
Abstract
Epigenetic mechanisms involving the modulation of gene activity without modifying the DNA bases are reported to have lifelong effects on mature neurons in addition to their impact on synaptic plasticity and cognition. Histone methylation and acetylation are involved in synchronizing gene expression and protein function in neuronal cells. Studies have demonstrated in experimental models of neurodegenerative disorders that manipulations of these two mechanisms influence the susceptibility of neurons to degeneration and apoptosis. In Alzheimer’s disease (AD), the expression of presenilin 1 (PSEN1) is markedly increased due to decreased methylation at CpG sites, thus promoting the accumulation of toxic amyloid-β (Aβ) peptide. In Parkinson’s disease (PD), dysregulation of α-synuclein (SNCA) expression is presumed to occur via aberrant methylation at CpG sites, which controls the activation or suppression of protein expression. Mutant Huntingtin (mtHTT) alters the activity of histone acetyltransferases (HATs), causing the dysregulation of transcription observed in most Huntington’s disease (HD) cases. Folate, vitamin B6, vitamin B12, and S-adenosylmethionine (SAM) are vital cofactors involved in DNA methylation modification; 5-azacytidine (AZA) is the most widely studied DNA methyltransferase (DNMT) inhibitor, and dietary polyphenols are DNMT inhibitors in vitro. Drug intervention is believed to reverse the epigenetic mechanisms to serve as a regulator in neuronal diseases. Nevertheless, the biochemical effect of the drugs on brain function and the underlying mechanisms are not well understood. This review focuses on further discussion of therapeutic targets, emphasizing the potential role of epigenetic factors including histone and DNA modifications in the diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many studies over the last decade have strongly implicated epigenetic mechanisms in the regulation of gene expression involved in the regulation of several ageing-related diseases, such as cancer and heart failure, and in promoting the alteration of gene expression responsible for the ageing process of different tissues. Therefore, alteration of the epigenetic mechanisms occurring during ageing renders cells more prone to the transcriptional changes responsible for ageing-related diseases (Lovrečić et al. 2013; Pagiatakis et al. 2019).
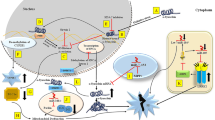
Neurodegeneration is a progressive loss of neuronal function that often results in cell death and brain dysfunction (Lu et al. 2013). The phenotypic effects are related to the specific areas encountering cell death (Armstrong and Barker 2001). Aberrant collaboration between proteins that bring about unusual intracellular and extracellular accumulation of self-aggregating misfolded proteins and the development of high-ordered insoluble fibrils are pathological features of many neurodegenerative diseases (Jellinger 2001). Important proteins involved in Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD) are amyloid precursor protein (APP), α-synuclein (SNCA), presenilin, tau, and Huntingtin (HTT) (Winner et al. 2011). SNCA is localized at the presynaptic terminal of neurons in the central nervous system (CNS) (Jakes et al. 1994) and dysfunction of this protein is a common hallmark in PD. The aggregation and deposition of these abnormal SNCA proteins in dopaminergic neurons have been postulated to be responsible for subsequent neurodegeneration (Recchia et al. 2004).
The presenilin 1 (PSEN1) gene is a key element in the creation of Aβ (De Strooper et al. 1998) via the proteolytic activity of γ-secretase (Vetrivel et al. 2006). Mutation in this gene alters the activity of the proteolytic enzyme and intensifies the accumulation of Aβ, which is generally found in AD patients. Tau proteins are crucial for maintaining normal cell function by interacting with tubulin in the formation of microtubules (Thomas and Fenech 2007). Hyperphosphorylation of tau protein leads to the formation of tangles, hence leading to the dissociation of the tau-tubulin complex and cell death (Thomas and Fenech 2007). Both Aβ and tangle formation are common in patients diagnosed with AD.
HTT is a multifunctional protein required for transcriptional modulation and intracellular transport and is associated with the endosome–lysosome pathway (Landles and Bates 2004). Mutant huntingtin (mtHTT) results in HD, as it causes bioenergetic failure, HD-linked neural dysfunction and cell death (Bossy-Wetzel et al. 2004).
Epigenetic processes that modulate gene action without modifying the DNA sequences have been proven to have lifelong effects on mature neurons (Tsankova et al. 2007) and exert their impact on synaptic plasticity and cognition (Abel and Zukin 2008). Histone methylation and acetylation are involved in the regulation of gene expression and protein function in neural stem cells (Mattson 2003). It has been proven in experimental models of neurodegenerative disorders that manipulations of both mechanisms influence the susceptibility of neurons to degeneration and apoptosis (Mattson 2003). In the postmortem AD human brain, higher presenilin 1 (PSEN1) expression during brain development and in disease progression is seen due to the reduced methylation mechanisms both at CpG and non-CpG sites (Monti et al. 2020), and promotes the accumulation of toxic Aβ peptide. In PD, dysregulation of SNCA expression is presumed to be caused by aberrant methylation at CpG sites, which control the activation or suppression of protein expression. The distribution of methylated CpG sites is related to anticipated transcription factor (TF) binding sites, suggesting that a decrease in methylation could stimulate SNCA expression in the PD brain (Jowaed et al. 2010). Cyclic adenosine monophosphate response element-binding protein (CREB) binding protein (CBP) functions as a histone acetyltransferase (HAT) as well as a transcriptional cofactor. CBP acts as a HAT in acetylating histones that contribute to transcription by restoring the chromatin configuration. Significantly, it has been known that sequestration of CBP by mtHTT prompts neuronal transcriptional dysfunction (Lee et al. 2013). MtHTT alters the activity of HATs and causes the transcription regulation anomalies observed in most HD patients (Sadri-Vakili and Cha 2006).
Restraint of histone deacetylase (HDAC) activity controls cellular and molecular capacities, triggering synaptic plasticity and neuronal apoptosis, limiting oxidative stress, activating transcription, and inducing modification of histone acetylation levels, eventually encouraging neuroprotection (Gupta et al. 2020). Folate, vitamin B6, vitamin B12, and S-adenosyl methionine (SAM) are vital cofactors involved in DNA methylation modification (Coppedè 2014); 5-azacytidine (AZA) is the most widely studied DNA methyltransferase (DNMT) inhibitor (Xu et al. 2012b), and dietary polyphenols are DNMT inhibitors in vitro (Fang et al. 2007).
This review is centred on the role of methylation in the pathogenesis of AD, PD, and HD and how methylation is associated with these neurodegenerative diseases. We will also discuss the therapeutic targets available to date highlighting the role of epigenetics in these diseases, including histone and DNA modifications.
Epigenetic Regulation in Neurodegenerative Diseases
Principles of Epigenetics
The mechanisms of epigenetic regulation, which include DNA methylation, chromatin remodelling, histone post-translational modifications, and non-coding RNAs (Pagiatakis et al. 2019), are involved in several aspects of neuronal function and advancement (Berson et al. 2018; Aristizabal et al. 2019). Epigenetic regulation has consequences in human wellbeing, with modifications in chromatin known to be associated with numerous illnesses, in which drugs that hinder DNA methylation and histone deacetylation have been approved for clinical use by the Food and Drug Administration (FDA) (Jones et al. 2016).
With explicit significance in the brain, changes in a few chromatin-related factors lead to neurological disorders, including autism spectrum disorder (ASD), mental retardation, intellectual disability, and epilepsy (Bourgeron 2015), highlighting the vital roles of epigenetic mechanisms for brain development and capacity (Wilson 2008). Methylation of the gene often gives rise to silencing, which will affect the phenotype, and these silenced states are inheritable during cellular division (Miranda and Jones 2007). Hypermethylated DNA retains its methylation and remains transcriptionally silent (McGarvey et al. 2007), hence allowing daughter cells to maintain the same expression pattern as the precursor cells (Miranda and Jones 2007).
