
Abstract
Collagens are the most abundant components of the extracellular matrix and many types of soft tissues. Elastin is another major component of certain soft tissues, such as arterial walls and ligaments. Many other molecules, though lower in quantity, function as essential components of the extracellular matrix in soft tissues. Some of these are reviewed in this chapter. Besides their basic structure, biochemistry and physiology, their roles in disorders of soft tissues are discussed only briefly as most chapters in this volume deal with relevant individual compounds. Fibronectin with its muldomain structure plays a role of “master organizer” in matrix assembly as it forms a bridge between cell surface receptors, e.g., integrins, and compounds such collagen, proteoglycans and other focal adhesion molecules. It also plays an essential role in the assembly of fibrillin-1 into a structured network. Laminins contribute to the structure of the extracellular matrix (ECM) and modulate cellular functions such as adhesion, differentiation, migration, stability of phenotype, and resistance towards apoptosis. Though the primary role of fibrinogen is in clot formation, after conversion to fibrin by thrombin, it also binds to a variety of compounds, particularly to various growth factors, and as such fibrinogen is a player in cardiovascular and extracellular matrix physiology. Elastin, an insoluble polymer of the monomeric soluble precursor tropoelastin, is the main component of elastic fibers in matrix tissue where it provides elastic recoil and resilience to a variety of connective tissues, e.g., aorta and ligaments. Elastic fibers regulate activity of TGFβs through their association with fibrillin microfibrils. Elastin also plays a role in cell adhesion, cell migration, and has the ability to participate in cell signaling. Mutations in the elastin gene lead to cutis laxa. Fibrillins represent the predominant core of the microfibrils in elastic as well as non-elastic extracellular matrixes, and interact closely with tropoelastin and integrins. Not only do microfibrils provide structural integrity of specific organ systems, but they also provide a scaffold for elastogenesis in elastic tissues. Fibrillin is important for the assembly of elastin into elastic fibers. Mutations in the fibrillin-1 gene are closely associated with Marfan syndrome. Fibulins are tightly connected with basement membranes, elastic fibers and other components of extracellular matrix and participate in formation of elastic fibers. Tenascins are ECM polymorphic glycoproteins found in many connective tissues in the body. Their expression is regulated by mechanical stress both during development and in adulthood. Tenascins mediate both inflammatory and fibrotic processes to enable effective tissue repair and play roles in pathogenesis of Ehlers-Danlos, heart disease, and regeneration and recovery of musculo-tendinous tissue. One of the roles of thrombospondin 1 is activation of TGFβ. Increased expression of thrombospondin and TGFβ activity was observed in fibrotic skin disorders such as keloids and scleroderma. Cartilage oligomeric matrix protein (COMP) or thrombospondin-5 is primarily present in the cartilage. High levels of COMP are present in fibrotic scars and systemic sclerosis of the skin, and in tendon, especially with physical activity, loading and post-injury. It plays a role in vascular wall remodeling and has been found in atherosclerotic plaques as well.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
The connective tissue in general is comprised of three groups of proteins: collagens, proteoglycans, and a variety of different glycoproteins. In addition to the main weight-bearing structural proteins of connective tissue – the fibril forming collagens (discussed in the Chap. 2 by Mienaltowski and Birk) – as well as the often hydrophilic role of proteoglycan proteins (discussed in the Chap. 4 on proteoglycans by Halper, and the Chap. 13 on Animal Models by Birk), growth factors (discussed in Chap. 5 by Halper), other proteins are also important for structure and signaling within the matrix tissue of the body. Several of these proteins are currently being identified as having several important functions in the developmental phase of the tissue, where these molecules can act as mediators of signaling or structural changes in the matrix tissue. Further, many of the glycoproteins have been demonstrated to play important roles not only during normal tissue physiology, but also in response to maintaining tissue homeostasis and responding and adapting to perturbations such as mechanical loading/unloading, or tissue damage and subsequent regeneration. Further, several of them are important for pathological tissue response, e.g., in cancer, fibrosis or connective tissue anomalies. Of interest as far as the adaptation of these glycoproteins is, that several of them – including collagens and proteoglycans – can be modulated in their level of expression and synthesis by the degree of mechanical loading that the specific tissue exposed to mechanical loading senses [1]. In the following pages some basic information about these glycoproteins is provided. However, as already mentioned above, because many of these glycoproteins are active participants in the pathogenesis of a variety of soft tissue diseases they will be discussed rather briefly in this chapter as they are also described in several chapters dealing with specific disorders of soft tissues.
3.1 Fibronectin
Fibronectin (FN) is a widely distributed multidomain glycoprotein present in most extracellular matrices (ECM). It has a molecular weight of 230–270 kD, and can, in addition to its presence in the extracellular matrix, also be detected at substantial concentrations in plasma. Fibronectin is composed of types I, II, and III repeating units (FNI, FNII and FNIII). Two intramolecular disulfide bonds are formed within type I and type II modules to stabilize the folded structure. Type III modules are formed by seven-stranded β-barrel structures that lack disulfides [2, 3]. The FN units or domains mediate self-assembly and ligand binding for collagen/gelatin, integrins, heparin, fibronectin, and other extracellular molecules [4]. The 500-kDa FN dimer is formed through a pair of anti-parallel disulfide bonds at the C terminus. FN exists in multiple isoforms generated by alternative splicing. The single FN gene transcript encodes 12 isoforms in rodents and cows, and 20 isoforms in humans. Alternative splicing occurs by exon skipping at EIIIA/EDA and EIIIB/EDB and by exon subdivision at the V region/IIICS. Fibronectin is secreted in the form of soluble inactive dimers with disulfide bonds that must be activated by interaction with α5β1 and other integrins [5, 6].
Fibronectin is widely expressed in embryos and adults, especially in regions of active morphogenesis, cell migration and inflammation. Tumor cells contain in general reduced levels of fibronectin, whereas fibronectin levels are high in tissues undergoing repair (i.e., wound healing) and/or fibrosis. In the process of matrix assembly, multivalent ECM proteins are induced to self-associate and to interact with other ECM proteins to form fibrillar networks. Matrix assembly is usually initiated by ECM glycoproteins binding to cell surface receptors, such as fibronectin dimers binding to α5β1 integrin. Receptor binding stimulates fibronectin self-association mediated by the N-terminal assembly domain and organizes the actin cytoskeleton to promote cell contractility. Fibronectin conformational changes expose additional binding sites that participate in fibril formation and in conversion of fibrils into a stabilized, insoluble form. Once assembled, the FN matrix impacts tissue organization by contributing to the assembly of other ECM proteins. Fibronectin plays an important role in fibrillogenesis in regard to initiation, progression and maturation of matrix assembly. The prominent role of fibronectin in matrix assembly lies in fibronectin ability, enabled by its multidomain structure, to bind simultaneously to cell surface receptors, e.g., integrins, and to collagen, proteoglycans and other focal adhesion molecules [7]. This property also makes it possible to mediate the assembly of several extracellular matrix protein, including type I and III collagen, thrombospondin-1 and microfibrils [4]. Fibronectin is also called a “master organizer” by some investigators [4, 8]. Perhaps more important in the context of this volume is to emphasize the role fibronectin plays in the assembly of fibrillin-1 into a structured network (see below).
3.2 Laminin
Laminins are a family of large multidomain, heterotrimeric glycoproteins with molecular weights of 500–800 kDa, located in the basement membrane. Sixteen trimeric isoforms have been described in mouse and human tissues, and these isoforms vary in their cell and tissue specificity. In general, each laminin isoform consists of three chains, α, β, and γ, and each isoform exists in five, four, and three genetically distinct forms, respectively [9–11]. Most vertebrates have five α, three γ and three to six β genes [11]. The large range in size is due to variability in the chain size: the α chains are the largest (Mr ~200–400 kDa), both the β and γ chains range in size from 120 to 200 kDa. In addition, all forms of these three chains are highly glycosylated, some have glycosaminoglycan chains attached [9, 11]. Homologous tandem repeats of structural motifs are incorporated in all laminins, with more similarities between β and γ chains. Laminins are cross- or T-shaped molecules with two or three short arms and one long arm. The short arms consist of N-terminal parts of one of the three chains and they contain laminin-type epidermal growth factor-like (LE) repeats [11] The long arm contains portions of all three chains [9]. Common to all laminins is a coiled-coil domain with about 80 heptad sequence repeats at or close to the C-terminal end. This coiled-coil domain bears homology to segments of β and γ chains and is responsible for proper assembly of the trimer [11, 12]. Assembly of the laminin molecule is also controlled to some extent by proteolytic processing prior to laminin binding to its receptors [11].
