
Abstract
X-linked adrenoleukodystrophy (X-ALD) is the most frequent peroxisomal disorder that is characterized by progressive demyelination of the white matter, adrenal insufficiency, and accumulation of very long-chain fatty acids in body fluid and tissues. This disorder is clinically heterogeneous with seven different phenotypes in male patients and five phenotypes in female carriers. An ultimate treatment for X-ALD is not available. Depending on the rate of the disease progression and the degree of an individual handicap, special needs and challenges vary greatly. The exact mechanisms underlying the pathophysiology of this multifactorial neurodegenerative disorder remains obscure. Previous studies has been related oxidative stress with the pathogenesis of several disease that affecting the central nervous system, such as neurodegenerative disease, epilepsy, multiple sclerosis, Alzheimer, and Parkinson diseases. In addition, oxidative damage has been observed in various in vivo and in vitro studies with inborn errors of metabolism, including X-ALD. In this context, this review is focused on oxidative stress in X-ALD, with emphasis on studies using biological samples from patients affected by this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
X-linked adrenoleukodystrophy (X-ALD, OMIM # 300100) is the most frequent peroxisomal disorder that occurs in approximately 1 in 17,000 live male births (Bezman et al. 2001). This inborn error of metabolism (IEM) is characterized by progressive demyelination of the white matter, adrenal insufficiency, and accumulation of very long-chain fatty acids (VLCFA), mainly hexacosanoic acid (C26:0) and tetracosanoic acid (C24:0) in body fluid and tissues (Moser et al. 2001). This disease is caused by mutation in ABCD1 gene located in Xq28 chromosome that provokes a loss of function of adrenoleukodystrophy protein (ALDP). ABCD1 encodes a peroxisomal membrane ATP-binding cassette (ABC) half-transporter, which imports cytosolic VLCFA or VLCFA-CoA esters into the peroxisome for degradation by homeostatic β-oxidation (Bezman et al. 2001; Moser et al. 2001, 2005a, b). The accumulation of these fatty acids may contribute to the pathogenesis of X-ALD; however, the role of VLCFA is poorly known. It was assumed that the excess of VLCFA was toxic to myelin and to adrenal cortex and testis, since it was observed the direct toxic effects of C26:0 on adrenocortical cells and the disruptive effects of C26:0 on cell membrane structure, stability, and function (Moser et al. 2001).
This disorder is clinically heterogeneous with seven different phenotypes in male patients and five phenotypes in female carriers—heterozygotes (HTZ). The phenotype cannot be predicted by VLCFA plasma concentration or by the nature of the mutation. There is no clear pattern of genotype-phenotype correlation since several phenotypes may occur in the same family, suggesting involvement of genetic modifiers and/or environmental, stochastic or epigenetic factors. In males, the different phenotypes forms are (1) childhood cerebral form (CCER), (2) juvenile cerebral form, (3) adult cerebral form, (4) adrenomyeloneuropathy (AMN), (5) Addison-only disease, (6) olivo-ponto-cerebellar, and (7) asymptomatic patients (Moser et al. 2001). The two most prevalent phenotypes are CCER form and AMN. The first represents 60 % of male X-ALD patients and it manifests most commonly before ten-year old (Berger et al. 2014). CCER involves a rapid progression of neurological symptoms, including visual and auditory disturbances, decreased school performance, adrenal insufficiency, walking difficulties, demyelination and total disability within three years. The second presents a late-onset (28 ± 9 years) and slowly progression, with peripheral neuropathy, distal axonopathy in spinal cord, sphincter disturbances, sexual dysfunction, impaired vibration, and position sense in the legs. AMN form affects 30–40 % of the total male population with X-ALD and some cases progress over decades. These patients show little or no inflammatory component, and they may be wrongly diagnosed with multiple sclerosis or spastic paraparesis (Moser et al. 2001; Smith et al. 1999; Engelen et al. 2012 Triantafyllou et al. 2014). The asymptomatic form includes individuals with biochemical or genetic abnormalities but without any manifestations of adrenal or cerebral involvement (Triantafyllou et al. 2014).