DNA methylation is involved in development and normal cell homeostasis by regulating cellular processes such as transcription, chromatin structure, chromosome stability, and genomic imprinting (Robertson 2005). DNA methylation controls transcription by adding a methyl group at the 5′ carbon of the cytosine ring situated in CpG dinucleotides via the action of DNMTs (Lu et al. 2013). CpG islands (CGIs) or unmethylated CpG dinucleotides are localized in tissue-specific genes and vital "housekeeping" genes engaged in routine maintenance and are expressed in most tissues (Rodenhiser and Mann 2006). Unmethylated CpGs promote transcription, and DNA silencing occurs upon methylation of these pairs (Rodenhiser and Mann 2006). Neuronal methylation, which consists of ~ 75% CpG and ~ 25% CpH (H = A/C/T) methylation, is conserved in the human brain, enriched in regions of low CpG density, used up at protein-DNA interaction sites and negatively correlated with gene expression (Guo et al. 2014). Previous reports found that 948 out of 27,578 CpG sites are involved in disease-associated methylation differences in DNA originating from the human prefrontal cortex (Bakulski et al. 2012). Three DNMT isoforms are involved in maintaining the methylation status within the genome: DNMT1, DNMT3a, and DNMT3b, and SAM functions as a methyl donor (Halušková 2010). DNMT1 is fundamental in methylation for appropriate neuronal and CNS functioning during early development (van Groen 2010). Apart from their involvement in synaptic plasticity, learning and memory, DNMT1 and DNMT3 are both required to conserve DNA methylation and to synchronize gene expression in the adult CNS (Tian et al. 2010) (Jin et al. 2011). In addition, the DNA hypomethylation of long interspersed element-1 (LINE-1) and Alu elements (Alu) in circulating blood has potential value for cancer diagnosis (Xu et al. 2012a). However, several studies have shown that there is no significant ageing-associated hypomethylation of LINE-1 and Alu retroelements in cellular DNA from peripheral blood (El-Maarri et al. 2011; Erichsen et al. 2018). Another recent study showed that some locus-specific DNA methylation changes are highly reproducible across aged people, independent of sex and tissue type which point to the existence of a programmed epigenetic reconfiguration during ageing and has given rise to the “epigenetic clock” theory (Ciccarone et al. 2018).
Therefore, further studies should aim to confirm whether DNA methylation also remains stable in older populations (60–80-year-old individuals). It is yet to be proven whether this epigenetic marker is the causative event or a consequence of the progression of these diseases. In addition, the fact that the methylation pattern of DNA varies among individuals and the large amounts of DNA input required to provide sufficient and representative data may limit findings.
Epigenetics and Alzheimer’s Disease
AD is a complex multifactorial disorder involving familial and sporadic forms. It is more prevalent in women than men, especially in people aged more than 80 years. Familial forms represent only a minority of the cases, ranging from 5 to 10% of the cases, compared with sporadic forms, which represent most cases and likely occur as a result of complex gene–gene and gene-environmental interplay (Migliore and Coppedè 2009). Familial early-onset AD begins before age 65 years and is mainly caused by point mutations in three genes, APP, PSEN1, and PSEN2 (Migliore and Coppedè 2009). These genes are involved in the APP processing pathway, the proteins of which are encoded on chromosome 21, chromosome 14, and chromosome 1. More than 150 mutations in PSEN1 and a few in PSEN2 have been reported, for a total of approximately 160 mutations in the PSEN genes alone (Migliore and Coppedè 2009). Mutation of PSEN1 fosters the deposition of Aβ by increasing extracellular cleavage, known as the amyloidogenic pathway, through the action of γ-secretase and β-secretase, releasing Aβ1-42 peptides, whereas the non-amyloidogenic pathway releases Aβ1-40 by the cleavage of α-secretase and γ-secretase. Research has shown an elevated ratio of Aβ1-42/Aβ1-40 in the brains of young transgenic animals co-expressing APP and mutant PS1 compared to the brains of transgenic mice co-expressing APP alone and transgenic mice co-expressing wild-type human PS1 and APP (Borchelt et al. 1996). Another study proved that PS1 mutants caused AD by modulating APP processing and favouring the production of Aβ1-42/43, while the loss of normal PS1 function did not lead to AD (Davis et al. 1998). Full-length Aβ1-42 and the modified pyroglutamate peptides Aβp3-42 and Aβp11-42 are found abundantly in the AD brain, but less study has been devoted to the truncated proteins than to the full-length proteins (Sanders et al. 2009). Recent findings proved that the truncated, modified peptides can inhibit the aggregation of full-length Aβ1-42 (Sanders et al. 2009). The latest studies proposed that cerebrospinal fluid biomarker profiles characterized by decreased Aβ peptide levels and increased total and phosphorylated tau levels at threonine 181 (pT181) can be used to discriminate between AD and other neurodegenerative diseases. However, these changes are not entirely specific to AD, and it is noteworthy that other phosphorylated isoforms of tau, possibly more specific for the disease process, have been described in the brain parenchyma of patients (NICE. and NICE 2018; Barthélemy et al. 2020).
DNA methylation plays a pivotal role in running the biochemical process in the higher organisms for normal development. These processes involve the accumulation of methyl group in the 5′ of the cytosine within the CpG dinucleotides. This accumulation is enhanced by DNA methyltransferases (DNMTs), which is heritable (Yi and Kim 2015).
In AD, DNA methylation has already been proven to be involved in modulating Aβ production by regulating the expression of PSEN1 and BACE1. Hypomethylation of both genes boosts their expression and gives rise to the mass build-up of Aβ peptides (Fuso et al. 2005). Fuso et al. (2011) reported a hypomethylated PSEN1 gene promoter in cell lines and transgenic mouse models of AD carrying APP gene mutations (Fuso et al. 2011). BACE1 contributes to synaptic functions by regulating the cAMP/PKA/CREB pathway, and alteration of its expression has been proven to have a significant effect on memory functions and cognitive deficits in transgenic mice (Chen et al. 2012). In late 2019, a case–control study was conducted in Colombia involving 50 individuals with late-onset Alzheimer disease (LOAD) and 50 age- and sex-matched controls to evaluate DNA methylation patterns in the BIN1 (bridging integrator 1) 3′ intergenic region. The findings of this study showed that loss of DNA methylation at CpGs in BIN1 might play an important role in the expression of BIN1 and may be a biomarker for identifying individuals at high risk of developing LOAD in the future (Salcedo-Tacuma et al. 2019).
In view of histone modifications in epigenetics, there is a significant increase in the levels of histone deacetylase 6, a modulator for tau phosphorylation and accumulation has been seen in the brain regions such as hippocampus tissues and cerebral cortical in Alzheimer's disease patients when compared with the control subjects (Ding et al. 2008). Another trait of AD that is most likely connected to memory impairment and cognitive decline is synaptic failure. APOE isoforms differentially manage synaptic plasticity and repair. It is intriguing to note that studies of one-month-old mice uncovered comparable outcomes, suggesting that APOE4-driven changes in neuronal circuitry occur early (Safieh et al. 2019). Approximately 20% of the individuals in a typical control population carry at least one APOE4 allele (Zhong and Weisgraber 2009). In addition to this major risk genetic factor, hyperhomocysteinemia (HHCys) has been correlated with AD and PD, especially in the late stages of the illnesses or after long-term levodopa treatment. A longitudinal study, lasting ≥ 8 years, which involved 1092 individuals with dementia (mean age = 76 years), disclosed that the risk of developing AD was doubled in patients with levels of HHCys > 14 μmol/l (Feligioni et al. 2019). In addition, low concentrations of dietary and circulating folate during gestation increased the risks of premature delivery, underweight infants, and foetal growth retardation (Hernández-Díaz et al. 2000), resulting in elevation of homocysteine levels and an increased risk of AD. This condition was found to stimulate neuronal degeneration of APP-mutant transgenic mice and increased Aβ-induced death in cultured hippocampal neurons (Kruman et al. 2002). In 2014, Lunnon et al. (2014) and De Jager et al. (2014) reported the results of the first two large-scale, epigenome-wide association studies (EWAS) in AD. These two-independent epigenome-wide association studies of AD cohorts have identified overlapping methylation signals in four loci, ANK1, RPL13, RHBDF2, and CDH23.