Laminins adhere to cells primarily via binding of the G domain of the α chains to integrins, dystroglycan, or sulfated glycolipids. The N-terminal globular domains of the α1 and α2 chains as well as the globular domains VI (LN) of the α5 chains can bind to several integrin isoforms (α1β1, α2β1, α3β1, and αVβ3). This process enables cell binding on both ends of laminins containing the three α chains. The laminin γ2 chain has been reported to bind α2β1 integrin. The N-terminal globular domains of some α-chains can also bind sulfatides. This type of binding may also link the laminin molecules to the cell surface. Laminins contribute to the structure of the ECM and influence associated cells in regards to adhesion, differentiation, migration, stability of phenotype, and resistance towards apoptosis. Laminin molecules interact not only with collagen type IV, integrins and dystroglycans but also with other components of the basal membrane matrix, and thus contribute to the overall structure. They can also interact with components in the underlying interstitial stroma. The cellular effects of laminins are mediated largely via ligand binding to cell membrane receptors, and this signaling can alter transcription levels of genes and even influence chromatin remodeling of gene promoters. The insoluble network formed by laminin and type IV collagen plays a structural and functional role in the basement membrane and cells associated with it. Though at this point we do not know to what extent, if any, laminins play a role in the pathogenesis of connective and soft tissue diseases it is clear that they contribute to normal function of tendons, blood vessels and other connective soft tissues. For example, this network participates in transmission of the contractile force from the skeletal muscle to the tendons [13]. A decrease in laminin in the basement membrane covering the outermost aspect of the tendon was identified in type IV collagen deficient mice. This was accompanied by formation of spontaneous tendon adhesions [14]. That laminins are, indeed, required for proper healing of tendons and other connective tissues, such as cornea, has been shown by Molloy et al. [15] and Sato et al. [16], respectively. There is some evidence indicating increased expression of β2 chain of laminin in ascending aorta in patients with Marfan syndrome [17].
Taken together laminins are not passive adhesion proteins, but rather, they actively modulate cell behavior; influence differentiation, migration, and phenotype stability. They also inhibit apoptosis by signaling via cell membrane receptors such as integrins and dystroglycan. However, the details of laminin signaling are still largely unexplored. Laminins constitute the first ECM component appearing in the developing early embryo, and embryonic laminins have found an important use as culture matrices for stem cells. Other laminins are crucial for normal function of numerous tissues and organs, e.g., nerve, epithelium, blood vessels, and kidney. The commercial unavailability of most laminin isoforms has hampered in vitro studies. However, many isoforms have been offered recently by several companies as recombinant proteins, which may enable deeper insight into functional properties. Laminins may find numerous new applications in cell biology and cell therapy research. The vast complexity of laminin effects cannot be explained solely by simple integrin binding and signaling [11].
3.3 Fibrinogen
Fibrinogen is a large, complex, fibrous glycoprotein with three pairs of polypeptide chains: Aα, Bβ and γ [18]. The chains are linked together by 29 disulfide bonds. Fibrinogen is 45 nm in length, with globular domains at each end and in the middle connected by α-helical coiled-coil rods and has Mr 340 kDa. The E-region consisting of N-terminal ends of the six chains and the D-regions consisting of the C-terminal ends of the Bβ and γ chains and a portion of the Aα chain are separated by 3-stranded α-helical coiled-coil regions [19]. Both strongly and weakly bound calcium ions are important for maintenance of fibrinogen structure and functions. The fibrinopeptides, which are in the central region, are cleaved by thrombin to convert soluble fibrinogen to insoluble fibrin polymer, via intermolecular interactions of the “knobs” exposed by fibrinopeptide removal with “holes” always exposed at the ends of the molecules. Fibrin monomers polymerize via these specific and tightly controlled binding interactions to make half-staggered oligomers that lengthen into protofibrils. The protofibrils aggregate laterally to make fibers, which then branch to yield a three-dimensional network-the fibrin clot-essential for hemostasis. X-ray crystallographic structures of portions of fibrinogen have provided some details on how these interactions occur. Finally, a transglutaminase, Factor XIIIa, covalently binds specific glutamine residues in one fibrin molecule to lysine residues in another fibrin molecule via isopeptide bonds, stabilizing the clot against mechanical, chemical, and proteolytic insults [20]. The gene regulation of fibrinogen synthesis and its assembly into multichain complexes proceed via a series of well-defined steps. Alternate splicing of two of the chains yields common variant molecular isoforms. The mechanical properties of clots, which can be quite variable, are essential to fibrin’s functions in hemostasis and wound healing [21]. The fibrinolytic system, with the zymogen plasminogen binding to fibrin together with tissue-type plasminogen activator to promote activation to the active enzyme plasmin, results in digestion of fibrin at specific lysine residues. Fibrin(ogen) also specifically binds a variety of other proteins, including fibronectin, albumin, thrombospondin, von Willebrand factor, fibulin, fibroblast growth factor-2, vascular endothelial growth factor, and interleukin-1. Though its ability to bind to a variety of compounds, particularly to various growth factors makes fibrinogen a player in cardiovascular and extracellular matrix physiology [18, 22–25], fibrinogen does not appear to play a specific role in pathogenesis of disorders discussed in this volume.
Studies of naturally occurring dysfibrinogenemias and variant molecules have increased our understanding of fibrinogen’s functions. Fibrinogen binds to activated αIIbβ3 integrin on the platelet surface, forming bridges responsible for platelet aggregation in hemostasis, and also has important adhesive and inflammatory functions through specific interactions with other cells [26]. Fibrinogen-like domains originated early in evolution, and it is likely that their specific and tightly controlled intermolecular interactions are involved in other aspects of cellular function and developmental biology.
3.4 Elastin
Elastin is an insoluble polymer of the monomeric soluble precursor tropoelastin. Elastin is the main component of elastic fibers in matrix tissue, and as such it is the main contributor to the elasticity of these fibers [27, 28]. Tropoelastin is encoded by a single human gene and is secreted as an ~60 kDa unglycosylated protein by a variety of cells, including fibroblasts, endothelial and smooth muscle cells, chondrocytes and keratinocytes [28]. The splicing of the primary tropoelastin transcript is tissue-specific, and thus allows for conformational and functional adjustment for each location [29]. The primary tropoelastin sequence is an arrangement of hydrophobic domains rich in valine, proline and glycine, providing elasticity to the final product, elastin. These hydrophobic domains alternate with hydrophilic domains which contain lysine residues whose role it is to stabilize elastin microfibrils by cross-linking [30–32]. However, before this can occur tropoelastin units are chaperoned to the extracellular surface [33] where they coacervate [34] into protein-dense spherules [35] which then undergo cross-linking and fibril assembly. Ninety per cent of the final product, i.e., of an elastic fiber, consists of a central amorphous core of elastin surrounded by a layer of microfibrils composed mostly of glycoprotein fibrillin, but also of many other proteins, among them fibulins, collagen VIII, and emilins with microfibrils as well [29, 36]. Proteoglycans, including biglycan [37] and glycosaminoglycan heparan sulfate [38] have been detected within the elastic core. Moreover, it has been shown that the presence of sulfated proteoglycans within the extracellular matrix regulates elastin assembly [39]. In addition, water plays an important role not just in the three dimensional organization of elastin molecules but also in the final degree of hydration and elasticity [38]. Elastic fibers form an interconnecting fenestrated network of lamellae in the arterial media. The lamellae are layers of elastic fibers surrounded by circumferentially oriented smooth muscle cells and collagen fibers [40].
The high content of hydrophobic amino acids makes elastin one of the most chemically resistant and durable proteins in the entire body [41]. It is distributed throughout the body in the form of tissue-specific elastic networks [28]. Elastin containing fibers provide elastic recoil in tissues where repetitive distention and relaxation is a requirement for their function, and is found typically in skin, lungs, ligaments, tendons and vascular tissues [42]. The relative content of elastin can vary from around a few percent in skin, to more than 70 % in some ligament structures in the animal kingdom. Elastic fibers are essential for proper function of at least three areas. As a major structural component elastic fibers provide elastic recoil and resilience to a variety of connective tissues, e.g., aorta and ligaments. Elastic fibers regulate activity of TGFβs through their association with fibrillin microfibrils. In addition, elastin also plays a role in cell adhesion, cell migration, survival and differentiation, and can, to some extent, act as a chemotactic agent [27, 29]. Elastin, and for that matter tropoelastin as well, is also a signaling molecule. Tropoelastin inhibits proliferation of arterial smooth muscle cells, induces the formation and organization of actin stress fibers and acts as a chemotactic agent [43].
