
Abstract
Breast cancer is the second leading cause of cancer death in women and metastasizes to bone in greater than 80 % of advanced-disease patients. Once breast cancer bone metastases are established, the disease is incurable and drives numerous complications that increase morbidity and diminish patients’ quality of life. Many mechanisms have been implicated in bone metastases of breast cancer. The critical role of Wnt signalling pathway inhibition in initiating bone lesions has been demonstrated in a variety of bone diseases and tumours. Overexpression of dickkopf-1 (Dkk1) protein, a negative regulator of the Wnt/β-catenin pathway, has been found in breast cancer cell lines that form osteolytic metastases preferentially and in serum from breast cancer patients with osteolytic bone metastases. Further understanding of the mechanistic role of Dkk1 as a link between primary breast tumours and secondary osteolytic bone metastases may facilitate development of anti-Dkk1 antibody therapeutic tools.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Breast cancer continues to be the most frequently diagnosed malignancy among women, with an estimated 1.38 million new cases being diagnosed worldwide each year. There are 458,000 deaths per year from breast cancer, making it the most common cause of female cancer death [1]. Bone is the preferred site of metastatic recurrence, arising in greater than 80 % of patients with advanced breast cancer [2]. Complications resulting from bone metastases include pain, reduced mobility, and reduced quality of life. In addition, patients are at considerable risk of skeletal related events (SREs), such as hypercalcemia, fracture, and spinal cord compression, and often require surgery, radiotherapy or both [3]. Metastatic sequelae account for approximately two-thirds of the costs associated with breast cancer treatment [4].
Elucidation of the fundamental mechanisms responsible for breast cancer metastasis to bone, including identifying specific biomarkers of inter-/intra-tumour spatial and metastatic potential, is required to improve patient risk stratification such that the best current therapies for particular patients can be selected. Moreover, a better mechanistic understanding may reveal promising new therapeutic targets. This review discusses the available evidence for the role of the Dkk1 protein, a Wnt signalling inhibitor, in breast cancer-induced bone metastasis and the potential therapeutic benefits of Dkk1 antibody therapy as a new strategy for decreasing breast cancer burden in osteolytic bone metastases.
Pathophysiology of bone metastases
Physiologic bone homeostasis is the result of the coordinated activities of three separate cell lineages: the haematopoietic stem cell lineage, which leads to the formation of bone-resorbing osteoclasts, the mesenchymal stem cell lineage, which leads to the formation of bone-forming osteoblasts, and the bone-maintaining osteocytes [5]. Osteoblasts and their precursors express several mediators that regulate osteoclastogenesis and osteoclast activity. The receptor activator of nuclear factor kappa B ligand (RANKL), a member of the tumor necrosis factor (TNF) receptor superfamily, is produced both in membrane-bound and soluble forms. Binding of RANKL to transmembrane receptor activator of nuclear factor kappa B (RANK) on the cell surface of osteoclasts and osteoclast precursors promotes their proliferation and maturation. Osteoprotegerin (OPG), another member of the TNF receptor superfamily, functions as a potent anti-osteoclastogenic cytokine by acting as a competitive decoy receptor for RANKL, thereby inhibiting RANK-RANKL interaction [6, 7]. The OPG/RANKL/RANK triad of proteins has been shown in genetic and pharmacologic studies to play a critical role in bone resorption [8]. Furthermore, macrophage colony stimulating factor, which is produced by several cell types including osteoblasts and stromal cells, activates an intracellular cascade upon binding its receptor c-fms, which is expressed on the surface of osteoclastic cells. This binding leads to proliferation and differentiation of osteoclast precursors and survival of mature osteoclasts [9]. Other osteoblast-produced cytokines including TNF, interleukin (IL)-1, IL-6 and IL-7 have been shown to play important roles in amplifying osteoclastogenesis and intensifying osteoclastic resorption [10].
The coupling of bone resorption to bone formation is essential for the correct function and maintenance of the skeletal system. The arrival of cancer cells within the bone microenvironment perturbs the resorption-formation balance, leading to excess bone loss or formation. The ability of cancer cells to metastasize is characteristic of advanced disease and occurs only after the gradual accumulation of a necessary set of pro-metastatic mutations [11]. Primary breast tumours are heterogeneous in nature, and cancer cells with vastly distinct capacities can exist within a single tumour. Large scale gene expression analyses and microarrays have identified several gene signatures that can distinguish between non-metastatic and metastatic cells derived from the same primary tumour [12, 13], and have identified genes that cooperate in breast cancer metastasis to bone [14, 15]. Cancer cell metastasis is a multistep process consisting of local invasion and intravasation at the primary site, survival in systemic circulation, and extravasation and colonization at the distant sites [16]. Once distant from the primary tumour site and resident in the bone marrow, cancer cells and other cell types within the bone microenvironment establish tumour cell micrometastatic foci within the marrow or the so-called bone metastatic niche [17]. Resident metastatic cells secrete various factors that promote the release of growth factors from the bone matrix, creating a vicious cycle that renders bone metastases incurable [18].