Likewise, Aβ-induced phosphorylation of tau protein and its resultant inability to bind microtubules will lead to the formation of intracellular tangles known as neurofibrillary tangles (NFTs), the presence of which has been reported to correlate with protein phosphatase 2A (PP2A), which modulates tau phosphorylation by reverting it (Cleveland et al. 1977), (Xu et al. 2006). Increased demethylation of PP2A at the L309 site is mediated by Aβ overexpression, giving rise to compromised dephosphorylation of abnormally hyperphosphorylated tau (Zhou et al. 2008). The aforementioned study also conveys a subunit-specific reduction in PP2A catalytic and regulatory mRNA, resulting in diminished protein expression and phosphatase activity, hyperphosphorylation of tau, the formation of NFTs and neuronal degeneration (Vogelsberg-Ragaglia et al. 2001). There have likewise been reports of overexpression of endogenous inhibitors of PP2A, such as inhibitor 2, along with their cleavage and redistribution, and when overexpressed in an in vivo model, these inhibitors resulted in key features of AD, including amyloid-β deposition, tau hyperphosphorylation, neurodegeneration and cognitive deficits (Voronkov et al. 2011). In some postmortem brains or neuronal cells of AD individuals, a lot of work has been done to find more specific evidence. Wang et al. (2008) had demonstrated that patients with LOAD have a larger epigenetic distance from the normal in brain tissue. It has been compared with controls and that the epigenetic distance increases with age. Hence, these findings supporting the role of epigenetic effects in the development of the disease. Some genes that play central roles in amyloid-β processing are PSEN1 and APOE, and methylation homeostasis such as MTHFR and DNMT1 also show a significant interindividual epigenetic variability, which may contribute to AD predisposition. In cortical neurons of a postmortem AD brain, immunoreactivity for 5-methylcytosine (5-mC) was decreased compared to the control (Mastroeni et al. 2010).
Epigenetics and Parkinson’s Disease
Mutations in SNCA, PARK2, LRRK2, PINK1, UCHL1, or DF1 have been reported to be the causative genes and found occasionally in most of the sporadic cases of PD, although there is no evidence in the majority of the cases (Matsumoto et al. 2010). Less than 1% of PD patients in the general population have gene mutations in SNCA, while the majority have abnormal aggregation of SNCA protein (Maraganore et al. 2006).
The brain contains an abundance of synuclein protein, consisting of three members: α-synuclein, β-synuclein, and γ-synuclein (Goedert 2001). Among these, α-synuclein (SNCA), which is the main component of Lewy bodies and Lewy neuritis and makes up the filamentous inclusions of system atrophy, is the cardinal neuropathological marker in PD and other neurodegenerative disorders (Spillantini et al. 1998). SNCA also plays a critical role in PD pathogenesis. Both duplication or triplication and point mutations (e.g. A30P, E46K, and A53T) in SNCA1 are associated with autosomal dominant familial PD (Huang et al. 2019). Extracellular aggregated SNCA induced microglial activation and later enhanced dopaminergic neurodegeneration in a mesencephalic neuron-glia culture system (Zhang et al. 2005). Lewy bodies, which are related to nerve loss are more abundant in patients with mild and moderate neuronal loss than in those with severe neuronal loss (Schulz-Schaeffer 2010).
Current studies have reported that there is a decrease in DNA methylation in the regulatory regions of specific genes in PD brains. Decreased levels of nuclear DNMT1 are mediated by SNCA, leading to global DNA hypomethylation, which was observed in postmortem human brain samples and the brains of SNCA transgenic mouse models (Desplats et al. 2011). Findings in other studies indicated that mRNA expression of SNCA is decreased but the protein levels are maintained at high levels due to the regulation of post-translational stabilization of the proteins (Li et al. 2004). Hypomethylation in the DNA from the substantia nigra, putamen, and cortex region of sporadic PD patients has been reported (Jowaed et al. 2010). A study on methylation of SNCA and leucine-rich repeat kinase 2 (LRRK2) showed significant hypomethylation in certain CpG sites in leukocyte DNA from PD patients compared to controls (Tan et al. 2014). In contrast, analysis of the SNCA methylation level on CpG islands located in intron 1 of PD patients showed no significant differences between PD patients and controls (Song et al. 2014b).
Oxidative stress by free radicals is one of the significant causes of PD. It is currently well established that free radicals play a vital role in ageing (Hamilton et al. 2001), and PD has been regarded as one of the greatest risk factors in ageing. Initially, the proof for the presence of oxidative stress in PD originated from reports dependent on postmortem examinations of brain samples from patients with PD that exhibited elevated degrees of oxidized proteins, lipids, and nucleic acids (Kumar et al. 2012).
Genome-wide methylation analysis of sporadic PD patients discovered a single hypomethylated gene, CYP2E1, in both putamen and cortex regions (Kaut et al. 2012), indicating that epigenetic variants in this gene contribute to PD susceptibility.
Epigenetics and Huntington’s Disease
Although the loss of neurons in many brain areas has been accounted for in HD, the selective neurodegeneration of the γ-aminobutyric acid-releasing spiny-projection neurons of the striatum is predominant (Landles and Bates 2004). Damage of brain striatal neurons is a consequence of the extension of CAG repeats in the HTT protein, which lead to HD characteristics (Steffan et al. 2000). HD pathogenesis includes cytoplasmic cleavage of HTT, which releases an amino-terminal fragment capable of nuclear localization (Steffan et al. 2000). Individuals with normal amounts of CAG repeats have 7–34 repeats, whereas mutations in exon 1 of this gene extend the cytosine-adenine-guanine trinucleotide repeats, which code for the polyglutamine (polyQ) moiety in the HTT protein (Sadri-Vakili and Cha 2006).
However, the age of onset of symptoms depends on whether the gene is passed through the paternal or maternal germline. The gene itself becomes modified differently, and paternal transmission has been shown to involve DNA methylation that can lead to higher or earlier expression of the gene (Reik 1988). There is proof for a relationship between male exposure to various drugs/toxins and increased mutations, including numerical and basic chromosomal abnormalities, point mutations, copy number variations (CNVs), and duplications/deletion of microsatellite regions (Curley et al. 2011).
Transcriptional impairment has been proven to be involved in HD (Ferrante et al. 2003). Genome-wide chromatin immunoprecipitation sequencing (ChIP-Seq) confirmed that AP-1 and SOX2 are transcriptional regulators associated with methylation changes in regions of low CpG content due to the presence of mtHTT in cell lines derived from mouse striatal neurons (Ng et al. 2013). Specific and significant losses of acetylated essential histone (AcH2A, AcH2B, AcH3, and AcH4) expression in cells in the caudate nucleus and Purkinje cells of the cerebellum were observed in HD compared with patients with frontotemporal lobar degeneration and control subjects, while the level of HDAC5 was elevated in these cells (Yeh et al. 2013). The epigenetic changes that occur in AD, PD and HD is summarized in Table 1.
Therapeutic Interventions for Neurodegenerative Diseases
Epigenetics and Therapeutic Approaches
Mounting evidence of the involvement and importance of epigenetic alterations in neurodegenerative disorders has presented new therapeutic interventions for these disorders. Experiments in animal models of neurodegenerative disorders have confirmed the possible role of epigenetic drugs, together with inhibitors of histone deacetylases and methyl donor compounds (Adwan and Zawia 2013). Small drugs such as histone deacetylase inhibitors (HDACi) can cross the blood–brain barrier (BBB), thus slowing the initiation and development of symptoms in animal models of neurodegenerative diseases (Coppedè 2014).
