/10.1016/j.jconrel.2015.04.002
Chan N, Willis A, Kornhauser N, Ward MM, Lee SB, Nackos E, Seo BR, Chuang E, Cigler T, Moore A, Donovan D, Vallee Cobham M, Fitzpatrick V, Schneider S, Wiener A, Guillaume-Abraham J, Aljom E, Zelkowitz R, Warren JD, Lane ME, Fischbach C, Mittal V, Vahdat L (2017) Influencing the tumor microenvironment: a phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin Cancer Res 23(3):666–676. https://doi.org/10.1158/1078-0432.CCR-16-1326
Hecht JR, Benson AB 3rd, Vyushkov D, Yang Y, Bendell J, Verma U (2017) A phase II, randomized, double-blind, placebo-controlled study of simtuzumab in combination with FOLFIRI for the second-line treatment of metastatic KRAS mutant colorectal adenocarcinoma. Oncologist 22(3):243-e223. https://doi.org/10.1634/theoncologist.2016-0479
Benson AB 3rd, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, Bendell J, Kudrik F (2017) A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist 22(3):241-e215. https://doi.org/10.1634/theoncologist.2017-0024
Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, Mikels A, Vaysberg M, Ghermazien H, Wai C, Garcia CA, Velayo AC, Jorgensen B, Biermann D, Tsai D, Green J, Zaffryar-Eilot S, Holzer A, Ogg S, Thai D, Neufeld G, Van Vlasselaer P, Smith V (2010) Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med 16(9):1009–1017. https://doi.org/10.1038/nm.2208
Rodriguez HM, Vaysberg M, Mikels A, McCauley S, Velayo AC, Garcia C, Smith V (2010) Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J Biol Chem 285(27):20964–20974. https://doi.org/10.1074/jbc.M109.094136
Rasmussen HS, McCann PP (1997) Matrix metalloproteinase inhibition as a novel anticancer strategy: a review with special focus on batimastat and marimastat. Pharmacol Ther 75(1):69–75
Cathcart J, Pulkoski-Gross A, Cao J (2015) Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis 2(1):26–34. https://doi.org/10.1016/j.gendis.2014.12.002
Rao BG (2005) Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr Pharm Des 11(3):295–322
Au JL, Yeung BZ, Wientjes MG, Lu Z, Wientjes MG (2016) Delivery of cancer therapeutics to extracellular and intracellular targets: determinants, barriers, challenges and opportunities. Adv Drug Deliv Rev 97:280–301. https://doi.org/10.1016/j.addr.2015.12.002
Rodriguez-Cabello JC, Arias FJ, Rodrigo MA, Girotti A (2016) Elastin-like polypeptides in drug delivery. Adv Drug Deliv Rev 97:85–100. https://doi.org/10.1016/j.addr.2015.12.007
Arosio D, Casagrande C (2016) Advancement in integrin facilitated drug delivery. Adv Drug Deliv Rev 97:111–143. https://doi.org/10.1016/j.addr.2015.12.001
Multhaupt HA, Leitinger B, Gullberg D, Couchman JR (2016) Extracellular matrix component signaling in cancer. Adv Drug Deliv Rev 97:28–40. https://doi.org/10.1016/j.addr.2015.10.013
Hinderer S, Layland SL, Schenke-Layland K (2016) ECM and ECM-like materials—biomaterials for applications in regenerative medicine and cancer therapy. Adv Drug Deliv Rev 97:260–269. https://doi.org/10.1016/j.addr.2015.11.019
Celia-Terrassa T, Kang Y (2018) Metastatic niche functions and therapeutic opportunities. Nat Cell Biol 20(8):868–877. https://doi.org/10.1038/s41556-018-0145-9
Ordonez-Moran P, Huelsken J (2014) Complex metastatic niches: already a target for therapy? Curr Opin Cell Biol 31:29–38. https://doi.org/10.1016/j.ceb.2014.06.012
Horton ER, Astudillo P, Humphries MJ, Humphries JD (2016) Mechanosensitivity of integrin adhesion complexes: role of the consensus adhesome. Exp Cell Res 343(1):7–13. https://doi.org/10.1016/j.yexcr.2015.10.025
Murphy DA, Courtneidge SA (2011) The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol 12(7):413–426. https://doi.org/10.1038/nrm3141