Bone metastases in breast cancer are characterized primarily by increased osteoclast activity and bone destruction [19]. Osteolytic bone metastases are present in 80 % of patients with stage IV disease [20]. Metastatic breast cells secrete factors that are capable of both RANKL-dependent and -independent activation of osteoclast formation, leading to bone resorption. Tumour-derived Jagged1 engages Notch pathway receptors in pre-osteoclasts directly, promoting their differentiation into mature, multinucleated osteoclasts [21]. On the other hand, parathyroid hormone-related protein (PTHrP) increases bone resorption indirectly by stimulating RANKL expression and inhibiting OPG expression by osteoblasts and bone stromal cells [22]. Inhibition of PTHrP with neutralizing antibodies has been shown to reduce osteolytic lesions produced by MDA-MB-231, a subpopulation of breast cancer cells that have the potential to induce osteolytic bone, in mouse models [23]. Elevated expression of both PTHrP and CXCR4A (a member of the chemokine superfamily that regulates cell migration and targeting) were identified in a study of breast cancer patients who developed skeletal metastases [24]. Li et al. 23–25 demonstrated that PTHrP ablation not only delays breast cancer initiation and primary tumour progression, but also inhibits expression of the metastasis marker CXCR4 in primary breast tumours. IL-6 also increases osteoclast formation and activity via the RANK ligand pathway, whereas IL-8 acts directly and indirectly on osteoclasts [26, 27].
As bone is resorbed, growth factors that are stored in bone matrix, such as insulin like-growth factor-I and transforming growth factor-β (TGF-β), are released and stimulate the proliferation of breast cancer cells. TGF-β is released in its active form during osteoclastic resorption and stimulates PTHrP production by tumour cells [28]. In addition, TGF-β regulates several genes that are responsible for enhanced bone metastases such as IL-11 and connective tissue growth factor [29]. Bone loss can result from increased bone resorption as well as decreased bone formation. Wnt proteins and bone morphogenetic proteins (BMPs) are important regulators of osteoblast activity and proliferation [30, 31]. Gregory et al. proved that the injection of the MDA-MB-231 human breast cancer cells into bone tissue of immunodeficient mice caused a significant down-regulation of osteoblast activity in the bone remodeling cycle. mRNA expression of dickkopf-1 (Dkk1) and Noggin, inhibitors of Wnt signalling and BMP signalling, respectively, was confirmed in the MDA-MB-231 cell line [32].
Although ongoing clinical trials targeting breast cancer bone metastases may identify effective treatments, further study of the molecular interactions between invading tumour cells and host bone cells is required to inform the development of new effective treatments for this challenging clinical problem. This review is focused on the role of Wnt signalling in breast cancer-induced bone metastases with special attention being given to its inhibitor, the Dkk1 protein.
Wnt signalling in breast cancer and bone metastases
Wnt signalling
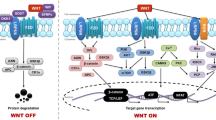
The Wnt family consists of 19 members that share a signal sequence of approximately 350 amino acids with a conserved pattern of 23–24 cysteine residues [33]. They act in a variety of cellular processes in both the adult organism and the developing embryo, including cell growth, cell proliferation and motility, generation of cell polarity, and apoptosis [34, 35]. Historically, Wnt proteins have been grouped into two classes, canonical and noncanonical, based on their activity in cell lines or in vivo assays. Noncanonical Wnts activate the planar cell polarity pathway (PCP) and the Wnt/Ca+2 pathway [36]. Canonical Wnts stabilize β-catenin, thereby activating transcription of T cell factor/lymphoid enhancing factor (TCF/LEF). In the absence of Wnts, glycogen synthase kinase (GSK3β), axin, adenomatous polyposis coli (APC), and casein kinase I (CKI) form the β-catenin destruction complex, which phosphorylates β-catenin, enabling it to be degraded by proteasomes. In the ‘off state’, extracellular Wnt ligands can interact with various secreted antagonists and cells maintain low cytoplasmic and nuclear levels of β-catenin. When Wnt concentrations exceed the buffering capacity of Wnt inhibitors, Wnt signalling is initiated by binding of the Wnt protein to one of the 10 members of the frizzled (FZD) receptor family; Wnt-FZD then binds low-density lipoprotein-related protein-5 (LRP5) or LRP6. The resultant complex activates Dishevelled (Dvl), a protein that draws Axin away from the destruction complex and antagonizes its ability to phosphorylate β-catenin, thereby preventing β-catenin destruction. Thus the ‘on state’ involves increasing the post-translational stability of β-catenin. As β-catenin levels rise, the protein accumulates and translocates to the nucleus, where it interacts with TCF/LEF transcription factors and enhances expression of their target genes (Fig. 1) [37]. The relative activation of the canonical or noncanonical signalling pathways depends on the receptor complement [38, 39].

Canonical Wnt/β-catenin signalling pathway. In the on state, canonical Wnt signalling is activated. Wnt protein binds to one of the 10 members of the frizzled receptor family; Wnt-FZD then binds LRP5 or LRP6. The resultant complex activates Dvl, a protein that draws axin away from the destruction complex and antagonizes its ability to phosphorylate β-catenin, thereby preventing β-catenin degredation. If β-catenin is not degraded, it accumulates and translocates to the nucleus where it binds to the TCF/LEF transcription factor and enhances target gene expression. In the off state, canonical Wnt signalling is inactivated. In the presence of Wnt antagonists (Dkk1, SFRP or WIF), the phosphorylation complex (GSK3β, CK1, Axin and APC kinases) becomes active and phosphorylates β-catenin
Wnt pathway activation is modulated by several secreted protein families that can be divided into two functional classes. The first class includes the frizzled related protein (sFRP) family and Wnt inhibitory protein (WIF) family, which bind directly to Wnts, thereby functioning as Wnt antagonists for both canonical and noncanonical signalling [40]. The second class includes the dickkopf (Dkk) family and the sclerostin (SOST) family that bind to the LRP5/LRP6 component of the Wnt receptor complex, inhibiting Wnt/β-catenin signalling [41].