HDACs are separated into four different classes; HDACIs, HDACIIs, HDACIIIs, and HDACIV (Dokmanovic et al. 2007). HDACs are potential therapeutic targets in various chronic diseases, including cancer and fibrotic disorders (Pang and Zhuang 2010). HDAC-induced histone hypoacetylation is associated with gene silencing; thus, altered expression and mutations in genes encoding HDACs have been correlated with tumour development (Ropero and Esteller 2007). HDAC3 deficiency increases collagen deposition in atherosclerotic lesions and induces the stable plaque phenotype observed in transplanted atherosclerosis-susceptible mice upon HDAC3 deletion (Hoeksema and de Winther 2016). HDACi stimulates growth arrest, inhibits differentiation, induces apoptosis of tumour cells, and affects the acetylation status and function of non-histone proteins, with minimal effects on normal tissue (Kim et al. 2006; Lane and Chabner 2009). Although the mechanisms of HDACi are not completely elucidated, they are believed to be able to cause a build-up of acetylated histones and many non-histone proteins that are implicated in the regulation of gene expression, cell proliferation, cell migration, and cell death, and a wide diversity of altered cells are sensitive to inhibitor-induced cell death (Dokmanovic et al. 2007). Several structural classes of HDACi are under clinical trial for various diseases, including hydroxamic acids (vorinostat), cyclic peptides (depsipeptide or romidepsin), benzamides, and aliphatic acids (valproic acid (VA)) (Adwan and Zawia 2013).
Nutrition and dietary compounds, for instance, vitamin B, folate, and methionine, are known to affect epigenetic regulation and mechanisms, especially DNA methylation and one-carbon metabolism. In addition, dietary compounds can modulate the activity of protein-related methylation; for example, they can inhibit DNMTs. Thus, both nutriepigenetic or nutriepigenomic molecules, which influence basic human wellbeing, have been developed as another promising field in current research. Polyphenols, as an example, highlight the unique cooperation between the genome and the environment, particularly at physiological concentrations (Remely et al. 2015). They induce a significant effect only if consumed in large amounts or if the levels of methyl donors are limited; however, but possible toxicity resulting from oxidation of polyphenols might occur, and precautions need to be taken upon consuming these compounds (Fang et al. 2007).
Folate supplementation, for instance, can inhibit the adverse effects of ageing, for example, uracil misincorporation, DNA methylation, protein methylation, mitochondrial deletion, and critical gene expression, and was observed to protect against colon cancer (Jang et al. 2005; Kim 2005). However, severe folate insufficiency generated hypomethylation (by 40%) within a mutation hot spot (exons 6–7), but not in exon 8, of the p53 tumour suppressor gene, despite a 56% increase in genomic DNA methylation in the rat liver. This finding increases the likelihood that the impact of folate insufficiency on DNA methylation might be site- and gene-specific and proposes that the progression of genomic and site-specific DNA methylation because of folate deficiency may vary (Mierzecki et al. 2015).
5-Azacytidine or azacitidine (AzaC) is one of the most widely studied DNMT inhibitors (DNMTi) and is mainly used in managing myelodysplastic syndrome (MDS), in which it functions as a methyl donor remover in vitro to weaken the gene silencing effect upon methylation. A 67-year-old man with high-risk MDS exhibited haematological improvement three months after the first cycle of AzaC therapy (Takaoka et al. 2014).
AD and Epigenetic Therapy
The current drugs agreed upon by the FDA for the treatment of AD are acetylcholinesterase inhibitors such as donepezil, rivastigmine, galantamine, and tacrine along with the N-methyl-D-aspartate (NMDA) receptor antagonist memantine, but they do not impede the progression of AD and instead merely target symptoms. It has been demonstrated on AD models that some epigenetic drugs might be beneficial in protecting against neurodegeneration and improving cognitive function (Adwan and Zawia 2013; Cacabelos and Teijido 2018). Moreover, the donepezil-treated group revealed a considerably lower incidence of cardiac rupture during the acute phase of myocardial infarction than the untreated group, an effect that occurred via the inhibition of metalloproteinase-9-related acute inflammatory tissue injury (Arikawa et al. 2011). Combined administration of donepezil and memantine deters the diminution of cerebral blood flow in the prefrontal area and improves clinical symptoms of overall cognitive function and behavioural and psychological symptoms of dementia in AD patients (Araki et al. 2014). Nasal administration enables the potential absorption of the drug into the CNS by bypassing the BBB and ensures targeting of therapeutics to the CNS with rapid achievement of sufficient drug levels in the target tissue and low systemic exposure (Sharma et al. 2015). Recently, a nanoemulsion (NE) loaded with memantine was studied. The finalized NE showed a particle size of ~ 11 nm and a percentage transmittance of ~ 99%. The in vitro release studies showed 80% drug release in simulated nasal fluid. The developed NE loaded with memantine could be used for intranasal administration to enhance the effect on AD (Kaur et al. 2020).
Galantamine had an apparent effect on the preservation of memory in patients with mild-to-moderate AD during 26 weeks (Song et al. 2014a). The one and only drug that actively promoted for the treatment of AD with proven activity as an allosteric modulator of nicotinic acetylcholine receptors (nAChRs), a competitive inhibitor of acetylcholinesterase (AChE) and reversible is galantamine. With galantamine, it helps to decrease in activity and expression of nAChRs promote a huge reduction in central cholinergic neurotransmission in patients with AD (Lilienfeld 2002). Tacrine-6-ferulic acid (T6FA) significantly inhibits autoaggregation and acetylcholinesterase-induced aggregation of Aβ1–40 in vitro, blocks cell death induced by Aβ1–40 in PC12 cells, and improves cognitive ability in an AD mouse model (Pi et al. 2012). Hybrid tacrine-8-hydroxyquinoline (IQM-622) decreases the deposition of Aβ in APP/PS1 mice, promotes the degradation of intracellular Aβ in astrocytes and protects against Aβ toxicity in cultured astrocytes and neurons (Antequera et al. 2012). Patients that treated with tacrine required multiple-dosage regimen to maintain the therapeutic level in given of its short half-life and adverse effect. These patients also need to undergo regular blood monitoring due to the hepatotoxicity of the drugs (Watkins et al. 1994). Up to date, tacrine was discontinued due to the liver toxicity and many side effects (Sharma 2019). Significant improvements with the rivastigmine patch and capsule were observed in patients with advanced dementia stage, yet no significant improvements were noted in patients with mild-to-moderate AD (Farlow et al. 2011).
Cognitive capacities in the neurodegenerating brain are not lost but merely impaired due to epigenetic blockade mediated by HDAC2, which can potentially be reversed (Gräff et al. 2012). Treatments with sodium valproate, sodium butyrate, or vorinostat completely restored contextual memory in APPswe/PS1dE9 mice and inhibited HDACI isoforms, indicating that HDACI inhibition is plausible for treating cognitive deficits associated with early-stage AD (Kilgore et al. 2010). Valproic acid (VPA) decreased Aβ production by halting the GSK-3β-mediated γ-secretase cleavage of APP, reduced neuritic plaque formation, and alleviated memory deficits in APP23 mice carrying the human Swedish mutant APP751 (Qing et al. 2008). VPA alleviated p65 NF-κB phosphorylation and boosted the levels of acetyl-H3, Bcl-2, and GSK-3β in the hippocampus of APPswe/PS1dE9 (APP/PS1) transgenic mice (Xuan et al. 2015).
In addition, ageing-related memory impairments can also be affected by sodium butyrate via its influence on the early consolidation phase of memory formation. However, sodium butyrate showed no effect in younger rats with normal memory retention (Reolon et al. 2011). Sodium butyrate improves associative memory by elevating hippocampal histone acetylation and increasing the expression of genes implicated in associative learning in APPPS1-21 mice even when administered at an advanced stage of pathology (Govindarajan et al. 2011).