Elastin and collagen are the dominant components of the ECM in large elastic arteries, such as aorta [40]. The two compounds play different, but complementary roles in arterial physiology: reversible extensibility during cycling loading is provided by elastin [40, 44], whereas strength and the ability to withstand high pressure is the responsibility of collagen [40, 45]. The assembly of elastic fibers proceeds only during tissue development, and cedes with maturation so older tendons contain less elastin then young tendons [46, 47]. In effect that means that with aging the stiffness of arterial wall increases due to degradation and fragmentation of elastic fibers [40, 48]. Matrix metalloproteinases (MMPs) are just some of the proteases participating in this destructive process [40, 49]. Increased levels of MMP-1 and MMP-9 have been detected in aortic aneurysms [50]. Local blockage of MMP activities in animal models either by TIMP-1 [51], inhibition of MMP-2 by calpain-1 inhibition [52], or by doxycycline, an inhibitor of MMPs [53] shows potential treatment venues. Whether they can be utilized for treatment of even prevention of complications of Marfan syndrome or related disorders remains to be seen. It is thought that production of collagen increases to compensate for the elastin deficit, however, this pushes the arterial wall towards increased stiffness [40]. Increased elastin production has been documented in some animal models of hypertension, but it is either not high enough [54] or the new elastin fibers are not assembled properly [55].
Elastin gene mutations can be divided into two groups [40]. Autosomal dominant supravalvular aortic stenosis is a representative of the first group. Besides aortic stenosis, patients develop hypertension, increased arterial stiffness leading to congestive heart failure [40]. Hypertrophy and hyperplasia of smooth muscle cells in the media of the affected arteries is due to fragmentation of elastic lamellae and changes in ECM composition [56]. This pathology is due to loss of function mutations in the elastin (ELN) gene [57]. Consequently, the mutant elastin protein is nonfunctional and does not interfere with the production and assembly of normal, functional elastin in heterozygous individuals who are then less affected than homozygous people [40].
An autosomal dominant form of cutis laxa belongs to the second group which encompasses disorders resulting from missense mutation, usually near the 3′ end of the transcript [40, 58, 59]. Cutis laxa and related disorders are described in more detail in Chap. 11 by Eva Morava et al. The mutant elastin interferes with normal assembly, metabolism and function of elastic fibers [59].
Lack of elastin in the body is fatal. Elastin knockout mice (Eln−/−) die shortly after birth with subendothelial cells accumulation blocking blood flow and with markedly increased arterial stiffness [40, 60]. The presence of additional lamellar units in heterozygous Eln+/− mice indicates an attempt to compensate and to remodel in a response to increased hemodynamic stress during development [61]. Fibrillin-1 hypomorphic mice (mgR/mgR) serve as a model of Marfan syndrome because of aneurysm formation in the ascending aorta and elastolysis in all segments of aorta [62].
3.5 Fibrillins
Because of close association of mutated fibrillin-1 with Marfan syndrome which is being discussed in detail in Chap. 6 by Cook and Ramirez, only a brief description of fibrillins is provided in this chapter. Fibrillins are a group of large extracellular glycoproteins (~350 kDa) [29] that consists of three isoforms, fibrillin-1, -2, and -3. Fibrillin molecules contain 40–80 amino acid residues, several calcium-binding epidermal growth factor (cbEGF)-domains interspersed with several eight-cysteine-containing (TB) motifs binding TGFβ [4, 29, 63]. No other extracellular proteins contain that much cysteine as fibrillins [42]. Whereas fibrillin-2 and fibrillin-3 are mostly expressed in embryonic tissues with the exception of peripheral nerves and, to lesser degree, skin and tendon [64, 65] fibrillin-1 is a protein appearing in both embryonic and adult tissues [65–67].
Fibrillins represent the predominant core of the microfibrils in elastic as well as non-elastic extracellular matrixes, and interact closely with tropoelastin and integrins, e.g., through direct bindings. Not only do microfibrils provide structural integrity of specific organ systems, but they also provide a scaffold for elastogenesis in elastic tissues such as skin, lung, and vessels [46]. Thus, fibrillin is important for the assembly of elastin into elastic fibers. The precise arrangement of fibrillin within microfibrils is a matter of speculation; several working models have been suggested to explain the architecture of microfibrils [67]. It is known that different mutations in different regions, including the propeptide sequence encoded by the C-terminal domain, of the fibrillin-1 gene lead to impaired assembly of microfibrils in individuals with Marfan syndrome [67–69]. Robinson et al. provide an excellent, more comprehensive review of these issues, including review of self-assembly of fibrillins and cross-link formation in fibrillin assembly [67]. Besides fibrillin and elastin, the two major components, many other proteins participate in the makeup of microfibrils. As noted above fibronectin in particular plays an essential role in this process, more specifically, through binding of a C-terminal fibrillin-1 region with the fibronectin gelatin-binding region [8]. It is interesting to note that homocysteinylation of fibronectin in homocystinuria reduces fibronectin dimers to monomers, and, as a consequence, impairs assembly of fibrillin and microfibrils. Similar impairment is the result of homocysteinylation of fibrillin-1 [70].
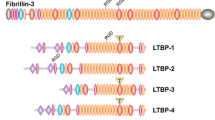
As already mentioned above, fibrillins contain several TGFβ-binding motifs, this feature makes their structure, and, in part, their function. similar to that of latent-TGFβ-binding proteins (or LTBPs) (see more in Chap. 6), [67].
Mutations in genes for fibrillin-1 and -2 lead to several disorders in people: mutation in fibrillin-1 can result in autosomal dominant Marfan and Weill-Marchesani syndromes, mutation in fibrillin-2 leads to Beal syndrome [4, 67].
3.6 Fibulins
Fibulins are a group of seven glycoproteins that are expressed and secreted by many cell types and tissues, and that are tightly connected with basement membranes, elastic fibers and other components of extracellular matrix. The members of the fibulin family are divided into class I and II, based on their length and domain structures [71]. Class II consists of short fibulins 3, 4, 5 and 7. Fibulins 3–5 participate in elastic fiber formation and are expressed during embryonic development, especially in skeletal and cardiovascular tissues [71]. This is facilitated by Ca2+ [72]. Fibulin-3 is predominantly found in mesenchyme that develops into cartilage and bone, fibulin-4 is markedly expressed in heart muscle, fibulin-5 highly in vasculature, and fibulin-7 is highly expressed in teeth, placenta, hair follicles and cartilage. The molecules of short fibulins contain tandem repeats of six cbEGF domains that are connected by one amino acid in a pattern similar to the one found in fibrillin-1 [73]. Fibulin 5 contains an arginine-glycine-aspartic acid (RGD) motif which mediates binding to integrin receptors on endothelial cells and vascular smooth muscle cells [74]. This step is necessary for elastic fiber assembly [71]. Fibulin-5 also inhibits α5β1 and α4β1 fibronectin receptor-mediated downstream signaling [71]. The C-terminal fibulin module (which, by the way, is present in all fibulins) contains an elastic-binding domain in fibulin-5 [75]. The same module in fibulin-5 also interacts with lysyl oxidase-like 1, 2 and 4 (Loxl 1, Loxl 2 and Loxl4), enzymes playing crucial role in cross-linking [76, 77] whereas it is the N-terminal domain responsible for binding to Lox in fibulin-4 [78]. Lysyl oxidases, including those binding to fibulin-5 and -4 mediate crosslinking of tropoelastin monomers into insoluble elastin polymer [79]. The binding between the C-terminal module of fibulin-3 and tissue inhibitor of metalloproteinase 3 is another example of close relationship between a short fibulin and connective tissue metabolism [80]. The level of fibulin-5 is particularly high in the cardiovascular system and lung, though fibulin-4 is expressed in the outer layer of media of large blood vessels, and fibulin-3 appears in capillaries, skin and the basement membrane [71]. The participation of fibulin-5 in elastogenesis is solely due to its exclusive binding to tropoelastin but not to polymerized elastin in vitro [75]. Its role is inhibition of excessive tropoelastin coacervation into large aggregates, and consequently this allows for integration of microassembles of tropoelastin into the microfibril scaffolding [71]. In addition to fibrillins-1 and -2, as also discussed above earlier in this chapter under Elastin, fibulins are present in microfibrils of scaffolding for elastic fibers as well [81].