Female HTZ for X-ALD can present a wide range of five phenotypes namely: asymptomatic, mild myelopathy, moderate to severe myelopathy, cerebral involvement, and adrenal insufficiency (Moser et al. 2001). Approximately 50 % of female carriers over 40-year old may develop mild neurological abnormalities such as an impaired vibration sense and hyperreflexia in the legs as age advances, and 20 % of carriers over the age of 40 years develop an AMN-like syndrome (Moser et al. 2001; Maier et al. 2002). In contrast, the majority of the HTZ younger than 30-year old are asymptomatic. Cerebral involvement and adrenal insufficiency are rare (1 %) (Moser et al. 2005a; Berger and Gärtner 2006).
The plasmatic determination of VLCFA is still the best initial biomarker of X-ALD (Kemp and Wanders 2010). Therefore, the diagnosis of X-ALD hemizygote and female carriers consists in the increased concentrations of C26:0 and C24:0 in serum, even as by high C24:0/C22:0 and C26:0/C22:0 ratios (Moser and Moser 1991). The mutation analysis is the best method to establish the carrier status in women, since false-negative results may occur in 15–20 % of the VLCFA determinations for HTZ (Maier et al. 2002). For HTZ pregnancies, in whom the risk of having an affected male is 25 % (or 50 % if the fetus is known to be male), prenatal diagnosis is achieved by determination of VLCFA levels in cultured amniocytes or cultured chorionic villus cells and by mutation analysis (Wanders et al. 1993; Moser et al. 1999; Kemp et al. 2012). Brain magnetic resonance imaging (MRI) is always abnormal in neurologically symptomatic males which are approximately 85 % of affected individuals, showing a characteristic pattern of symmetric enhanced T-2 signal in the parieto-occipital region with contrast enhancement at the advancing margin. MRI findings often provide the first clue to diagnosis, have great value in assessing prognosis, and help to selection and evaluation of therapeutic approaches (Moser et al. 2001).
An ultimate treatment for X-ALD is not available. Depending on the rate of the disease progression and the degree of an individual handicap, special needs and challenges vary greatly. At this time, current treatment is limited on three modes of therapy in childhood form: adrenal hormone replacement for adrenal insufficiency (present in about 70 % of male patients), Lorenzo’s Oil (LO) therapy for asymptomatic X-ALD boys who have normal cerebral MRI, and hematopoietic stem cell or bone marrow transplantation (BMT) for boys with early stage of cerebral involvement (Moser et al. 2005b; Berger and Gärtner 2006; Moser 2006). However, for symptomatic AMN and HTZ, patients no reasonable treatment is accessible. Most X-ALD patients, specially AMN and HTZ symptomatic patients, need the care of a multi-professional team which includes physiotherapy, urologic consultation for erectile dysfunction, impaired bladder and bowel control, prevention and treatment of urinary infections, endocrinological consultation for the detection and management of adrenal insufficiency, and psychological counseling (Moser et al. 2001; Moser 2006, Engelen et al. 2012). Emerging therapies for X-ALD, such as gene therapy, histone acetyl transferases inhibitors and antioxidant strategies, have been proposed (Berger et al. 2010). A clinical trial with antioxidants in X-ALD is ongoing in Spain, since antioxidants reduce oxidative stress biomarkers and axonal degeneration in the spinal cord of Abcd1 knockout mice (Galea et al. 2012).
The mechanisms that lead to cerebral demyelination, axonal degeneration in spinal cord, and adrenal insufficiency remain obscure, as well as the role played by the main accumulated VLCFA product (C26:0). Singh and Pujol (2010) have proposed a hypothesis for neuropathology in X-ALD enclosing metabolic disease and axonal degeneration (excess of VLCFA, lower plasmalogens, and oxidative stress), induction of inflammation and general loss of peroxisomal function and neurodegeneration. Galea et al. (2012) have come up with the idea that X-ALD is pathogenic continuum of increased severity where multiple noxious factors accumulate in a highly interactive manner to cause CCER or AMN and VLCFA-elicited oxidative stress would be the first hit (reactive oxygen and nitrogen species generation), followed by inflammation leading to CCER and perhaps to AMN.