Another study reported that high consumption of vitamins C, E, B6, and B12, folate, unsaturated fatty acids, and fish can decrease AD risk (Luchsinger and Mayeux 2004). Another study showed that SAM, exerts a neuroprotective function on both methylation and oxidation metabolism by preventing oxidative stress and lipid peroxidation and modulating glutathione metabolism through superoxide dismutase and glutathione S-transferase activity (Cavallaro et al. 2010). Patients receiving vitamin or nutraceutical formulations (folate, vitamin B6, alpha-tocopherol, SAM, N-acetyl cysteine, and acetyl-l-carnitine) had improved the domains of the Neuropsychiatric Inventory (NPI) and maintenance of performance in the Alzheimer's Disease Cooperative Study-Activities of Daily Living (ADL) when compared with patients receiving naproxen, rofecoxib, or placebo. These studies also showed equivalent or more profound effects than previous studies using donepezil and galantamine (Chan et al. 2009). Rofecoxib (Vioxx), however, was removed from the market due to the increased risk of cardiovascular events.
PD and Epigenetic Therapy
Dopamine replacement therapies (DRTs) have improved PD management, yet these treatments cause weakening when used in the long term and do not protect deteriorating neurons against death (Harrison and Dexter 2013). Impulse control disorders (ICDs) are prevalent among patients with PD receiving dopaminergic medications and begin after initiation of dopamine (DA) agonist therapy and cease upon its discontinuation (Ambermoon et al. 2011). A correlation analysis in a cohort study uncovered that increasing doses of DA were related with decreasing performance on a pattern recognition task in treated patients with PD, showing that DRT improves frontal lobe function (strategizing) yet degrades temporal lobe function (visual memory) (Miah et al. 2012).
There is increasing evidence that there is a pathological disparity in PD among the deacetylation and acetylation of histone proteins, and therefore, the utilization of histone deacetylase-inhibiting agents has been proposed to improve this pathological imbalance (Harrison and Dexter 2013). In a study of novel recognition tests, the time spent discovering new objects by mice was markedly improved and there was a SIRT2-induced decrease in cell proliferation and neuroblast differentiation in the dentate gyrus after treatment with sodium butyrate (Yoo et al. 2015). Trichostatin A (TSA) increases astrocytic glutamate uptake when neurotoxicity occurs, thus enhancing glutamate uptake to decrease the MPP-induced elevation of glutamate in the medium, which might partially prevent the downregulation of GluTs. This finding might be a new mechanism implicated in the neuroprotection of HDACIs (Wu et al. 2008). Nevertheless, TSA treatment influences PD pathogenesis by decreasing cell survival and increasing apoptosis in dopaminergic neuronal cells (Wang et al. 2009).
Apart from the anticonvulsant drug sodium valproate, also known as Epilim, valproate also likely acts as a brain-penetrant and has been well tested and used clinically (Nalivaeva et al. 2009). It is noteworthy that in the action of mood stabilizers, epigenetics may propose a crucial role. As one of the histone deacetylase (HDAC) inhibitor, sodium valproate probably has a downstream epigenetics action (Lee et al. 2015). It is widely known that HDAC inhibitors are one of the epigenetic regulators with numerous beneficial effects at both systemic and cellular levels (Hull et al. 2016). Nonetheless, accumulating reports suggest that valproate leads to adverse effects such as organ failure, birth defects if consumed during gestation, and a decline in IQ in children. Administration of valproate to eight patients with PD with defects in GABA metabolism did not markedly alter any of the disease features; instead, it increased dyskinesia in the patients (Nutt et al. 1979). However, a recent study reported that a combination of valproate and lithium carbonate ameliorated the loss of dihydroxyphenyl acetic acid and rescued dopaminergic neurons following MPTP treatment in male C57BL/6 mice with PD (Li et al. 2012). Apart from that, given the progressive degeneration of dopaminergic neurons and aggregation of a-synuclein that correlated with PD pathophysiology, it is reported that HDAC6 help to relieve polyglutamine-mediated neurodegeneration via autophagy (Shukla and Tekwani 2020).
The DNMT inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) induced transcriptional upregulation of tyrosine hydroxylase and SNCA, thus increasing the vulnerability of dopaminergic neurons to neurotoxic damage (Wang et al. 2013). 5-Aza-dC restored retinoic acid receptor-β2 (RAR-β2) inducibility by all-trans-retinoic acid (ATRA) in some cell lines with a hypermethylated RAR-β2 promoter (Youssef et al. 2004).
HD and Epigenetic Therapy
Drugs aimed at correcting epigenetic alterations, including histone modifications and DNA modifications, have shown promise in treating HD (Wang et al. 2014). Potential disease-modifying therapeutics for HD include histone deacetylase inhibitors, such as sodium phenylbutyrate (SPB) or sodium butyrate (Hu et al. 2011). Sodium butyrate amplified histone and specificity protein-1 acetylation, protected against 3-nitropropionic acid neurotoxicity, and limited neuropathological sequelae in the R6/2 transgenic mouse model of HD (Ferrante et al. 2003). Administration of phenylbutyrate increased brain histone acetylation, decreased histone methylation levels, and exerted neuroprotective effects in the N171-82Q transgenic mouse model of HD (Gardian et al. 2005).
The HDACi 4b and 136 presented a strong ability to inhibit HDAC3 and were most effective in limiting the expression of genes relevant to HD, including Ppp1r1b, in R6/2 transgenic mice (Jia et al. 2012). SIRT2 enhances motor function, extends survival, and reduces brain atrophy and is associated with a decrease in aggregated mtHTT in two genetic mouse models of HD (Chopra et al. 2012). SIRT2-specific inhibitor AK-1 treatment-induced proteasomal degradation of the Snail transcription factor resulted in the upregulation of p21, a cyclin-dependent kinase inhibitor, leading to G1 arrest, slow proliferation, and slow wound-healing activity in HCT116 human colon cancer cells (Cheon et al. 2015).
Vorinostat or suberoylanilide hydroxamic acid (SAHA), an inhibitor of HDACIs and HDACIIs, can cross the BBB and increase histone acetylation in the brain and can be administered orally by drinking water when complexed with cyclodextrins (Hockly et al. 2003). Prolonged SAHA treatment causes degradation of HDAC4 in the cortex and brain stem but not in the hippocampus and decreases HDAC2 levels without affecting their transcript levels in vivo (Mielcarek et al. 2011). SAHA, on the other hand, increases vesicular transport of BDNF by inhibiting HDAC6, thereby increasing acetylation at lysine 40 of α-tubulin in HD brains (Dompierre et al. 2007). The Epigenetic intervention in AD, PD and HD is summarized in Table 2.
Conclusion
Epigenetic changes play an important role in the progression of AD, PD and HD. Thus, it is important to develop and improve techniques to examine chromatin structure and function to understand epigenetic regulation in normal ageing and neurodegenerative diseases. More studies should be conducted aiming at restoring chromatin dynamics and therefore proper gene expression, which may provide novel therapeutic strategies if applied early and in combination with other therapies addressing all aspects of these diseases.