Fibulins serve not only as structural ECM components, but also act as mediators of several cellular processes, such as cell growth, differentiation, angiogenesis and tumor growth. Thus they serve as modulators of cellular behavior and function [82]. The molecular mechanism of fibulin activity is not fully explained, but high levels of fibulin are often observed in cartilage, especially during development. Fibulin-1 (molecular weight around 100 kDa) was originally thought to be an intracellular molecule linking cytoskeletal components to β integrins, but later it was shown that fibulin-1 was also present in fibril matrix surrounding fibroblasts in culture as well as in embryos [83]. Fibulin-2 demonstrates some overlap with fibulin-1, but its expression is more prominent in the developing heart, both aortic and coronary vessels [84]. Studies in animals lacking fibulins demonstrate importance of these glycoproteins in pathogenesis of a variety of developmental and pathological processes, e.g., impaired tissue elasticity, altered vision and reduced vascular formation.
The role of fibulins and elastin in several human diseases is being discussed in several chapters of this volume.
3.7 Tenascins
Tenascins are ECM polymorphic glycoproteins with high molecular weights between 150 and 380 kDa. They are a family of multimeric proteins labeled as tenascin-C, -R, -W, -X and -Y [85–87]. Tenascins are composed of identical subunits built from variable numbers of repeated domains, including heptad repeats, EGF-like repeats, fibronectin type III domains and a C-terminal globular domain similar to that seen in fibrinogens. Whereas tenascin-R is predominantly found in the central nervous system, the other members of the tenascin family are found more widespread in connective and soft tissues in the body. With regards to tenascin-R, its expression is predominantly present during development of the CNS. Tenascin-X and –Y are predominantly seen in skeletal muscle connective tissue, and tenascin-C and –W have both been observed in a variety of developing tissues, and a large interest has been invested in these tenascins in relation to tumor development and growth, where they play important roles.
The first described tenascin was the C isomer. Tenascin-C is a large monomer of Mr 300 kDa, assembled into a hexamer. As other tenascins the molecule consists of a N-teminal domain, EGF-like repeats, several fibronectin type II domains and a C-teminal fibrinogen-like globular domain [87]. Tenascin-C is transiently expressed in the mesenchyme around developing organs such as kidney, teeth and mammary glands. It is present in the periostium, ligaments, tendons, myo-tendinous junctions, smooth muscle and perichondrium both during embryonic development and in adult tissues. However, expression of tenascin-C in the adult tissue is generally low, only to be transiently elevated upon tissue injury and often down-regulated again after tissue repair is complete. Although tenascin-C shares structural relationship to fibronectin, it differs in adhesive function. Where fibronectin is adhesive in nature, tenascin-C is only weakly adhesive – if at all – for most cells, and it does in fact limit the fibronectin-mediated cell spreading when the two proteins are combined [88]. Tenascin-C interferes with cell spreading by inhibiting binding of fibronectin to its co-receptor syndecan-4, and integrin α5β1 signaling to FAK and RhoA is also impaired whereby focal adhesions are diminished [89–92].
The expression of tenascin-C is regulated by mechanical stress both during development and in adulthood, and its expression is predominantly present in tissues experiencing high tensile stress, such as ligaments, tendons and smooth muscle [93]. Mechanical loading of muscle induces tenascin-C mRNA and protein in endomysial fibroblasts of the affected holding muscle [94]. Tenascin-C was over-expressed in hypertensive rat arterial smooth muscle [95] and in the periosteum of rat ulnae loaded in vivo, but tenascin-C expression was low in the osteotendinous interphase of immobilized rat legs [94]. Interestingly, elevated levels of tenascin-C were found in the blood of human patients with rheumatoid arthritis [96], and in synovial fluid after injury to the human and canine knee [97].
In relation to ECM tissue damage, tenascin-C has been demonstrated to play different roles that can mediate both inflammatory and fibrotic processes to enable effective tissue repair. For example, tenascin-C makes a prominent appearance in pathological heart conditions. Though barely expressed in the normal adult heart its level increases in the heart after myocardial infarction, during myocarditis, hypertensive heart disease, to name just a few examples [87]. According to the current hypothesis tenascin-C is directly involved in ventricular remodeling through releasing cardiomyocytes from the adherence to the extracellular matrix and through upregulation of matrix metalloproteinases [87, 98]. A high level of expression of tenascin-C in cardiac tissues correlates with poor patient prognosis [99]. Interestingly, tenascin-C was found in calcified aortic valve, together with matrix metalloproteinase-12 where they likely contribute to the fragmentation of elastic fibers [100].
Tenascin-X differs from tenascin-C and -R in that it is less glycosylated, and that it is present in almost all tissues, and especially widely expressed in developing fetal tissues. It is highly expressed in skeletal muscle, heart, tendon and skin. It plays an interesting role in relation to physical activity, as it is known to be upregulated in skeletal muscle in relation to acute mechanical loading and known to be present in tissues that are subjected to high stress [101, 102]. Tenascin-Y is an avian equivalent of tenascin-X [85, 103].
Overall, tenascin proteins are found to be dys-regulated in many pathological conditions like cancer, heart- and vessel disease, as well as in connective tissue diseases with manifestations in skin, tendon and muscle like e.g., special forms of Ehlers-Danlos syndrome (more discussed in Chap. 9) and Dupuytren disease [104]. Further, tenascins have been shown to be important in regeneration and recovery of musculo-tendinous tissue, in that they possess a de-adhesive effect whereby they potentially can contribute to a coordinated tissue reorganization and build-up [105]. It has been suggested that they “orchestrate” muscle build up after injury [106]. Thus it is likely that tenascins are important for ensuring mechanical properties of weight bearing ECM as well as ensuring an optimal recovery of ECM after mechanical injury.
3.8 Thrombospondins
Thrombospondins (TSPs) are a group of five modular glycoproteins, each one of them encoded by a separate gene [107–109]. TSP-1 and TSP-2 form group A, and TSPs 3–5 are in group B. Their binding to various components of the extracellular matrix, such as heparan sulfate proteoglycans, and to numerous cell membrane receptors enables TSPs to modulate cell functions in a variety of tissues [107]. They are considered to be “adhesion-modulating” components of the extracellular matrix [110].
In particular, we will discuss TSP-1 and TSP-5 as their involvement in metabolism of the extracellular matrix is pertinent to issues discussed in this volume. The activation of latent TGFβ by TSP-1 plays an important role in wound healing, and also in pathogenesis of fibrotic processes in kidney and heart in diabetes [111, 112]. Increased expression of TSP-1 (accompanied by increased TGFβ activity) was observed in fibrotic skin disorders such as keloids [113] and scleroderma [114].
TSP-1, normally stored in platelets, is released from their α-granules upon injury so it can participate in tissue repair [115]. It is a homotrimer of three 150 kDa subunits. Each unit is composed of N-terminal laminin G-like domain, and in the last 650 amino acids, of several EGF-like domains, 13 calcium-binding repeats and a globular L-type lectin-like domain. These regions in the last 650 amino acids are usually referred to as the C-terminal or “signature” region [110]. With glycosylation the size of TSP-1 balloons to staggering Mr ~450 kDa [116]. Its expression in adult organism is minimal (except for storage pool in platelets) and is upregulated only as a result of injury [117] and/or chronic disease [116, 118]. TSP1 binds to many cell membrane receptors, including CD47 [116], integrins [119], heparan sulfate and LDL [120]. TSP-1 not only binds to latent TGFβ through thrombospondin repeats, but it also activates this growth factor [121]. It is thought that TSP-1 facilitates presentation of TGFβ to the TGFβ receptor [115]. TSP-1 was shown to upregulate type I collagen expression through its N- and C-terminal domains which may explain the sometimes opposing cellular responses stimulated by TSP-1 [115, 122]. TGFβ activity induced by TSP-1 is a normal process during early tissue repair, however, if TSP-1 expression persists in later stages of wound healing fibrosis may prevail [115]. In addition, TSP-1 regulates activity of several other growth factors, most notably, VEGF, EGF and PDGF. In particular, TSP-1 plays an important role in transactivation of EGF receptors in epithelial and endothelial cells, and thus can disrupt endothelial barrier [123]. Though TSP-1 has hypertensive effect on cardiovascular system and is known to play a role in pathogenesis of atherosclerosis and peripheral vascular disease [124], the activity is mediated through control of nitric oxide synthesis (and thus increasing arterial resistance), rather than through an impact on or binding to a structural component of the blood vessel wall [124]. TSP-2 is involved in collagen fibril assembly and is capable of inhibition of angiogenesis and protease activity, but unlike TSP-1 it does not activate TGFβ [87].