Wnt signalling and bone homeostasis
Wnts play a central role in controlling embryonic bone development and bone mass [42]. They are also essential in postnatal bone regenerative processes, such as ectopic bone formation and fracture repair [43]. Osteoblasts are the main cellular targets of Wnt actions in bone. Canonical Wnt signalling controls osteoblasts on several levels. Firstly, Wnt signalling can affect osteoblast commitment by blocking adipogenesis and directing mesenchymal progenitors to become either osteoblasts or chondrocytes [44]. Secondly, Wnt signalling modulates osteoblast proliferation. Specifically, the Wnt pathway components, Dkk1 and sFRPs, are upregulated markedly during the late phase of osteoblast differentiation, suggesting that a negative Wnt feedback loop may control the last steps of osteoblast maturation [45]. Thirdly, Wnt signalling affects osteoblast function. LRP5-deficient mice display a decrease in bone matrix deposition [46], and osteoblasts overexpressing a constitutively active mutant of β-catenin show an increase expression of the collagens type I α1 and α2 genes. Moreover, Wnt signalling increases the expression of OPG in osteoblasts and stromal cells, whereas β-catenin-deficient osteoblasts exhibit elevated expression of RANKL and diminished expression of OPG [47]. Thus, osteoblast-selective deficiency of β-catenin affects bone resorption as well as bone formation.
The Wnt/β-catenin pathway has previously been shown to be critically involved in other forms of bone malignancy, including multiple myeloma (MM) [48] and prostate cancer bone metastasis [49]. Hence, it is reasonable to suspect that this pathway may be important in breast cancer-bone metastasis as well.
Wnt signalling in breast cancer and its metastasis into bone
Wnt signalling has emerged as a critical mediator of cell–cell singling events during both embryogenesis and adult tissue maintenance, and the association of deregulated Wnt/β-catenin signalling with cancer has been well documented. β-catenin is a multifunctional protein involved in both cell–cell adhesion and signal transduction [50]. Constitutive activation of β-catenin signalling leads to excessive stem cell renewal/proliferation that predisposes cells to tumorigenesis [51]. Wnt signalling appears to be involved in numerous aspects of mammary development, including cell fate determination, maintenance of mammary progenitor cell populations, branching morphogenesis and alveolar differentiation [52, 53].
The identification of Wnt1 as a key oncogene in naturally occurring mouse mammary tumours [54] led to intense investigation of Wnts and the potential involvement of their signalling in breast cancer over the last three decades. Mutation of APC and overexpression of a stabilized mutant of β-catenin induced mammary tumorigenesis in a mouse model [55]. Wnt/β-catenin pathway dysregulation, as evidenced by abnormal Wnt expression, Wnt antagonist secretion, and APC inactivation, has been observed in human breast cancer [56–58]. Moreover, with respect to β-catenin itself, nuclear β-catenin has been observed in as many as 63 % of breast cancers [59]. Nuclear staining of β-catenin and overexpression of its downstream target cyclin D1 have been associated with a worse prognosis of breast cancer and with metastasis [60, 61]. Moreover, Chen et al. showed that TM40D-MB breast cancer cells, which have a high potential for bone metastasis, exhibit significantly higher endogenous β-catenin signalling activity than TM40D cells, which do not metastasize to bone. In addition, they observed that inactivation of the β-catenin pathway inhibited osteoblast differentiation in a tumour-bone co-culture system, as indicated by decreased alkaline phosphatase activity [62].
Although there is strong evidence, reviewed above, indicating that Wnt signalling in cancerous breast tissue can drive tumour cell growth and invasiveness, the underlying mechanisms mediating these effects remain unclear. Johnson et al. showed that induction of Wnt/β-catenin signalling in highly metastatic breast cancer cells significantly increased Gli2 and PTHrP gene expression and promoter activity [63]. PTHrP promotes the release of TGF-β, which in turn upregulates tumour-derived Gli2 and PTHrP expression and stimulates tumour cell proliferation in bone [64].
Epithelial-mesenchymal transition (EMT) is an essential developmental process that enables reprogramming of polarized epithelial cells towards a motile, mesenchymal phenotype. Aberrant EMT activation can endow cancer cells with the migratory and invasive capabilities associated with metastatic competence [65]. Moreover, tumour progression is driven by a small subpopulation of cancer cells termed cancer stem cells (CSCs) that exhibit the ability to self-renew and to regenerate the phenotypic heterogeneity of the parental tumour [66]. Mani et al. found that induction of EMT also generates cells with stem-cell-like properties [67]. In addition, studies suggest that Wnt signalling contributes to the induction and maintenance of CSC states activated by the EMT program [68]. Lamb et al. found that Wnt pathway gene expression was increased in malignant breast tissue compared to normal breast tissue, and this expression was predictive for recurrence within subtypes of breast cancer. Furthermore, activation of Wnt signalling was significantly higher in breast cancer stem cell-enriched populations than in normal breast stem cell-enriched populations [69]. These findings suggest that Wnt activation might be limited to a subpopulation of cancer initiating stem cells.