Leitinger B, Hohenester E (2007) Mammalian collagen receptors. Matrix Biol 26(3):146–155. https://doi.org/10.1016/j.matbio.2006.10.007
Torres PH, Sousa GL, Pascutti PG (2011) Structural analysis of the N-terminal fragment of the antiangiogenic protein endostatin: a molecular dynamics study. Proteins 79(9):2684–2692. https://doi.org/10.1002/prot.23096
Oudart JB, Monboisse JC, Maquart FX, Brassart B, Brassart-Pasco S, Ramont L (2017) Type XIX collagen: a new partner in the interactions between tumor cells and their microenvironment. Matrix Biol 57–58:169–177. https://doi.org/10.1016/j.matbio.2016.07.010
Nagase H, Fields GB (1996) Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Biopolymers 40(4):399–416. https://doi.org/10.1002/(SICI)1097-0282(1996)40:4%3C399:AID-BIP5%3E3.0.CO;2-R
Mithieux SM, Weiss AS (2005) Elastin. Adv Protein Chem 70:437–461. https://doi.org/10.1016/S0065-3233(05)70013-9
Wells JM, Gaggar A, Blalock JE (2015) MMP generated matrikines. Matrix Biol 44–46:122–129. https://doi.org/10.1016/j.matbio.2015.01.016
Cain SA, Mularczyk EJ, Singh M, Massam-Wu T, Kielty CM (2016) ADAMTS-10 and -6 differentially regulate cell-cell junctions and focal adhesions. Sci Rep 6:35956. https://doi.org/10.1038/srep35956
Bax DV, Mahalingam Y, Cain S, Mellody K, Freeman L, Younger K, Shuttleworth CA, Humphries MJ, Couchman JR, Kielty CM (2007) Cell adhesion to fibrillin-1: identification of an Arg-Gly-Asp-dependent synergy region and a heparin-binding site that regulates focal adhesion formation. J Cell Sci 120(Pt 8):1383–1392. https://doi.org/10.1242/jcs.003954
Jovanovic J, Iqbal S, Jensen S, Mardon H, Handford P (2008) Fibrillin-integrin interactions in health and disease. Biochem Soc Trans 36(Pt 2):257–262. https://doi.org/10.1042/BST0360257
Joshi R, Goihberg E, Ren W, Pilichowska M, Mathew P (2017) Proteolytic fragments of fibronectin function as matrikines driving the chemotactic affinity of prostate cancer cells to human bone marrow mesenchymal stromal cells via the α5β1 integrin. Cell Adh Migr 11(4):305–315. https://doi.org/10.1080/19336918.2016.1212139
White ES, Baralle FE, Muro AF (2008) New insights into form and function of fibronectin splice variants. J Pathol 216(1):1–14. https://doi.org/10.1002/path.2388
Faron G, Balepa L, Parra J, Fils JF, Gucciardo L (2018) The fetal fibronectin test: 25 years after its development, what is the evidence regarding its clinical utility? A systematic review and meta-analysis. J Matern Fetal Neonatal Med. https://doi.org/10.1080/14767058.2018.1491031
Sawicka KM, Seeliger M, Musaev T, Macri LK, Clark RA (2015) Fibronectin interaction and enhancement of growth factors: importance for wound healing. Adv Wound Care (New Rochelle) 4(8):469–478. https://doi.org/10.1089/wound.2014.0616
Wang Y, Ni H (2016) Fibronectin maintains the balance between hemostasis and thrombosis. Cell Mol Life Sci 73(17):3265–3277. https://doi.org/10.1007/s00018-016-2225-y
Mercuri FA, Maciewicz RA, Tart J, Last K, Fosang AJ (2000) Mutations in the interglobular domain of aggrecan alter matrix metalloproteinase and aggrecanase cleavage patterns Evidence that matrix metalloproteinase cleavage interferes with aggrecanase activity. J Biol Chem 275(42):33038–33045
Viapiano MS, Hockfield S, Matthews RT (2008) BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J Neurooncol 88(3):261–272. https://doi.org/10.1007/s11060-008-9575-8
Demircan K, Topcu V, Takigawa T, Akyol S, Yonezawa T, Ozturk G, Ugurcu V, Hasgul R, Yigitoglu MR, Akyol O, McCulloch DR, Hirohata S (2014) ADAMTS4 and ADAMTS5 knockout mice are protected from versican but not aggrecan or brevican proteolysis during spinal cord injury. Biomed Res Int 2014:693746. https://doi.org/10.1155/2014/693746