However, there is at least one syndrome where a mutation in a gene encoding an enzyme responsible for proper TSP-1 function leads to structural changes which form the basis of the so called Peters Plus syndrome. This syndrome is an autosomal recessive disorder phenotypically characterized by eye defects, short stature, developmental delay and cleft lip due to a mutation of a gene encoding a β1,3-glucosyltransferase which adds a glucose to O-linked fucose (and producing a rare glucose-β 1,3-fucose disaccharide) and which is responsible for glycosylation of thrombospondin type 1 repeats [125, 126]. Beside TSP-1, properdin, F-spondin, some members of a-disintegrin-and-metalloproteinase-with-thrombospondin-like-motifs family (ADAMTS-13 and ADAMTSL-1) carry the same disaccharide [125, 126]. Heart defects, such as hypoplastic left heart syndrome [127], patent ductus arteriosus, and atrial septal defect are present is some variants [128]. Though the eye involvement is usually characterized by anterior eye chamber defects leading to glaucoma [125, 128], corneal pathology has been recognized in some cases as well, and then it consists of intracorneal fibrosis [129] and keratolenticular adhesions [125, 128].
3.9 Cartilage Oligomeric Matrix Protein (COMP) or Thrombospondin-5
COMP or thrombospondin-5 belongs to the family of 5 extracellular calcium- and glycosaminoglycan-binding proteins that play a role predominantly during development, angiogenesis and wound healing. It consists of five identical subunits that are linked together at their N-terminal pentamerization end to result in an almost “star-like” structure and has Mr ~524 kDa [130]. COMP shares a conserved multidomain architecture in its C-terminal region with TSP-1 [110]. It also contains eight calmodulin units, four EGF-like repeats, and a globular C-terminal domain [130, 131], and the 5 “arms” have on their C-terminal end high affinity binding sites for type I, II and IX collagen [132, 133], and for fibronectin [134]. Thrombospondin-5/COMP is present primarily in cartilage, and has been suggested to be important in relation to cartilage turnover and pathogenesis of osteoarthritis [135]. It is also expressed in other connective tissues like tendon, especially if the tissue has undergone strenuous mechanical loading [136, 137]. The exact role of COMP in the fibril formation and assembly in the extracellular matrix is becoming better understood, and it is thought that COMP facilitates the joining of collagen molecules during formation of fibril structures [137, 138]. It has been shown that high levels of COMP are present in fibrotic scars and systemic sclerosis of the skin [136, 139]. It has been suggested that a very high concentration of COMP can in fact inhibit collagen fibril formation [115].
COMP is expressed in normal tendon where its mRNA is confined to tenocytes and the protein itself was located in the normally aligned fiber structures together with type I collagen. Virtually no COMP (and no type I collagen), but only type III collagen was found in the normal endotenon [137]. Physical activity leads to increased expression of COMP, at least in the equine tendon [136], as do pathological processes. High levels of COMP were identified in the synovial fluid obtained from the sheaths of the equine superficial digital flexor tendons diagnosed with synovitis [140]. Likewise, injury to superficial digital flexor tendons leads to increased expression of COMP, and type I and III collagens in the endotenon and high levels of all three molecules can be visualized in the injured and granulation tissue [137]. Rock et al. have shown that COMP promotes attachment of ligament cells and chondrocytes to components of the extracellular matrix using two mechanisms which involve CD47 and integrins. Such data indicate an important role for COMP in formation of structural scaffolding, an essential step in cell attachment to the extracellular matrix and in matrix-cell signaling [131].
In addition, new data indicate that COMP, and its degradation by ADAMTS-7, plays an important role in vascular remodeling [141]. COMP has been found in atherosclerotic plaques and lesions forming in arteries undergoing re-stenosis [142], together with SLRPs, such as decorin [143]. It has been suggested that COMP promotes differentiation of vascular smooth muscle cells and that binding and degradation of COMP by ADAMTS-7 in injured arteries enables migration of vascular smooth muscle cells and neointima formation. The hope is that ADAMTS-7 may be suitable as a therapeutic agent in combating restenosis of atherosclerotic blood vessels after angioplasties and related procedures [141]. More recent study from the same laboratory shows that COMP inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2 and that the COMP in atherosclerotic arteries story is a little bit more complicated than initially thought [144].
Though COMP has been involved in metabolism of multiple tissues, including cartilage, tendons and blood vessels the only mutations in the COMP gene known to be responsible for pathological conditions so far identified are those affecting the skeleton, such as pseudoachondroplasia and multiple epiphyseal dysplasia [131, 145].
References
Kjaer M (2004) Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol Rev 84:649–698
Leahy DJ, Aukhil I, Erickson HP (1996) 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84:155–164
Potts JR, Campbell ID (1994) Fibronectin structure and assembly. Curr Opin Cell Biol 6:648–655
Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP (2009) Fibrillin assembly requires fibronectin. Mol Biol Cell 20:846–858
Mao Y, Schwarzbauer J (2005) Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol 24:389–399
Takahashi S, Leiss M, Moser M, Ohashi T, Kitao T, Heckmann D, Pfeifer A, Kessler H, Takagi J, Erickson HP, Fässler R (2007) The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J Cell Biol 178:167–178
Singh P, Schwarzbauer JE (2012) Fibronectin and stem cell differentiation – lessons from chondrogenesis. J Cell Sci 125:3703–3712
Dallas SL, Chen Q, Sivakumar P (2006) Dynamics of assembly and reorganization of extracellular matrix proteins. Curr Top Dev Biol 75:1–24
Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JCR, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki M, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD (2005) A simplified laminin nomenclature. Matrix Biol 24:326–332
Miner JH, Yurchenco PD (2004) Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol 20:255–284
Domogatskaya A, Rodin S, Tryggvason K (2012) Functional diversity of laminins. Annu Rev Cell Dev Biol 28:523–553
MacDonald PR, Lustig A, Steinmetz MO, Kammerer RA (2010) Laminin chain assembly is regulated by specific coiled-coil interactions. J Struct Biol 170:398–405
Grounds MD, Sorokin L, White J (2005) Strength at the extracellular matrix-muscle interface. Scand J Med Sci Sports 15:381–391
Taylor SH, Al-Youha S, Van Agtmael T, Lu Y, Wong J, McGrouther DA, Kadler KE (2011) Tendon is covered by a basement membrane epithelium that is required for cell retention and the prevention of adhesion formation. PLoS One 6:e16337
Molloy TJ, de Bock CE, Wang Y, Murrell GA (2006) Gene expression changes in SNAP-stimulated and iNOS-transfected tenocytes–expression of extracellular matrix genes and its implications for tendon-healing. J Orthop Res 24:1869–1882
Sato N, Nakamura M, Chikama T, Nishida T (1999) Abnormal deposition of laminin and type IV collagen at corneal epithelial basement membrane during wound healing in diabetic rats. Jpn J Ophthalmol 43:343–347