Conversely, other studies failed to find evidence for Wnt pathway activation in human breast tumours [60, 70], or even an association between β-catenin expression and outcome or metastasis [71, 72]. This discrepancy between findings may be due to tumours not being classified into molecular subtypes. Understanding the molecular mechanisms by which Wnt/β-catenin signalling components can act in the bone-tumour microenvironment is important, biologically as well as clinically, for the future development of anti metastatic strategies.
Dkk1 in breast cancer and bone metastases
Dkk1 and bone homeostasis
The Dkk family consists primarily of four secreted proteins in vertebrates (Dkk1, 2, 3, 4) [73]. The most studied member of the family is Dkk1 protein which was discovered due to its ability of blocking Wnt signalling required for head induction during early Xenopus embryogenesis [74]. Dkk1 prevents activation of Wnt signalling by binding to the Wnt co-receptor LRP5/6 [75]. Dkk1 also interacts with the single-pass transmembrane receptor proteins Kremen1 and Kremen2 [76] (Fig. 1). Thus, Dkk1 forms a ternary Dkk1/LRP6/Kremen complex, which promotes endocytosis of LRP, making it unavailable for interaction with Wnt. This Wnt signalling modulation can be achieved by Dkk1, 2, and 4 proteins which bind the same effectors (LRPs or Kremens), but not Dkk3, which does not block canonical Wnt signalling. Dkk4 appears to be functionally indistinguishable from Dkk1, whereas Dkk2 appears to be a poor inhibitor of Wnt signalling [77], perhaps in part, because Dkk2 cannot be expressed at the same high levels achieved by Dkk1. In addition, Dkk2 seems to activate Wnt/β-catenin signalling in Xenopus embryos [78, 79]. Therefore, we will focus on the involvement of Dkk1 in breast cancer-bone metastasis.
Binding of Dkk1 to LRP5 is a key regulator for bone mass. In humans, LRP5 gain-of-function mutations are associated with high bone mass [80], whereas loss-of-function mutations lead to osteoporosis pseudoglioma syndrome, which is characterised by low bone density [81]. Reduced expression of Dkk1 in mice haploinsufficient for the Dkk1 gene results in a high-bone-mass phenotype, whereas transgenic mice overexpressing Dkk1 exhibit osteopenia [82, 83]. Finally, diverse evidence indicates that Dkk1 is also a major determinant of bone and joint pathology in inflammatory arthritis. Dkk1 neutralisation in TNF transgenic mice was found to provide complete protection from inflammatory bone loss by preventing TNF-mediated functional impairment of osteoblast and enhanced osteoclast activity [84]. Weng et al. showed that attenuation of Dkk1 expression in cartilage and subchondral bone tissue promotes expression of β-catenin and survival of chondrocytes and osteoblasts in the osteoarthritic joint microenvironment [85].
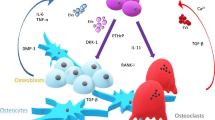
Several lines of evidence demonstrate that Dkk1 counteracts Wnt-mediated effects on bone via stimulation of osteoclast activity and inhibition of osteoblast formation and differentiation (Fig. 2). Constitutive expression of Dkk1 has been shown to promote adipogenic differentiation in 3T3-L1 preadipocytes [86]. In vitro examination of C3H10T1/2 osteoprogenitor cells revealed that Msx2, a homeodomain transcription factor first identified in osteoblasts, inhibited Dkk1 promotor activity and reduced RNA polymerase association with Dkk1 chromatin [87]. In addition, Dkk1-mediated inhibition of Wnt signalling was found to limit OPG expression, thereby shifting the OPG:RANKL ratio in favour of bone resorption [88].

Wnt signalling and Dkk1 in bone development. Wnt signalling enhances bone formation by directing the developmental program of mesenchymal stem cells toward osteoblast formation. Mature osteoblasts upregulate OPG, which blocks RANKL-induced osteoclastogenesis, resulting in inhibition of bone resorption. Dkk1 inhibits osteoblast formation and differentiation by diverting progenitors toward adipogenesis. Preosteoblasts enhance bone resorption by boosting RANKL-induced osteoclastogenesis
Dkk1 in breast cancer and its metastasis into bone
Dkk1 overexpression in solid tumours is associated with worse survival [89]. However, the significance of Dkk1 expression in breast cancer progression and prognosis remains inconclusive. Some studies have shown that Dkk1 acts as a putative tumour suppressor in breast cancer cells via the suppression of Wnt signalling [90, 91] or via mechanisms independent of β-catenin-dependent transcription [92]. As mentioned above, the Wnt pathway influences self-renewal in the context of stem cells and cancer. Agur et al. showed that high concentrations of Dkk1 decreased mammosphere formation in both primary breast cancer cells and breast cancer cell lines by diverting proliferating cancer stem cells toward differentiation. Consequently, Dkk1 represents a potential target for ablation in differentiation therapy [93]. On the other hand, Sato et al. observed elevated expression of Dkk1 in four out of six human breast cancer types and found that 65.1 % (110/169) of breast cancer patients examined in their study had Dkk1 positive serum [94]. Forget et al. showed that Dkk1 is preferentially expressed in ER and PR-negative tumours, in tumours from women with a family history of breast cancer, and in primary tumours from patients with axillary lymph node invasion [95]. In addition, increased expression of Dkk1 was confirmed in hormone-resistant breast cancer cell lines, and Dkk1 expression in triple negative cancers was associated with poor outcome in these patients [96]. Moreover, elevated Dkk1 levels in the serum of breast cancer patients have been associated with shorter overall survival and relapse-free survival [97]. These contradicting observations may be due to differences in tumour type, tumour stage, tissue origin (epithelial or mesenchymal), or cellular subtypes.