Li H, Leung TC, Hoffman S, Balsamo J, Lilien J (2000) Coordinate regulation of cadherin and integrin function by the chondroitin sulfate proteoglycan neurocan. J Cell Biol 149(6):1275–1288
Mohan V, Wyatt EV, Gotthard I, Phend KD, Diestel S, Duncan BW, Weinberg RJ, Tripathy A, Maness PF (2018) Neurocan inhibits semaphorin 3F induced dendritic spine remodeling through NrCAM in cortical neurons. Front Cell Neurosci 12:346. https://doi.org/10.3389/fncel.2018.00346
Wu Y, Chen L, Zheng PS, Yang BB (2002) β 1-Integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J Biol Chem 277(14):12294–12301. https://doi.org/10.1074/jbc.M110748200
Overall CM (2002) Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol Biotechnol 22(1):51–86. https://doi.org/10.1385/MB:22:1:051
Iozzo RV, Moscatello DK, McQuillan DJ, Eichstetter I (1999) Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem 274(8):4489–4492
Moreth K, Iozzo RV, Schaefer L (2012) Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 11(11):2084–2091. https://doi.org/10.4161/cc.20316
Goldoni S, Humphries A, Nystrom A, Sattar S, Owens RT, McQuillan DJ, Ireton K, Iozzo RV (2009) Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol 185(4):743–754. https://doi.org/10.1083/jcb.200901129
Khan GA, Girish GV, Lala N, Di Guglielmo GM, Lala PK (2011) Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol 25(8):1431–1443. https://doi.org/10.1210/me.2010-0426
Hausser H, Wedekind P, Sperber T, Peters R, Hasilik A, Kresse H (1996) Isolation and cellular localization of the decorin endocytosis receptor. Eur J Cell Biol 71(4):325–331
Nastase MV, Young MF, Schaefer L (2012) Biglycan: a multivalent proteoglycan providing structure and signals. J Histochem Cytochem 60(12):963–975. https://doi.org/10.1369/0022155412456380
Grindel B, Li Q, Arnold R, Petros J, Zayzafoon M, Muldoon M, Stave J, Chung LW, Farach-Carson MC (2016) Perlecan/HSPG2 and matrilysin/MMP-7 as indices of tissue invasion: tissue localization and circulating perlecan fragments in a cohort of 288 radical prostatectomy patients. Oncotarget 7(9):10433–10447. https://doi.org/10.18632/oncotarget.7197
Eble JA, Wucherpfennig KW, Gauthier L, Dersch P, Krukonis E, Isberg RR, Hemler ME (1998) Recombinant soluble human α3β1 integrin: purification, processing, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry 37(31):10945–10955. https://doi.org/10.1021/bi980175+
Kaasboll OJ, Gadicherla AK, Wang JH, Monsen VT, Hagelin EMV, Dong MQ, Attramadal H (2018) Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J Biol Chem 293(46):17953–17970. https://doi.org/10.1074/jbc.RA118.004559
Su JL, Chiou J, Tang CH, Zhao M, Tsai CH, Chen PS, Chang YW, Chien MH, Peng CY, Hsiao M, Kuo ML, Yen ML (2010) CYR61 regulates BMP-2-dependent osteoblast differentiation through the αvβ3 integrin/integrin-linked kinase/ERK pathway. J Biol Chem 285(41):31325–31336. https://doi.org/10.1074/jbc.M109.087122
Crockett JC, Schutze N, Tosh D, Jatzke S, Duthie A, Jakob F, Rogers MJ (2007) The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of αvβ3 and αvβ5. Endocrinology 148(12):5761–5768. https://doi.org/10.1210/en.2007-0473
Chen CC, Young JL, Monzon RI, Chen N, Todorovic V, Lau LF (2007) Cytotoxicity of TNFα is regulated by integrin-mediated matrix signaling. EMBO J 26(5):1257–1267. https://doi.org/10.1038/sj.emboj.7601596
Tsai HC, Chang AC, Tsai CH, Huang YL, Gan L, Chen CK, Liu SC, Huang TY, Fong YC, Tang CH (2019) CCN2 promotes drug resistance in osteosarcoma by enhancing ABCG2 expression. J Cell Physiol 234(6):9297–9307. https://doi.org/10.1002/jcp.27611