Della Corte A, De Santo LS, Montagnani S, Quarto C, Romano G, Amarelli C, Scardone M, De Feo M, Cotrufo M, Caianiello G (2006) Spatial patterns of matrix protein expression in dilated ascending aorta with aortic regurgitation: congenital bicuspid valve versus Marfan’s syndrome. J Heart Valve Dis 15:20–27
Fish RJ, Neerman-Arbez M (2012) Fibrinogen gene regulation. Thromb Haemost 108:419–426
Doolittle RF, Goldbaum DM, Doolittle LR (1978) Designation of sequences involved in the “coiled-coil” interdominal connections in fibrinogen: constructions of an atomic scale model. J Mol Biol 120:311–325
Ariens RA, Lai TS, Weisel JW, Greenberg CS, Grant PJ (2002) Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood 100:743–754
Cilia La Corte AL, Philippou H, Ariëns RA (2011) Role of fibrin structure in thrombosis and vascular disease. Adv Protein Chem Struct Biol 83:75–127
Sahni A, Francis CW (2000) Vascular endothelial growth factor binds to fibrinogen and fibrin and stimulates endothelial cell proliferation. Blood 96:3772–3778
Sahni A, Odrljin T, Francis CW (1998) Binding of basic fibroblast growth factor to fibrinogen and fibrin. J Biol Chem 273:7554–7559
Clark RA, Lanigan JM, DellaPelle P, Manseau E, Dvorak HF, Colvin RB (1982) Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound reepithelialization. J Invest Dermatol 79:264–269
Donaldson DJ, Mahan JT, Amrani D, Hawiger J (1989) Fibrinogen-mediated epidermal cell migration: structural correlates for fibrinogen function. J Cell Sci 94:101–108
Armstrong PC, Peter K (2012) GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thromb Haemost 107:808–814
Muiznieks LD, Weiss AS, Keeley FW (2010) Structural disorder and dynamics of elastin. Biochem Cell Biol 88:239–250
Mithieux SM, Wise SG, Weiss AS (2013) Tropoelastin – a multifaceted naturally smart material. Adv Drug Deliv Rev 65:421–428
Kielty CM (2006) Elastic fibres in health and disease. Expert Rev Mol Med 8:1–23
Csiszar K (2001) Lysyl oxidases: a novel multifunctional amine oxidase family. Prog Nucleic Acid Res Mol Biol 70:1–32
Lee JE, Kim Y (2006) A tissue-specific variant of the human lysyl oxidase-like protein 3 (LOXL3) functions as an amine oxidase with substrate specificity. J Biol Chem 281:37282–37290
Kim YM, Kim EC, Kim Y (2011) The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol Biol Rep 38:145–149
Hinek A, Rabinovitch M (1994) 67-kD elastin-binding protein is a protective “companion” of extracellular insoluble elastin and intracellular tropoelastin. J Cell Biol 126:563–574
Yeo GC, Keeley FW, Weiss AS (2011) Coacervation of tropoelastin. Adv Colloid Interface Sci 167:94–103
Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP (2006) Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol 207:87–96
Berk DR, Bentley DD, Bayliss SJ, Lind A, Urban Z (2012) Cutis laxa: a review. J Am Acad Dermatol 66:842.e1–842.e17
Baccarani-Contri M, Vincenzi D, Cicchetti F, Mori G, Pasquali-Ronchetti I (1990) Immunocytochemical localization of proteoglycans within normal elastin fibers. Eur J Cell Biol 53:305–312
Gheduzzi D, Guerra D, Bochicchio B, Pepe A, Tamburro AM, Quaglino D, Mithieux S, Weiss AS, Pasquali Ronchetti I (2005) Heparan sulphate interacts with tropoelastin, with some tropoelastin peptides and is present in human dermis elastic fibers. Matrix Biol 24:15–25
Kozel BA, Ciliberto CH, Mecham RP (2004) Deposition of tropoelastin into the extracellular matrix requires a competent elastic fiber scaffold but not live cells. Matrix Biol 23:23–34
Wagenseil JE, Mecham RP (2012) Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5:264–273
Mithieux SM, Weiss AS (2005) Elastin. Adv Protein Chem 70:437–461
Chung MI, Miao M, Stahl RJ, Chan E, Parkinson J, Keeley FW (2006) Sequences and domain structures of mammalian, avian, amphibian, and teleost tropoelastins: clues to the evolutionary history of elastin. Matrix Biol 25:495–504
Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY (2003) A critical role for elastin signaling in vascular morphogenesis and disease. Development 130:411–423
Mecham RP (1998) Overview of extracellular matrix. In: Current protocols in cell biology. Wiley, New York
Fung YC (1993) Biomechanics: mechanical properties of living tissues, 2nd edn. Springer, New York
Wagenseil JE, Mecham RP (2009) Vascular extracellular matrix and arterial mechanics. Physiol Rev 89:957–989
Kostrominova TY, Brooks SV (2013) Age-related changes in structure and extracellular matrix protein expression levels in rat tendons. Age 35:2203–2214
Greenwald SJ (2008) Ageing of the conduit arteries. J Pathol 211:157–172
Li Z, Froehlich J, Galis ZS, Lakatta EG (1999) Increased expression of matrix metalloproteinase-2 in the thickened intima of aged rats. Hypertension 33:116–123
Tamarina NA, McMillan WD, Shively VP, Pearce WH (1999) Expression of matrix metalloproteinases and their inhibitors in anuerysm and normal aorta. Surgery 122:264–271
Allaire E, Forough R, Clowes M, Starcher B, Clowes AW (1998) Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest 102:1413–1420
Jiang L, Wang M, Zhang J, Monticone RE, Telljohann R, Spinnetti G, Pintus G, Lakatta EG (2008) Increased calpain-1 activity mediates age-associated angiotensin II signaling of vascular smooth muscle cells. PLoS One 3:e2231
Castro MM, Rizzi E, Figueiredo-Lopes L, Fernandes K, Bendhack LM, Pitol DL, Gerlach RF, Tanus-Santos JE (2008) Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis 198:320–331
Wolinsky H (1970) Response of the rat aortic media to hypertension. Morphological and chemical studies. Circ Res 26:507–522
Todorovich-Hunter L, Johnson D, Ranger P, Keeley F, Rabinovitch M (1988) Altered elastin and collagen synthesis associated with progressive pulmonary hypertension induced by monocrotaline. A biochemical and ultrastructural study. Lab Invest 58:184–195
O’Connor WN, Davis JB Jr, Geissler R, Cottrill CM, Noonan JA, Todd EP (1985) Supravalvular aortic stenosis. Clinical and pathological observations in six patients. Arch Pathol Lab Med 109:179–185
Urban Z, Michels VV, Thibodeau SN, Davis EC, Bonnefont J-P, Munnich A, Eyskens B, Gewillig M, Devriendt K, Boyd CD (2000) Isolated supravalvular aortic stenosis: functional haploinsufficiency of the elastin gene as a result of nonsense-mediated decay. Hum Genet 106:577–588
Rodriguez-Revenga L, Iranzo P, Badenas C, Puig S, Carrio A, Mila M (2004) A novel elastin gene mutation resulting in an autosomal dominant from of cutis laxa. Arch Dermatol 149:1135–1139
Tassabehji M, Metcalfe K, Hurst J, Ashcroft GS, Kielty C, Wilmot C, Donnai D, Read AP, Jones CJP (1998) An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autodomal dominant cutis laxa. Hum Mol Genet 7:1021–1028
Li DY, Brooke D, Davis EC, Mecham RP, Sorensen LK, Boak KK, Eichwald E, Keating MT (1998) Elastin is an essential determinant of arterial morphogenesis. Nature 393:276–289
Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, MacLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP (2003) Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112:1419–1428
Schwill S, Seppelt P, Grünhagen J, Ott CE, Jugold M, Ruhparwar A, Robinson PN, Karck M, Kallenbach K (2013) The fibrillin-1 hypomorphic mgR/mgR murine model of Marfan syndrome shows severe elastolysis in all segments of the aorta. J Vasc Surg 57:1628–1636
Kielty CM, Sherratt MJ, Marson A, Baldock C (2005) Fibrillin microfibrils. Adv Protein Chem 70:405–436
Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, Mecham RP, Ramirez F (1994) Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J Cell Biol 124:855–863
Charbonneau NL, Dzamba BJ, Ono RN, Keene DR, Corson GM, Reinhardt DP, Sakai LY (2003) Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences. J Biol Chem 278:2740–2749
Cain SA, Morgan A, Sherratt MJ, Ball SG, Shuttleworth CA, Kielty CM (2006) Proteomic analysis of fibrillin-rich microfibrils. Proteomics 6:111–122