Bone is the most frequent site of metastasis for several forms of cancer, including breast cancer. Evidence suggests that Wnt signalling and Dkk1 are involved in bone metastasis. Breast cancer-induced bone metastases are typically osteolytic, but occasionally osteoblastic lesions can occur. The most extensive data suggesting that Dkk1 promotes osteolytic metastasis come from studies of MM-associated bone disease [98, 99]. Few studies have investigated the role of Dkk1 in bone metastasis secondary to breast cancer showed that Dkk1 serum levels in women with breast cancer Voorzanger-Rousselot et al. and bone metastasis were higher than those in healthy age-matched controls and in women with metastases at sites other than bone [100]. Dkk1 was expressed by osteolytic breast cancer cell lines but not by osteoblastic lines. A subpopulation of breast cancer MDA-MB-231 cells known as the MDA-MB-231/bone cell line (MDA-231-BO) metastasize exclusively in bone and produce larger osteolytic lesions than the parental line [101]. Mice inoculated with MDA-231-BO cells, which developed radiologic and histologic evidence of skeletal lesions, had six-fold higher bone marrow levels of Dkk1 than control non-inoculated mice [102]. Moreover, Bu et al. demonstrated that MDA- MDA-231-BO cells exhibit increased levels of Wnt/β-catenin signalling and Dkk1 expression compared with MDA-MB-231 cells, and these changes were associated with inhibition of osteoblast differentiation and OPG expression. These effects could be neutralized by a specific anti-Dkk1 antibody [103]. Taken together, these observations indicate that Dkk1 may have a pathophysiological role in skeletal metastasis of breast cancer.
Some studies have demonstrated that Dkk1 is a downstream target of β-catenin-mediated transcriptional activity in several cell lines [104]. Nevertheless, because Dkk1 is relatively recently discovered inhibitor of Wnt/β-catenin signalling, the mechanisms by which breast cancer cells can avoid Dkk1 inhibition have to be identified. Menezes et al. postulated that excessive Dkk1 may accumulate in cancer cells but be unable to regulate the Wnt pathway due to malfunction of some component downstream of Dkk1. However, in surrounding bone cells with intact Wnt/β-catenin signalling, Dkk1 appears to function normally when it is up-regulated in a paracrine fashion [105]. If this hypothesis is confirmed experimentally, new therapeutic strategies to neutralize Dkk1 might be used in the treatment of breast cancer-induced bone metastasis.
The specific targeting of Wnt activation in bone may be achievable by targeting Dkk1 or SOST. Like Dkk1, SOST inhibits osteoblast differentiation by binding the Wnt co-receptors LRP5/6 on the surface of osteoblasts [106]. Indeed, SOST-neutralizing antibodies have been shown to have a strong anabolic effect in osteoporotic patients [109]. Mendoza-Villanueva et al. 109 showed that the Runt-related transcription factor 2, contributes to the formation of osteolytic bone metastases in breast cancer through induction of SOST expression [83], which is restricted to osteocytes [107]. Likewise, although Dkk1 is expressed widely during development, it is relatively restricted to bone (osteoblasts and osteocytes) in adult mice [108]. Thus, systemic administration of Dkk1 or SOST antagonists, may affect bone tissue selectively, favouring endogenous Wnt signalling-mediated increase in bone formation without affecting Wnt signalling in other tissues. There have not yet been clinical trials testing SOST antibody effects in cancer-induced bone diseases. Therefore, further studies are required to determine whether SOST contributes to the development of bone metastases in vivo.
Therapeutic approaches
Currently, approved pharmaceutical approaches for targeting bone metastases are limited to agents that interfere with osteoclasts-mediated bone resorption, including bisphosphonates and anti-RANKL antibody denosumab [110]. However, the discovering that osteolytic lesions result not only from enhanced osteoclast-mediated bone resorption, but also from inhibition of bone formation led to the development of therapeutic strategies aimed at restoring osteoblast function. A number of preclinical in vitro and in vivo studies have defined the role of Dkk1 antibodies as a potential therapy for MM-associated bone disease [111]. In addition, two different anti-Dkk1 antibodies are being tested clinically in phase 1 and 2 trials, respectively, in patients with MM. Although, final results from these studies have not been published yet, preliminarily, they indicate a favourable safety profile and proof of anabolic activity [112, 113]. In contrast to myeloma, the role of Dkk1 antibodies in the treatment of breast cancer bone metastasis is less well characterised.
In a study by Rachner et al., treatment of breast cancer cells with zoledronic acid regulated alkaline phosphatase and OPG production arising from Dkk1 suppression via inhibition of protein geranylgeranylation. In line with the in vitro data, breast cancer patients receiving adjuvant zoledronic acid exhibited a 60 % decrease in serum Dkk1 levels after 12 months of treatment [114]. Another recent study showed that postmenopausal breast cancer patients treated with aromatase inhibition had a modest decrease in Dkk1 serum levels which correlated with increased bone mineral density of the femoral neck and the total hip [115]. Experimental stimulation of bone turnover has been shown to increase skeletal metastases in several animal models, suggesting that high bone turnover should be countered with anti-resorptive drugs [116]. Taken together, the literature suggests that Dkk1 is a mediator of malignant bone disease and that further studies concerning its potential as a novel therapeutic target are warranted.