Abbreviations
- AD:
-
Alzheimer’s disease
- PSEN1:
-
Presenilin 1
- PD:
-
Parkinson’s disease
- SNCA:
-
α-Synuclein
- mtHTT:
-
Mutant Huntingtin
- HD:
-
Huntington’s disease
- HDACs:
-
Histone deacetylases
- SAM:
-
S-Adenosylmethionine
- AZA:
-
5-Azacytidine
- DNMT:
-
DNA methyltransferase
- HTT:
-
Huntingtin
- TF:
-
Transcription factor
- CREB:
-
Cyclic adenosine monophosphate response element-binding protein
- CBP:
-
CREB binding protein
- HATs:
-
Histone acetyltransferases
- FDA:
-
Food and Drug Administration
- ASD:
-
Autism spectrum disorder
- CGIs:
-
CpG islands
- HHCys:
-
Hyperhomocysteinemia
- NFTs:
-
Neurofibrillary tangles
- PP2A:
-
Protein phosphatase 2A
- SVs:
-
Synaptic vesicles
- LRRK2:
-
Leucine-rich repeat kinase 2
- CNV:
-
Copy number variant
- ChIP-Seq:
-
Chromatin immunoprecipitation sequencing
- HDACi:
-
Histone deacetylase inhibitors
- BBB:
-
Blood–brain barrier
- VA:
-
Valproic acid
- FA:
-
Folic acid
- AzaC:
-
Azacitidine
- DNMTi:
-
DNMT inhibitors
- MDS:
-
Myelodysplastic syndrome
- NMDA:
-
N-Methyl-D-aspartate
- T6FA:
-
Tacrine-6-ferulic acid
- NPI:
-
Neuropsychiatric inventory
- ADL:
-
Alzheimer’s Disease Cooperative Study-Activities of Daily Living
- DRTs:
-
Dopamine replacement therapies
- ICDs:
-
Impulse control disorders
- 5-aza-dC:
-
5-Aza-2′-deoxycytidine
- RAR-β2 :
-
Retinoic acid receptor-β2
- ATRA:
-
All-trans-retinoic acid
- SPB:
-
Sodium phenylbutyrate
- SAHA:
-
Suberoylanilide hydroxamic acid
- BDNF:
-
Brain-derived neurotrophic factor
References
Abel T, Zukin RS (2008) Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol 8(1):57–64
Adwan L, Zawia NH (2013) Epigenetics: a novel therapeutic approach for the treatment of Alzheimer’s disease. Pharmacol Ther 139(1):41–50
Ambermoon P, Carter A, Hall WD, Dissanayaka NN, O’Sullivan JD (2011) Impulse control disorders in patients with Parkinson’s disease receiving dopamine replacement therapy: evidence and implications for the addictions field. Addiction 106(2):283–293
Antequera D, Bolos M, Spuch C, Pascual C, Ferrer I, Fernandez-Bachiller MI, Rodríguez-Franco MI, Carro E (2012) Effects of a tacrine-8-hydroxyquinoline hybrid (IQM-622) on Aβ accumulation and cell death: involvement in hippocampal neuronal loss in Alzheimer’s disease. Neurobiol Dis 46(3):682–691
Araki T, Wake R, Miyaoka T, Kawakami K, Nagahama M, Furuya M, Limoa E, Liaury K, Hashioka S, Murotani K (2014) The effects of combine treatment of memantine and donepezil on Alzheimer’s disease patients and its relationship with cerebral blood flow in the prefrontal area. Int J Geriatr Psychiatry 29(9):881–889
Arikawa M, Kakinuma Y, Handa T, Yamasaki F, Sato T (2011) Donepezil, anti-Alzheimer’s disease drug, prevents cardiac rupture during acute phase of myocardial infarction in mice. PLoS ONE 6(7):20629
Aristizabal MJ, Anreiter I, Halldorsdottir T, Odgers CL, McDade TW, Goldenberg A, Mostafavi S, Kobor MS, Binder EB, Sokolowski MB (2019) Biological embedding of experience: a primer on epigenetics. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1820838116
Armstrong RJ, Barker RA (2001) Neurodegeneration: a failure of neuroregeneration? The Lancet 358(9288):1174–1176
Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS (2012) Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimer’s Dis 29(3):571–588
Barthélemy NR, Bateman RJ, Hirtz C, Marin P, Becher F, Sato C, Gabelle A, Lehmann S (2020) Cerebrospinal fluid phospho-tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer’s disease and PET amyloid-positive patient identification. Alzheimer’s Res Therapy 12(1):1–11
Berson A, Nativio R, Berger SL, Bonini NM (2018) Epigenetic regulation in neurodegenerative diseases. Trends Neurosci 41(9):587–598
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D (1996) Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17(5):1005–1013
Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat Med 10(7):S2–S9
Bourgeron T (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 16(9):551–563
Cacabelos R, Teijido O (2018) Epigenetic drug discovery for Alzheimer’s disease. In: Epigenetics of aging and longevity. Elsevier, New York, pp 453–495
Cavallaro RA, Fuso A, Nicolia V, Scarpa S (2010) S-adenosylmethionine prevents oxidative stress and modulates glutathione metabolism in TgCRND8 mice fed a B-vitamin deficient diet. J Alzheimer’s Dis 20(4):997–1002
Chan A, Paskavitz J, Remington R, Rasmussen S, Shea TB (2009) Efficacy of a vitamin/nutriceutical formulation for early-stage Alzheimer’s disease: a 1-year, open-label pilot study with an 16-month caregiver extension. Am J Alzheimer’s Dis Other Demen 23(6):571–585
Chen Y, Huang X, Zhang Y-w, Rockenstein E, Bu G, Golde TE, Masliah E, Xu H (2012) Alzheimer’s β-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of β-amyloid. J Neurosci 32(33):11390–11395
Cheon MG, Kim W, Choi M, Kim J-E (2015) AK-1, a specific SIRT2 inhibitor, induces cell cycle arrest by downregulating Snail in HCT116 human colon carcinoma cells. Cancer Lett 356(2):637–645
Chopra V, Quinti L, Kim J, Vollor L, Narayanan KL, Edgerly C, Cipicchio PM, Lauver MA, Choi SH, Silverman RB (2012) The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep 2(6):1492–1497
Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M (2018) DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev 174:3–17
Cleveland DW, Hwo S-Y, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 116(2):227–247
Coppedè F (2014) The potential of epigenetic therapies in neurodegenerative diseases. Front Genet 5:220
Curley JP, Mashoodh R, Champagne FA (2011) Epigenetics and the origins of paternal effects. Horm Behav 59(3):306–314
Davis JA, Naruse S, Chen H, Eckman C, Younkin S, Price DL, Borchelt DR, Sisodia SS, Wong PC (1998) An Alzheimer’s disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron 20(3):603–609
De Jager PL et al (2014) Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 17(9):1156–1163
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391(6665):387–390
Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E (2011) α-Synuclein sequesters Dnmt1 from the nucleus a novel mechanism for epigenetic alterations in lewy body diseases. J Biol Chem 286(11):9031–9037
Ding H et al (2008) Histone deacetylase 6 interacts with the microtubule-associated protein tau. J Neurochem 106(5):2119–2130
Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5(10):981–989
Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci 27(13):3571–3583
El-Maarri O, Walier M, Behne F, van Üüm J, Singer H, Diaz-Lacava A, Nüsgen N, Niemann B, Watzka M, Reinsberg J (2011) Methylation at global LINE-1 repeats in human blood are affected by gender but not by age or natural hormone cycles. PLoS ONE 6(1):e16252
Erichsen L, Beermann A, Arauzo-Bravo MJ, Hassan M, Dkhil MA, Al-Quraishy S, Hafiz TA, Fischer JC, Santourlidis S (2018) Genome-wide hypomethylation of LINE-1 and Alu retroelements in cell-free DNA of blood is an epigenetic biomarker of human aging. Saudi J Biol Sci 25(6):1220–1226
Fang M, Chen D, Yang CS (2007) Dietary polyphenols may affect DNA methylation. J Nutr 137(1):223S-228S
Farlow MR, Grossberg GT, Meng X, Olin J, Somogyi M (2011) Rivastigmine transdermal patch and capsule in Alzheimer’s disease: influence of disease stage on response to therapy. Int J Geriatr Psychiatry 26(12):1236–1243
Feligioni M, Tinelli C, Di Pino A, Ficulle E, Marcelli S (2019) Hyperhomocysteinemia as a risk factor and potential nutraceutical target for certain pathologies. Front Nutr 6:49
Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 23(28):9418–9427
Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, Scarpa S (2011) Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging 32(2):187–199
Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S (2005) S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci 28(1):195–204
Gardian G, Browne SE, Choi D-K, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ (2005) Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington’s disease. J Biol Chem 280(1):556–563
Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2(7):492–501
Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A (2011) Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J Alzheimer’s Dis 26(1):187–197
Gräff J, Rei D, Guan J-S, Wang W-Y, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483(7388):222–226
Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G (2014) Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci 17(2):215
Gupta R, Ambasta RK, Kumar P (2020) Pharmacological intervention of histone deacetylase enzymes in the neurodegenerative disorders. Life Sci 243:117278
Halušková J (2010) Epigenetic studies in human diseases. Folia Biol (Praha) 56(3):83–96
Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A (2001) Does oxidative damage to DNA increase with age? Proc Natl Acad Sci 98(18):10469–10474
Harrison IF, Dexter DT (2013) Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol Ther 140(1):34–52
Hernández-Díaz S, Werler MM, Walker AM, Mitchell AA (2000) Folic acid antagonists during pregnancy and the risk of birth defects. N Engl J Med 343(22):1608–1614
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci 100(4):2041–2046
Hoeksema MA, de Winther MP (2016) Epigenetic regulation of monocyte and macrophage function. Antioxid Redox Signal 25(14):758–774
Hu Y, Chopra V, Chopra R, Locascio JJ, Liao Z, Ding H, Zheng B, Matson WR, Ferrante RJ, Rosas HD (2011) Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc Natl Acad Sci 108(41):17141–17146
Huang M, Wang B, Li X, Fu C, Wang C, Kang X (2019) α-Synuclein: a multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front Neurosci 13:28
Hull EE, Montgomery MR, Leyva KJ (2016) HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and inflammatory diseases. BioMed Res Int 2016:1–6
Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345(1):27–32
Jang H, Mason JB, Choi S-W (2005) Genetic and epigenetic interactions between folate and aging in carcinogenesis. J Nutr 135(12):2967S-2971S
Jellinger K (2001) Cell death mechanisms in neurodegeneration. J Cell Mol Med 5(1):1–17
Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S (2012) Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis 46(2):351–361
Jin B, Li Y, Robertson KD (2011) DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2(6):607–617
Jones PA, Issa J-PJ, Baylin S (2016) Targeting the cancer epigenome for therapy. Nat Rev Genet 17(10):630
Jowaed A, Schmitt I, Kaut O, Wüllner U (2010) Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci 30(18):6355–6359
Kaur A, Nigam K, Srivastava S, Tyagi A, Dang S (2020) Memantine nanoemulsion: a new approach to treat Alzheimer’s Disease. J Microencapsul 37:355–365
Kaut O, Schmitt I, Wüllner U (2012) Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 13(1):87–91
Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G (2010) Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35(4):870–880
Kim T-Y, Bang Y-J, Robertson KD (2006) Histone deacetylase inhibitors for cancer therapy. Epigenetics 1(1):15–24
Kim Y-I (2005) Nutritional epigenetics: impact of folate deficiency on DNA methylation and colon cancer susceptibility. J Nutr 135(11):2703–2709
Kruman II, Kumaravel T, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP (2002) Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci 22(5):1752–1762
Kumar H, Lim H-W, More SV, Kim B-W, Koppula S, Kim IS, Choi D-K (2012) The role of free radicals in the aging brain and Parkinson’s disease: convergence and parallelism. Int J Mol Sci 13(8):10478–10504
Landles C, Bates GP (2004) Huntingtin and the molecular pathogenesis of Huntington’s disease. EMBO Rep 5(10):958–963
Lane AA, Chabner BA (2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27(32):5459–5468
Lee J, Hwang YJ, Kim KY, Kowall NW, Ryu H (2013) Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 10(4):664–676
Lee R, Pirooznia M, Guintivano J, Ly M, Ewald E, Tamashiro K, Gould T, Moran TH, Potash JB (2015) Search for common targets of lithium and valproic acid identifies novel epigenetic effects of lithium on the rat leptin receptor gene. Transl Psychiatry 5(7):e600–e600
Li W, Lesuisse C, Xu Y, Troncoso JC, Price DL, Lee MK (2004) Stabilization of α-synuclein protein with aging and familial Parkinson’s disease-linked A53T mutation. J Neurosci 24(33):7400–7409
Li X-z, Chen X-p, Zhao K, Bai L-m, Zhang H, Zhou X-p (2012) Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: possible mediation through enhanced autophagy. Int J Neurosci 123(2):73–79
Lilienfeld S (2002) Galantamine—a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev 8(2):159–176
Lovrečić L, Maver A, Zadel M, Peterlin B (2013) The role of epigenetics in neurodegenerative diseases. Neurodegener Dis. https://doi.org/10.5772/54744
Lu H, Liu X, Deng Y, Qing H (2013) DNA methylation, a hand behind neurodegenerative diseases. Front Aging Neurosci 5:85
Luchsinger JA, Mayeux R (2004) Dietary factors and Alzheimer’s disease. Lancet Neurol 3(10):579–587
Lunnon K et al (2014) Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci 17(9):1164–1170
Maraganore DM, De Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ (2006) Collaborative analysis of α-synuclein gene promoter variability and Parkinson disease. JAMA 296(6):661–670
Mastroeni D et al (2010) Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging 31(12):2025–2037
Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, Iwata A (2010) CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 5(11):15522
Mattson MP (2003) Methylation and acetylation in nervous system development and neurodegenerative disorders. Ageing Res Rev 2(3):329–342
McGarvey KM, Greene E, Fahrner JA, Jenuwein T, Baylin SB (2007) DNA methylation and complete transcriptional silencing of cancer genes persist after depletion of EZH2. Can Res 67(11):5097–5102
Miah I, Olde Dubbelink K, Stoffers D, Deijen J, Berendse H (2012) Early-stage cognitive impairment in Parkinson’s disease and the influence of dopamine replacement therapy. Eur J Neurol 19(3):510–516
Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP (2011) SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 6(11):e27746
Mierzecki A, Makarewicz-Wujec M, Kłoda K, Kozłowska-Wojciechowska M, Pieńkowski P, Naruszewicz M (2015) Influence of folic acid supplementation on coagulation, inflammatory, lipid, and kidney function parameters in subjects with low and moderate content of folic acid in the diet. Kardiologia Polska (Polish Heart J) 73(4):280–286
Migliore L, Coppedè F (2009) Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res/Fundam Mol Mech Mutagen 667(1–2):82–97
Miranda TB, Jones PA (2007) DNA methylation: the nuts and bolts of repression. J Cell Physiol 213(2):384–390
Monti N, Cavallaro RA, Stoccoro A, Nicolia V, Scarpa S, Kovacs GG, Fiorenza MT, Lucarelli M, Aronica E, Ferrer I (2020) CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 15(8):781–799
Nalivaeva NN, Belyaev ND, Turner AJ (2009) Sodium valproate: an old drug with new roles. Trends Pharmacol Sci 30(10):509–514
Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, Labadorf A, Housman DE, Fraenkel E (2013) Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci 110(6):2354–2359
NICE (2018) Dementia: assessment, management and support for people living with dementia and their carers. National Institute for Health and Care Excellence, London
Nutt J, Williams A, Plotkin C, Eng N, Ziegler M, Calne DB (1979) Treatment of Parkinson’s disease with sodium valproate: clinical, pharmacological, and biochemical observations. Can J Neurol Sci 6(3):337–343
Pagiatakis C, Musolino E, Gornati R, Bernardini G, Papait R (2019) Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res. https://doi.org/10.1007/s40520-019-01430-0
Pang M, Zhuang S (2010) Histone deacetylase: a potential therapeutic target for fibrotic disorders. J Pharmacol Exp Ther 335(2):266–272