Robinson PN, Arteaga-Solis E, Baldock C, Collod-Béroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA, Judge DP, Kielty CM, Loeys B, Milewicz DM, Ney A, Ramirez F, Reinhardt DP, Tiedemann K, Whiteman P, Godfrey M (2006) The molecular genetics of Marfan syndrome and related disorders. J Med Genet 43:769–787
Milewicz DM, Grossfield J, Cao SN, Kielty C, Covitz W, Jewett T (1995) A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J Clin Invest 95:2373–2378
Raghunath M, Putnam EA, Ritty T, Hamstra D, Park ES, Tschodrich-Rotter M, Peters P, Rehemtulla A, Milewicz DM (1999) Carboxy-terminal conversion of profibrillin to fibrillin at a basic site by PACE/furin-like activity required for incorporation in the matrix. J Cell Sci 112:1093–1100
Hubmacher D, Sabatier L, Annis DS, Mosher DF, Reinhardt DP (2011) Homocysteine modifies structural and functional properties of fibronectin and interferes with the fibronectin-fibrillin-1 interaction. Biochemistry 50:5322–5332
Yanagisawa H, Davis EC (2010) Unraveling the mechanism of elastic fiber assembly: the roles of short fibulins. Int J Biochem Cell Biol 42:1084–1093
Wachi H, Nonaka R, Sato F, Shibata-Sato K, Ishida M, Iketani S, Maeda I, Okamoto K, Urban Z, Onoue S, Seyama Y (2008) Characterization of the molecular interaction between tropoelastin and DANCE/fibulin-5. J Biochem 143:633–639
Hambleton S, Valeyev NV, Muranyi A, Knott V, Werner JM, McMichael AJ, Handford PA, Downing AK (2004) Structural and functional properties of the human notch-1 ligand binding region. Structure 12:2173–2183
Yanagisawa H, Schluterman MK, Brekken RA (2009) Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal 3:337–347
Zheng Q, Davis EC, Richardson JA, Starcher BC, Li T, Gerard RD, Yanagisawa H (2007) Molecular analysis of fibulin-5 function during de novo synthesis of elastic fibers. Mol Cell Biol 27:1083–1095
Hirai M, Ohbayashi T, Horiguchi M, Okawa K, Hagiwara A, Chien KR, Kita T, Nakamura T (2007) Fibulin-5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo. J Cell Biol 176:1061–1071
Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li T (2004) Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet 36:178–182
Horiguchi M, Inoue T, Ohbayashi T, Hirai M, Noda K, Marmorstein LY, Yabe D, Takagi K, Akama TO, Kita T, Kimura T, Nakamura T (2009) Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A 106:19029–19034
Sato F, Wachi H, Ishida M, Nonaka R, Onoue S, Urban Z, Starcher BC, Seyama Y (2007) Distinct steps of cross-linking, self-association, and maturation of tropoelastin are necessary for elastic fiber formation. J Mol Biol 369:841–851
Klenotic PA, Munier FL, Marmorstein LY, Anand-Apte B (2004) Tissue inhibitor of metalloproteinase-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1): implications for macular degenerations. J Biol Chem 279:30469–30473
Ramirez F, Dietz HC (2007) Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. J Cell Physiol 213:326–330
DeVega A, Iwamoto T, Yamada Y (2009) Fibulins: multiple roles in matrix structures and tissue functions. Cell Mol Life Sci 66:1890–1902
Zhang HY, Timpl R, Sasaki T, Chu ML, Ekblom P (1996) Fibulin-1 and fibulin-2 expression during organogenesis in the developing mouse embryo. Dev Dyn 205:348–364
Tsuda T, Wang H, Timpl R, Chu ML (2001) Fibulin-2 expression marks transformed mesenchymal cells in developing cardiac valves, aortic arch vessels and coronary vessels. Dev Dyn 222:89–100
Tucker RP, Drabikowski K, Hess JF, Ferralli J, Chiquet-Ehrismann R, Adams JC (2006) Phylogenetic analysis of the tenascin gene family: evidence of origin early in the chordate lineage. BMC Evol Biol 6:60
Tucker RP, Chiquet-Ehrismann R (2009) The regulation of tenascin expression by tissue microenvironments. Biochim Biophys Acta 1793:888–892
Okamoto H, Imanaka-Yoshida K (2012) Matricellular proteins: new molecular targets to prevent heart failure. Cardiovasc Ther 30:e198–e209
Chiquet-Ehrismann R, Kalla P, Pearson CA, Beck K, Chiquet M (1988) Tenascin interferes with fibronectin action. Cell 53:383–390
Huang W, Chiquet-Ehrismann R, Moyano JV, Garcia-Pardo V, Orend G (2001) Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res 61:8586–8594
Midwood KS, Schwarzbauer JS (2002) Tenascin-C modulates matrix contraction via focal adhesion kinase- and Rho-mediated signaling pathways. Mol Biol Cell 13:3601–3613
Chiquet-Ehrismann R, Chiquet M (2003) Regulation and putative functions during pathological stress. J Pathol 200:488–499
Jones FS, Jones PL (2000) The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodeling. Dev Dyn 218:235–259
Kreja L, Liedert A, Schlenker H, Brenner RE, Fiedler J, Friemert B, Dürselen L, Ignatius A (2012) Effects of mechanical strain on human mesenchymal stem cells and ligament fibroblasts in a textured poly(L-lactide) scaffold for ligament tissue engineering. J Mater Sci Mater Med 23:2575–2582
Järvinen TA, Józsa L, Kannus P, Järvinen TL, Hurme T, Kvist M, Pelto-Huikko M, Kalimo H, Järvinen M (2003) Mechanical loading regulates the expression of tenascin-C in the myotendinous junction and tendon but does not induce de novo synthesis in the skeletal muscle. J Cell Sci 116:857–866
Mackie EJ, Scott-Burden T, Hahn AW, Kern F, Bernhardt J, Regenass S, Weller A, Bühler FR (1992) Expression of tenascin by vascular smooth muscle cells. Alterations in hypertensive rats and stimulation by angiotensin II. Am J Pathol 141:377–388
Page TH, Charles PJ, Piccinini AM, Nicolaidou V, Taylor PC, Midwood KS (2012) Raised circulating tenascin-C in rheumatoid arthritis. Arthritis Res Ther 14:R260
Chockalingam PS, Glasson SS, Lohmander LS (2013) Tenascin-C levels in synovial fluid are elevated after injury to the human and canine joint and correlate with markers of inflammation and matrix degradation. Osteoarthritis Cartilage 21:339–345
Imanaka-Yoshida K (2012) Tenascin-C in cardiovascular tissue remodeling: from development to inflammation and repair. Circ J 76:2513–2520
Midwood KS, Hussenet T, Langlois B, Orend G (2011) Advances in tenascin-C biology. Cell Mol Life Sci 68:3175–3199
Perrotta I, Russo E, Camastra C, Filice G, Di Mizio G, Colosimo F, Ricci P, Tripepi S, Amorosi A, Triumbari F, Donato G (2011) New evidence for a critical role of elastin in calcification of native heart valves: immunohistochemical and ultrastructural study with literature review. Histopathology 59:504–513
Flück M, Tunc-Civelek V, Chiquet M (2000) Rapid and reciprocal regulation of tenascin-C and tenascin-Y expression by loading of skeletal muscle. J Cell Sci 113:3583–3591
Chiquet M, Gelman L, Lutz R, Maier S (2009) From mechanostransduction to extracellular matrix gene expression in fibroblasts. Biochim Biophys Acta 1793:911–920
Hagios C, Koch M, Spring J, Chiquet M, Chiquet-Ehrismann R (1996) Tenascin-Y: a protein of novel domain structure is secreted by differentiated fibroblasts of muscle connective tissue. J Cell Biol 134:1499–1512
Berndt A, Kosmehl H, Katenkamp D, Tauchmann V (1994) Appearance of the myofibroblastic phenotype in Dupuytren’s disease is associated with a fibronectin, laminin, collagen type IV and tenascin extracellular matrix. Pathobiology 62:55–58
Mackey AL, Brandstetter S, Schjerling P, Bojsen-Moller J, Qvortrup K, Pedersen MM, Doessing S, Kjaer M, Magnusson SP, Langberg H (2011) Sequences response of extracellular matrix de-adhesion and fibrotic regulators after muscle damage is involved in protection against future injury in human skeletal muscle. FASEB J 25:1943–1959
Flück M, Mund SI, Schittny JC, Klossner S, Durieux AC, Giraud MN (2008) Mechano-regulated tenascin-C orchestrates muscle repair. Proc Natl Acad Sci U S A 105:13662–13667