References
Ferlay J, Shin HR, Bray F et al (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12):2893–2917
Kozlow W, Guise TA (2005) Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia 10:169–180
Mundy GR (2002) Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2:584–593
Lipton A (2005) Management of bone metastases in breast cancer. Cur Treat Options Oncol 6:161–171
Aubin JE, Triffitt J (2002) Mesenchymal stem cells and the osteoblast lineage. In: Principles of bone biology, 2nd edn. Academic Press, New York, pp 59–81
Suda T, Takahashi N, Udagawa N et al (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 20:345–357
Simonet WS, Lacey DL, Dunstan CR et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Boyce BF, Xing L (2008) Functions of RANKL/RANK/OPG in bone modelling and remodelling. Arch Biochem Biophys 473:139–146
Quinn JM, Saleh H (2009) Modulation of osteoclast function in bone by the immune system. Mol Cell Endocrinol 310:40–51
Weitzmann MN (2013) The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica. doi:10.1155/2013/125705
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3:1–6
Yang J, Mani SA, Donaher JL et al (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastases. Cell 117:927–939
Eckhardt BL, Parker BS, van Laar RK et al (2005) Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res 3:1–13
Bohn OL, Nasir I, Brufsky A et al (2009) Biomarker profile in breast carcinomas presenting with bone metastasis. Int J Clin Exp Pathol 3:139–146
Ota D, Mimori K, Yokobori T et al (2011) Identification of recurrence-related microRNAs in the bone marrow of breast cancer patients. Int J Oncol 38:955–962
Patel LR, Camacho DF, Shiozawa Y et al (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68:3645–3654
Onder TT, Gupta PB, Mani SA et al (2005) Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. Mammary Gland Biol Neoplasia 10:169–180
Guise TA, Kozlow WM, Heras-Herzig A et al (2005) Molecular mechanisms of breast cancer metastases to bone. Clin Breast Cancer 5:46–53
Kakonen SM, Mundy GR (2003) Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 97:834–839
Kozlow W, Guise TA (2005) Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. Mammary Gland Biol Neoplasia 10:169–180
Sethi N, Dai X, Winter CG, Kang Y (2011) Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19:192–205
Karaplis A, Goltzman D (2000) PTH and PTHrP effects on the skeleton. Rev Endocr Metab Disord 1:331–341
Li J, Karaplis AC, Huang DC et al (2011) PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Investig 121:4655–4669
Bohn OL, Nasir I, Brufsky A et al (2010) Biomarker profile in breast carcinomas presenting with bone metastasis. Int J Clin Exp Pathol 3:139–146
Guise TA, Yin JJ, Taylor SD et al (1996) Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Investig 98:1544–1549
de la Mata J, Uy HL, Guise TA et al (1995) Interleukin-6 enhances hypercalcemia and bone resorption mediated by parathyroid hormone-related protein in vivo. J Clin Investig 95:2846–2852
Bendre MS, Margulies AG, Walser B et al (2005) Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res 65:11001–11009
Dallas SL, Rosser JL, Mundy GR et al (2002) Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem 277:21352–21360
Kang Y, Siegel PM, Shu W et al (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3:537–549
Feeley BT, Gamradt SC, Hsu WK et al (2005) Influence of BMPs on the formation of osteoblastic lesions in metastatic prostate cancer. J Bone Miner Res 20:2189–2199
Dai J, Keller J, Zhang J et al (2005) Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res 65:8274–8285
Gregory LS, Choi W, Burke L et al (2013) Breast cancer cells induce osteolytic bone lesions in vivo through a reduction in osteoblast activity in mice. PLoS One. doi:10.1371/journal.pone.0068103
Milat F, Ng KW (2009) Is Wnt signalling the final common pathway leading to bone formation? Mol Cell Endocrinol 310:52–62
Gordon MD, Nusse R (2006) Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem 281:22429–22433
Cadigan KM, Liu YI (2006) Wnt signalling: complexity at the surface. J Cell Sci 119:395–402
Miller JR (2002) The Wnts. Genome Biol 3:3001.1–3001.15
Clevers H (2006) Wnt/β-Catenin signaling in development and disease. Cell 127:469–480
van Amerongen R, Mikels A, Nusse R (2008) Alternative Wnt signaling is initiated by distinct receptors. Sci Signal. doi:10.1126/scisignal.135re9
Bryja V, Andersson ER, Shambony A et al (2009) The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol Biol Cell 20:924–936
Bovolenta P, Esteve P, Ruiz JM et al (2008) Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci 121:737–746
He X, Semenov M, Tamai K et al (2004) LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling: arrows point the way. Development 131:1663–1677
Krishnan V, Bryant HU, Macdougald OA (2006) Regulation of bone mass by Wnt signaling. J Clin Investig 116:1202–1209
Yan C, Benjamin A (2009) Wnt pathway, an essential role in bone regeneration. J Cell Biochem 106:353–362
Hill TP, Später D, Taketo MM et al (2005) Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 8:727–738
Vaes BLT, Dechering KJ, van Someren EP et al (2005) Microarray analysis reveals expression regulation of Wnt antagonists in differentiating osteoblasts. Bone 36:803–811