Pi R, Mao X, Chao X, Cheng Z, Liu M, Duan X, Ye M, Chen X, Mei Z, Liu P (2012) Tacrine-6-ferulic acid, a novel multifunctional dimer, inhibits amyloid-β-mediated Alzheimer’s disease-associated pathogenesis in vitro and in vivo. PLoS ONE 7(2):e31921
Qing H, He G, Ly PT, Fox CJ, Staufenbiel M, Cai F, Zhang Z, Wei S, Sun X, Chen C-H (2008) Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med 205(12):2781–2789
Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P (2004) α-Synuclein and Parkinson’s disease. FASEB J 18(6):617–626
Reik W (1988) Genomic imprinting: a possible mechanism for the parental origin effect in Huntington’s chorea. J Med Genet 25(12):805–808
Remely M, Lovrecic L, De La Garza A, Migliore L, Peterlin B, Milagro F, Martinez A, Haslberger A (2015) Therapeutic perspectives of epigenetically active nutrients. Br J Pharmacol 172(11):2756–2768
Reolon GK, Maurmann N, Werenicz A, Garcia VA, Schröder N, Wood MA, Roesler R (2011) Posttraining systemic administration of the histone deacetylase inhibitor sodium butyrate ameliorates aging-related memory decline in rats. Behav Brain Res 221(1):329–332
Robertson KD (2005) DNA methylation and human disease. Nat Rev Genet 6(8):597–610
Rodenhiser D, Mann M (2006) Epigenetics and human disease: translating basic biology into clinical applications. CMAJ 174(3):341–348
Ropero S, Esteller M (2007) The role of histone deacetylases (HDACs) in human cancer. Mol Oncol 1(1):19–25
Sadri-Vakili G, Cha J-HJ (2006) Mechanisms of disease: histone modifications in Huntington’s disease. Nat Clin Pract Neurol 2(6):330–338
Safieh M, Korczyn AD, Michaelson DM (2019) ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med 17(1):1–17
Salcedo-Tacuma D, Melgarejo JD, Mahecha MF, Ortega-Rojas J, Arboleda-Bustos CE, Pardo-Turriago R, Arboleda H (2019) Differential methylation levels in CpGs of the BIN1 gene in Individuals With Alzheimer disease. Alzheimer Dis Assoc Disord 33(4):321–326
Sanders HM, Lust R, Teller JK (2009) Amyloid-beta peptide Aβp3-42 affects early aggregation of full-length Aβ1-42. Peptides 30(5):849–854
Schulz-Schaeffer WJ (2010) The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120(2):131–143
Sharma D, Sharma RK, Sharma N, Gabrani R, Sharma SK, Ali J, Dang S (2015) Nose-to-brain delivery of PLGA-diazepam nanoparticles. AAPS PharmSciTech 16(5):1108–1121
Sharma K (2019) Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol Med Rep 20(2):1479–1487
Shukla S, Tekwani BL (2020) Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front Pharmacol 11:537
Song J, Ahn IS, Kang HS, Myung W, Lee Y, Woo S-y, Ku HM, Hwang T-Y, Carroll BJ, Kim DK (2014) Cognitive subdomain responses to galantamine in Alzheimer’s disease. J Nerv Ment Dis 202(3):253–259
Song Y, Ding H, Yang J, Lin Q, Xue J, Zhang Y, Chan P, Cai Y (2014) Pyrosequencing analysis of SNCA methylation levels in leukocytes from Parkinson’s disease patients. Neurosci Lett 569:85–88
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251(3):205–208
Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu Y-Z, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci 97(12):6763–6768
Takaoka K, Hangaishi A, Ito A, Morioka T, Kida M, Usuki K (2014) Late hematological improvement of myelodysplastic syndrome following treatment with 5-azacitidine therapy. Intern Med 53(19):2241–2243
Tan Y-y, Wu L, Zhao Z-b, Wang Y, Xiao Q, Liu J, Wang G, Ma J-f (2014) Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Parkinsonism Relat Disord 20(3):308–313
Thomas P, Fenech M (2007) A review of genome mutation and Alzheimer’s disease. Mutagenesis 22(1):15–33
Tian F, Marini AM, Lipsky RH (2010) Effects of histone deacetylase inhibitor Trichostatin A on epigenetic changes and transcriptional activation of Bdnf promoter 1 by rat hippocampal neurons. Ann N Y Acad Sci 1199(1):186–193
Tsankova N, Renthal W, Kumar A, Nestler EJ (2007) Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8(5):355–367
van Groen T (2010) DNA methylation and Alzheimer’s disease. In: Tollefsbo T (ed) Epigenetics of aging. Springer, Berlin, pp 315–326
Vetrivel KS, Zhang Y-w, Xu H, Thinakaran G (2006) Pathological and physiological functions of presenilins. Mol Neurodegener 1(1):4
Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM-Y (2001) PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol 168(2):402–412
Voronkov M, Braithwaite SP, Stock JB (2011) Phosphoprotein phosphatase 2A: a novel druggable target for Alzheimer’s disease. Future Med Chem 3(7):821–833
Wang F, Fischhaber PL, Guo C, Tang T-S (2014) Epigenetic modifications as novel therapeutic targets for Huntington’s disease. Epigenomics 6(3):287–297
Wang SC et al (2008) Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 3(7):e2698
Wang Y, Wang X, Li R, Yang ZF, Wang YZ, Gong XL, Wang XM (2013) A DNA methyltransferase Inhibitor, 5-Aza-2′-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci Ther 19(3):183–190
Wang Y, Wang X, Liu L, Wang X (2009) HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci Lett 467(3):212–216
Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW (1994) Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 271(13):992–998
Wilson AG (2008) Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol 79:1514–1519
Winner B, Kohl Z, Gage FH (2011) Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 33(6):1139–1151
Wu J-Y, Niu F-n, Huang R, Xu Y (2008) Enhancement of glutamate uptake in 1-methyl-4-phenylpyridinium-treated astrocytes by trichostatin A. NeuroReport 19(12):1209–1212
Xu X, Gammon MD, Hernandez-Vargas H, Herceg Z, Wetmur JG, Teitelbaum SL, Bradshaw PT, Neugut AI, Santella RM, Chen J (2012) DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J 26(6):2657–2666
Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Jong WY, Strack S, Jeffrey PD, Shi Y (2006) Structure of the protein phosphatase 2A holoenzyme. Cell 127(6):1239–1251
Xu Z, Li H, Jin P (2012) Epigenetics-based therapeutics for neurodegenerative disorders. Curr Geriatr Rep 1(4):229–236
Xuan A-G, Pan X-B, Wei P, Ji W-D, Zhang W-J, Liu J-H, Hong L-P, Chen W-L, Long D-H (2015) Valproic acid alleviates memory deficits and attenuates amyloid-β deposition in transgenic mouse model of Alzheimer’s disease. Mol Neurobiol 51(1):300–312
Yeh HH, Young D, Gelovani JG, Robinson A, Davidson Y, Herholz K, Mann DM (2013) Histone deacetylase class II and acetylated core histone immunohistochemistry in human brains with Huntington’s disease. Brain Res 1504:16–24
Yi JM, Kim TO (2015) Epigenetic alterations in inflammatory bowel disease and cancer. Intest Res 13(2):112–121
Yoo DY, Kim DW, Kim MJ, Choi JH, Jung HY, Nam SM, Kim JW, Yoon YS, Choi SY, Hwang IK (2015) Sodium butyrate, a histone deacetylase Inhibitor, ameliorates SIRT2-induced memory impairment, reduction of cell proliferation, and neuroblast differentiation in the dentate gyrus. Neurol Res 37(1):69–76
Youssef EM, Lotan D, Issa J-P, Wakasa K, Fan Y-H, Mao L, Hassan K, Feng L, Lee JJ, Lippman SM (2004) Hypermethylation of the retinoic acid receptor-β2 gene in head and neck carcinogenesis. Clin Cancer Res 10(5):1733–1742
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong J-S (2005) Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19(6):533–542
Zhong N, Weisgraber KH (2009) Understanding the basis for the association of apoE4 with Alzheimer’s disease: opening the door for therapeutic approaches. Curr Alzheimer Res 6(5):415–418
Zhou X-W, Gustafsson J-Å, Tanila H, Bjorkdahl C, Liu R, Winblad B, Pei J-J (2008) Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiology of disease 31(3):386–394
Acknowledgements
This review is part of a research study financially supported by the Ministry of Higher Education Grant (FRGS/1/2019/SKK08/UKM/01/4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohd Murshid, N., Aminullah Lubis, F. & Makpol, S. Epigenetic Changes and Its Intervention in Age-Related Neurodegenerative Diseases. Cell Mol Neurobiol 42, 577–595 (2022). https://doi.org/10.1007/s10571-020-00979-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-020-00979-z