Murphy-Ullrich JE, Iozzo RV (2012) Thrombospondins in physiology and disease: new tricks for old dogs. Matrix Biol 31:152–154
Adams JC, Lawler J (2004) The thrombospondins. Int J Biochem Cell Biol 36:961–968
Adams JC, Lawler J (2011) The thrombospondins. Cold Spring Harb Perspect Biol 3:a00971
Mosher DF, Adams JC (2012) Adhesion-modulating/matricellular ECM protein families: a structural, functional and evolutionary appraisal. Matrix Biol 31:155–161
Lu A, Miao M, Schoeb TR, Agarwal A, Murphy-Ullrich JE (2011) Blockade of TSP-1 dependent TGF-beta activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol 178:2573–2586
Belmadani S, Bernal J, Wei CC, Pallero MA, Dell’italia L, Murphy-Ullrich JE, Brecek KH (2007) A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am J Pathol 171:777–789
Chipev CC, Simman R, Hatch G, Katz AE, Siegel DM, Simon M (2000) Myofibroblast phenotype and apoptosis in keloid and palmar fibroblasts in vitro. Cell Death Differ 7:166–176
Mimura Y, Ihn H, Jinnin M, Assano Y, Yamane K, Tamaki K (2005) Constitutive thrombospondin-1 overexpression contributes to autocrine transforming growth factor-beta signaling in cultured scleroderma fibroblasts. Am J Pathol 166:1451–1463
Sweetwyne MT, Murphy-Ullrich JE (2012) Thrombospondin1 in tissue repair and fibrosis: TGF-β-dependent and independent mechanisms. Matrix Biol 31:178–186
Rogers NM, Yao M, Novelli EM, Thomson AW, Roberts DD, Isenberg JS (2012) Activated CD47 regulates multiple vascular and stress responses: implications for acute kidney injury and its management. Am J Physiol Renal Physiol 303:F1117–F1125
Agah A, Kyriakides TR, Lawler J, Bornstein P (2002) The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP/2-null mice. Am J Pathol 161:831–839
Hohenstein B, Daniel C, Hausknecht B, Boehmer K, Riess R, Amann KU, Hugo CP (2008) Correlation of enhanced thrombospondin-1 expression, TGF-beta signalling, and proteinuria in human type-2 diabetic nephropathy. Nephrol Dial Transplant 23:3880–3887
Chandrasekaran S, Guo NH, Rodrigues RG, Kaiser J, Roberts DD (1999) Pro-adhesive and chemotactic activities of thrombospondin-1 for breast carcinoma cells are mediated by alpha3beta1 integrin and regulated by insulin-like growth factor-1 and CD98. J Biol Chem 274:11408–11416
Chen H, Sottile J, Strickland DK, Mosher DF (1996) Binding and degradation of thrombospondin-1 mediated through heparan sulfate proteoglucans and low-density-lipoprotein receptor-related protein: localization of the functional activity to the trimeric N-terminal heparin-binding region of thrombospondin-1. Biochem J 318:959–963
Murphy-Ullrich JE, Poczatek M (2000) Activation of latent TGF-beta by thrombospondin-1: mechanism and physiology. Cytokine Growth Factor Rev 11:59–69
Elzie CA, Murphy-Ullrich JE (2004) The N-terminus of thrombospondin: the domain stands apart. Int J Biochem Cell Biol 36:1090–1101
Goldblum SE, Young BA, Wang P, Murphy-Ullrich JE (1999) Thrombospondin-1 induces tyrosine phosphorylation of adherens junction proteins and regulates an endothelial paracellular pathway. Mol Biol Cell 10:1537–1551
Robert DD, Miller TW, Rogers NM, Yao M, Isenberg JS (2012) The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol 31:162–169
Hess D, Keusch JJ, Lesnik Oberstein SA, Hennekam RC, Hofsteenge J (2008) Peter plus syndrome is a new congenital disorder of glycosylation and involves defective O-glycosylation of thrombospondin type 1 repeats. J Biol Chem 283:7354–7360
Heinonen TY, Maki M (2009) Peters’-plus syndrome is a congenital disorder of glycosylation caused by a defect in the beta1,3-glucosyltransferase that modifies thrombospondin type 1 repeats. Ann Med 41:2–10
Shimizu R, Saito R, Hoshino K, Ogawa K, Negishi T, Nishimura J, Mitsui N, Osawa M, Ohashi H (2010) Severe Peters Plus syndrome-like phenotype with anterior eye staphyloma and hypoplastic left heart syndrome: proposal of a new syndrome. Congenit Anom (Kyoto) 50:197–199
Hanna NN, Eickholt K, Agamanolis D, Burnstine R, Edward DP (2010) Atypical Peters plus syndrome with new associations. J AAPOS 14:181–183
Eberwein P, Reinhard T, Agostini H, Poloschek CM, Guthoff R, Auw-Haedrich C (2010) Intensive intracorneal keloid formation in a case of Peters plus syndrome and in Peters anomaly with maximum manifestation. Ophthalmologe 107:178–181
Oldberg Å, Antonssen P, Lindholm K, Heinegård D (1992) COMP (cartilage oligomeric matrix protein) is structurally related to thrombospondins. J Biol Chem 267:22346–22350
Rock MJ, Holden P, Horton WA, Cohn DH (2010) Cartilage oligometric matrix protein promotes cell attachment via two independent mechanisms involving CD47 and αVβ3 integrin. Mol Cell Biochem 338:215–224
Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD (2001) Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem 276:6046–6055
Rosenberg K, Olsson H, Mörgelin M, Heinegård D (1998) Cartilage oligomeric matrix protein shows high affinity zinc-dependent interaction with triple helical collagen. J Biol Chem 273:20397–20403
Di Cesare P, Hauser N, Lehman D, Pasumarti S, Paulsson M (1994) Cartilage oligomeric matrix protein (COMP) is an abundant component of tendon. FEBS Lett 354:237–240
Heinegård D (2009) Proteoglycans and more – from molecules to biology. Int J Exp Path 70:575–586
Smith RKW, Zunino L, Webbon PM, Heinegård D (1997) The distribution of cartilage oligomeric matrix protein (COMP) in tendon and its variation with tendon site, age and load. Matrix Biol 16:255–271
Södersten F, Hultenby K, Heinegård D, Johnston C, Ekman S (2013) Immunolocalization of collagens (I and III) and cartilage oligomeric matrix protein in the normal and injured equine superficial digital flexor tendon. Connect Tissue Res 54:62–69
Halasz K, Kassner A, Morgelin M, Heinegård D (2007) COMP as a catalyst in collagen fibrillogenesis. J Biol Chem 282:31166–31173
Hesselstrand R, Kassner A, Heinegård D, Saxne T (2008) COMP: a candidate molecule in the pathogenesis of systemic sclerosis with a potential as a disease marker. Ann Rheum Dis 67:1242–1248
Smith MR, Wright IM, Minshall GJ, Dudhia J, Verheyen K, Heinegård D, Smith RK (2011) Increased cartilage oligomeric matrix protein concentrations in equine digital flexor tendon sheath synovial fluid predicts interthecal tendon damage. Vet Surg 40:54–58
Wang L, Wang X, Kong W (2010) ADAMTS-7, a novel proteolytic culprit in vascular remodeling. Sheng Li Xue Bao 62:285–294
Riessen R, Fenchel M, Chen H, Axel DL, Karsch KR, Lawler J (2001) Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 21:47–54
Riessen R, Isner JM, Blessing E, Loushin C, Nikol S, Wight TN (1994) Regional differences in the distribution of the proteoglycans biglycan and decorin in the extracellular matrix of atherosclerotic and restenotic human coronary arteries. Am J Pathol 144:962–974
Du Y, Wang Y, Wang L, Liu B, Tian Q, Liu CJ, Zhang T, Xu Q, Zhu Y, Ake O, Qi Y, Tang C, Kong W, Wang X (2011) Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2. Circ Res 108:917–928
Posey KL, Hecht JT (2008) The role of cartilage oligomeric matrix protein (COMP) in skeletal disease. Curr Drug Targets 9:869–877
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Halper, J., Kjaer, M. (2014). Basic Components of Connective Tissues and Extracellular Matrix: Elastin, Fibrillin, Fibulins, Fibrinogen, Fibronectin, Laminin, Tenascins and Thrombospondins. In: Halper, J. (eds) Progress in Heritable Soft Connective Tissue Diseases. Advances in Experimental Medicine and Biology, vol 802. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7893-1_3
Download citation
DOI: https://doi.org/10.1007/978-94-007-7893-1_3
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7892-4
Online ISBN: 978-94-007-7893-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)