Kato M, Patel MS, Levasseur R et al (2002) Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol 157:303–314
Glass DA 2nd, Bialek P, Ahn JD et al (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8:751–764
Gunn WG, Conley A, Deininger L et al (2006) A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells 24:986–991
Hall CL, Daignault SD, Shah RB et al (2008) Dickkopf-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate 68:1396–1404
Barth AL, Näthke IS, Nelson WJ (1997) Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol 9:683–690
Fuchs E (2009) The tortoise and the hair: slow-cycling cells in the stem cell race. Cell 137:811–819
Brennan KR, Brown AMC (2004) Wnt proteins in mammary development and cancer. J Mammary Gland Biol Neoplasia 9:119–131
Hatsell S, Rowlands T, Hiremath M et al (2003) Beta-catenin and Tcfs in mammary development and cancer. J Mammary Gland Biol Neoplasia 8:145–158
Rijsewijk F, Schuermann M, Wagenaar E et al (1987) The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 14:649–657
Prosperi JR, Khramtsov AI, Khramtsova GF et al (2011) Apc mutation enhances PyMT-induced mammary tumorigenesis. PLOS One. doi:10.1371/journal.pone.0029339
Klopocki E, Kristiansen G, Wild PJ et al (2004) Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int J Oncol 25:641–649
Ai L, Tao Q, Zhong S et al (2006) Inactivation of Wnt inhibitory factor-1 (WIF1) expression by epigenetic silencing is a common event in breast cancer. Carcinogenesis 27:1341–1348
Zardawi SJ, O’Toole SA, Sutherland RL et al (2009) Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol Histopathol 24:385–398
Ryo A, Nakamura M, Wulf G et al (2001) Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol 3:793–801
Dolled-Filhart M, McCabe A, Giltnane J et al (2006) Quantitative in situ analysis of beta-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res 66:5487–5494
Wong SC, Lo SF, Lee KC et al (2002) Expression of frizzled-related protein and Wnt-signalling molecules in invasive human breast tumours. Br J Cancer 87:1281–1286
Chen Y, Shi HY, Stock SR et al (2011) Regulation of breast cancer-induced bone lesions by β-catenin protein signaling. J Biol Chem 286:42575–42584
Johnson RW, Merkel AR, Page JM et al (2014) PTHrP Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin Exp Metastasis 31:945–959
Johnson RW, Mai PN, Susan SP et al (2010) TGF-β promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical hedgehog signaling. Cancer Res 71:822–831
Thiery JP, Acloque H, Huang RY et al (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890
McDermott SP, MS Wicha (2010) Targeting breast cancer stem cells. Mol Oncol 4:404–419
Mani SA, Guo W, Mai-Jing L et al (2008) The epithelial-mesenchymal transition generates cells wit properties of stem cells. Cell 133:704–715
Scheel C, Eaton EN, Li SH et al (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145:926–940
Lamb R, Ablett MP, Spence K et al (2013) Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS One 8(7):e67811
Pedersen KB, Nesland JM, Fodstad O et al (2002) Expression of S100A4. E-cadherin, alpha- and beta-catenin in breast cancer biopsies. Br J Cancer 87:1281–1286
Gillett CE, Miles DW, Ryder K et al (2001) Retention of the expression of E-cadherin and catenins is associated with shorter survival in grade III ductal carcinoma of the breast. J Pathol 193:433–441
Bukholm IK, Nesland JM, Karesen R et al (1998) E-cadherin and alpha-, beta-, and gamma-catenin protein expression in relation to metastasis in human breast carcinoma. J Pathol 185:262–266
Zorn AM (2001) Wnt signalling: antagonistic Dickkopfs. Curr Biol 11:592–595
Glinka A, Wu W, Delius H et al (1998) Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 391:357–362
Mao J, Wu W, Li Y et al (2001) LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411:321–325
Mao B, Niehrs C (2003) Kremen2 modulates Dickkopf2 activity during Wnt/LRP6 signalling. Gene 302:179–183
Mao B, Wu W, Davidson G et al (2002) Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nature 417:664–667
Wu W, Glinka A, Delius H et al (2000) Mutual antagonism between dickkopf1 and dickkopf2 regulates Wnt/β-catenin signalling. Curr Biol 10:1611–1614
Brott BK, Sokol SY (2002) Regulation of Wnt/LRP signaling by distinct domains of Dickkopf proteins. Mol Cell Biol 22:6100–6110
Boyden LM, Mao J, Belsky J et al (2002) High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 346:1513–1521
Gong Y, Slee RB, Fukai N et al (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107:513–523
Morvan F, Boulukos K, Clement-Lacroix P et al (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21:934–945
Li J, Sarosi I, Cattley RC et al (2006) Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 39:754–766
Heiland GR, Zwerinal K, Baum W et al (2010) Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis 69:2152–2159
Weng LH, Wang CJ, Ko JY et al (2010) Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage destruction, and subchondral bone deterioration in osteoarthritic knees. Arthritis Rheum 62:1393–1402
Christodoulides C, Scarda A, Granzotto M et al (2006) WNT10B mutations in human obesity. Diabetologia 49:678–684
Cheng SL, Shao JS, Cai J et al (2008) Msx2 exerts bone anabolism via canonical Wnt signaling. J Biol Chem 283:20505–20522
Diarra D, Stolina M, Polzer K et al (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13:156–163