Babic AM, Chen CC, Lau LF (1999) Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin αvβ3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol 19(4):2958–2966
Scherberich A, Tucker RP, Degen M, Brown-Luedi M, Andres AC, Chiquet-Ehrismann R (2005) Tenascin-W is found in malignant mammary tumors, promotes α8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-α induced expression in vitro. Oncogene 24(9):1525–1532. https://doi.org/10.1038/sj.onc.1208342
Martina E, Degen M, Ruegg C, Merlo A, Lino MM, Chiquet-Ehrismann R, Brellier F (2010) Tenascin-W is a specific marker of glioma-associated blood vessels and stimulates angiogenesis in vitro. FASEB J 24(3):778–787. https://doi.org/10.1096/fj.09-140491
Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD (2002) Periostin secreted by epithelial ovarian carcinoma is a ligand for αVβ3 and αVβ5 integrins and promotes cell motility. Cancer Res 62(18):5358–5364
Kakizaki Y, Makino N, Tozawa T, Honda T, Matsuda A, Ikeda Y, Ito M, Saito Y, Kimura W, Ueno Y (2016) Stromal fibrosis and expression of matricellular proteins correlate with histological grade of intraductal papillary mucinous neoplasm of the pancreas. Pancreas 45(8):1145–1152. https://doi.org/10.1097/MPA.0000000000000617
Thijssen VL, Rabinovich GA, Griffioen AW (2013) Vascular galectins: regulators of tumor progression and targets for cancer therapy. Cytokine Growth Factor Rev 24(6):547–558. https://doi.org/10.1016/j.cytogfr.2013.07.003
Mendez-Huergo SP, Blidner AG, Rabinovich GA (2017) Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr Opin Immunol 45:8–15. https://doi.org/10.1016/j.coi.2016.12.003
Agnihotri R, Crawford HC, Haro H, Matrisian LM, Havrda MC, Liaw L (2001) Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin). J Biol Chem 276(30):28261–28267. https://doi.org/10.1074/jbc.M103608200
Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW (2007) An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 26(44):6361–6371. https://doi.org/10.1038/sj.onc.1210463
Furger KA, Allan AL, Wilson SM, Hota C, Vantyghem SA, Postenka CO, Al-Katib W, Chambers AF, Tuck AB (2003) Β3 integrin expression increases breast carcinoma cell responsiveness to the malignancy-enhancing effects of osteopontin. Mol Cancer Res 1(11):810–819
Rangaswami H, Bulbule A, Kundu GC (2006) Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol 16(2):79–87. https://doi.org/10.1016/j.tcb.2005.12.005
Teramoto H, Castellone MD, Malek RL, Letwin N, Frank B, Gutkind JS, Lee NH (2005) Autocrine activation of an osteopontin-CD44-Rac pathway enhances invasion and transformation by H-RasV12. Oncogene 24(3):489–501. https://doi.org/10.1038/sj.onc.1208209
Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, Simon R, Li Y, Robles AI, Chen Y, Ma ZC, Wu ZQ, Ye SL, Liu YK, Tang ZY, Wang XW (2003) Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 9(4):416–423. https://doi.org/10.1038/nm843
Funding
To study different aspect of tumor biology, J.A.E. receives financial support from Deutsche Forschungsgemeinschaft (SFB1009 A09) (MMP14 in invadopodia), from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under the REA Grant agreement n° [316610] (CAF differentiation in tumor stroma), and from the Wilhelm Sander Stiftung (grant:2016.113.1 to J.A.E.) (Interactions between tumor and endothelial cells).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Eble, J.A., Niland, S. The extracellular matrix in tumor progression and metastasis. Clin Exp Metastasis 36, 171–198 (2019). https://doi.org/10.1007/s10585-019-09966-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-019-09966-1