Liu Y, Tang W, Xie L et al (2014) Prognostic significance of dickkopf-1 overexpression in solid tumors: a meta-analysis. Tumour Biol 35:3145–3154
Zhou XL, Qin XR, Zhang XD et al (2010) Downregulation of Dickkopf-1 is responsible for high proliferation of breast cancer cells via losing control of Wnt/beta-catenin signaling. Acta Pharmacol Sin 31:202–210
Kim HY, Park JH, Won HY et al (2015) CBX7 inhibits breast tumorigenicity through DKK-1-mediated suppression of the Wnt/β-catenin pathway. FASEB J 29:300–313
Mikheey AM, Mikheeva SA, Maxwell JP et al (2008) Dickkopf-1 mediated tumor suppression in human breast carcinoma cells. Breast Cancer Res Treat 112:263–273
Agur Z, Kirnasovsky OU, Vasserman G et al (2011) Dickkopf1 regulates fate decision and drives breast cancer stem cells to differentiation: an experimentally supported mathematical model. PLoS One 6(9):e24225
Sato N, Yamabuki T, Takano A et al (2010) Wnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapy. Cancer Res 70:5326–5336
Forget MA, Turcotte S, Beauseigle D et al (2007) The Wnt pathway regulator DKK1 is preferentially expressed in hormone-resistant breast tumours and in some common cancer types. Br J Cancer 96:646–653
Xu WH, Liu ZB, Yang C et al (2012) Expression of dickkopf-1 and beta-catenin related to the prognosis of breast cancer patients with triple negative phenotype. PLoS One. doi:10.1371/journal.pone.0037624
Zhou SJ, Zhou SR, Yang XQ et al (2014) Serum Dickkopf-1 expression level positively correlates with a poor prognosis in breast cancer. Diagn Pathol 9:161
Hideshima T, Mitsiades C, Tonon G et al (2007) Understanding multiple myloma pathogenesis in the bone marrow to identify new therapeutics targets. Nat Rev Cancer 7:585–595
Qiang YW, Chen Y, Stephens O et al (2008) Myeloma derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: a potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 112:196–207
Voorzanger-Rousselot N, Journe F, Doriath V et al (2009) Assessment of circulating Dickkopf-1 with a new two-site immunoassay in healthy subjects and women with breast cancer and bone metastases. Calcif Tissue Int 84:348–354
Yoneda T, Williams PJ, Hiraga T et al (2001) A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res 16:1486–1495
Voorzanger-Rousselot N, Goehrig D, Journe F et al (2007) Increased Dickkopf-1 expression in breast cancer bone metastases. Br J Cancer 97:964–970
Bu G, Lu W, Liu CC et al (2008) Breast cancer-derived Dickkopf1 inhibits osteoblast differentiation and osteoprotegerin expression: implication for breast cancer osteolytic bone metastases. Int J Cancer 123:1034–1042
Niida A, Hiroko T, Kasai M et al (2004) DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 23:8520–8526
Menezes ME, Devine DJ, Shevde LA et al (2012) Dickkopf1: a tumor suppressor or metastasis promoter? Int J Cancer 130:1477–1483
Ellies DL, Viviano B, McCarthy J et al (2006) Bone density ligand. Sclerostin directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. Bone Miner 21:1738–1749
Kawai M, Mödder UI, Khosia S et al (2011) Emerging therapeutic opportunities for skeletal restoration. Nat Rev Drug Discov 10:141–156
Mendoza-Villanueva D, Zeef L, Shore P (2011) Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2-CBFβ-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res 13:R106
van Bezooijen DL, Roelen BAJ, Visser A et al (2004) Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 6:805–814
Rachner TD, Hadji P, Hofbauer LC (2012) Novel therapies in benign and malignant bone diseases. Pharmacol Ther 134:338–344
Pinzone JJ, Hall BM, Thudi NK et al (2009) The role of Dikkopf-1 in bone development, homeostasis, and disease. Blood 113:517–525
Rachner TD, Göbel A, Benad-Mehner P et al (2014) Dickkopf-1 as a mediator and novel target in malignant bone disease. Cancer Lett 346:172–177
Iyer SP, Beck JT, Ak Stewart et al (2014) A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br J Haematol 167:366–375
Rachner TD, Göbel A, Thiele S (2014) Dickkopf-1 is regulated by the mevalonate pathway in breast cancer. Breast Cancer Res. doi:10.1186/bcr3616
Kyvernitakis I, Rachner TD, Urbschat A et al (2014) Effect of aromatase inhibition on serum levels of sclerostin and dickkopf-1, bone turnover markers and bone mineral density in women with breast cancer. J Cancer Res Clin Oncol 140:1671–1680
Coleman RE, Guise TA, Lipton A et al (2008) Advancing treatment for metastatic bone cancer: consensus recommendations from the second Cambridge Conference. Clin Cancer Res 14:6387–6395
Funding
All authors declare that they have received no funding concerning this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Erich-Franz Solomayer holds a consultancy position at Novartis and Amgen and received compensation from Novartis, Amgen and Roche. Mariz Kasoha, Ingolf Juhasz-Boess, Daniel Herr and Jasmin Teresa Ney declare that they have no conflict of interests.
Ethical approval
No ethical approval was required.
Rights and permissions
About this article

Cite this article
Mariz, K., Ingolf, JB., Daniel, H. et al. The Wnt inhibitor dickkopf-1: a link between breast cancer and bone metastases. Clin Exp Metastasis 32, 857–866 (2015). https://doi.org/10.1007/s10585-015-9750-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-015-9750-1