Previous studies have been related oxidative stress to pathogenesis of several disease that affecting the central nervous system (CNS), such as neurodegenerative disease, epilepsy, multiple sclerosis, Alzheimer, and Parkinson diseases (Halliwell 2006; Halliwell and Gutteridge 2007). In addition, oxidative damage in some inborn errors of metabolism has been observed in various in vivo and in vitro studies (Wajner et al. 2004; Barschak et al. 2006; Sirtori et al. 2005; Ribas et al. 2010; Negretto et al. 2014) as well as in X-ALD (Table 1). In the following sections of this review, we focused on oxidative stress in X-ALD, with emphasis on studies including X-ALD patients.
Free Radicals, Antioxidant Compounds, and Oxidative Stress
Free radical is a highly reactive molecule since it possesses an unpaired electron on the outer atomic or molecular orbitals, which has a strong tendency to initiate chain reactions to complete his own orbital (Halliwell 1994). The reactive oxygen and nitrogen species (ROS and RNS, respectively) comprise both free radicals, like superoxide anion (O •−2 ), hydroxyl radical (OH•) and nitric oxide (NO•), and other molecules, such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO−) (Fridovich 1999; Halliwell and Gutteridge 2007). Radicals derived from oxygen and nitrogen are products of normal cellular metabolism, being ROS the most important class of radical species generated in living systems (Miller et al. 1990).
Beneficial effects of ROS occur at low/moderate production and involve physiological roles in cellular responses against infectious agents, in cellular signaling systems and in gene transcription (Zheng and Storz 2000; Halliwell and Gutteridge 2007; Valko et al. 2007). In addition, NO• produced by endothelial cells and acts as a relevant oxidative biological signaling, like immune response. This abundant RNS is crucial to vasoregulation, leukocyte adhesion, platelet aggregation, angiogenesis, and neurotransmission (Tarpey and Fridovich 2001; Halliwell and Gutteridge 2007). However, the overproduction of free radicals is deleterious and can cause potential oxidative damage in biomolecules (lipids, proteins, carbohydrate, and DNA), culminating in cell injury and death.
Lipids can be oxidized, chlorinated, and nitrated by a range of reactive species (RS) (Halliwell and Gutteridge 2007). Lipid peroxidation is a complex process and comprises three distinct mechanisms: free radical-mediated oxidation, non-enzymatic oxidation, and enzymatic oxidation (Shichiri 2014; Niki et al. 2005). Lipid peroxidation occurs through a chain reaction, which one initiator free radical can oxidize either lipid molecules in biological membranes or low-density lipoproteins (LDL). Lipid peroxidation is commonly regarded to harmful process leading to membrane structural modification like changes in permeability, fluidity, integrity, and functional loss/modification of biomembranes, lipoproteins, proteins, and DNA as well as is usually associated with cellular dysfunction, generating potentially toxic products (Halliwell and Gutteridge 2007).
Oxidative damage to proteins can have wide-ranging damaging effects such as affecting the function of receptors, enzymes, and transport proteins, and perhaps, generating new antigens that can provoke immune responses and can contribute to secondary damage to other biomolecules, for example, inactivation of DNA repair enzymes and loss of fidelity of damaged DNA polymerases in replicating DNA (Halliwell and Gutteridge 2007). Protein oxidative damage results in the formation of carbonyl groups by oxidation of protein side chains, especially proline, arginine, lysine, and threonine and reduction of sulfhydryl groups of susceptible amino acids (Stadman and Levine 2003; Levine et al. 1990). The majority of cellular sulfhydryl groups are protein-bound and the oxidation of these groups from specific cysteine residues to form disulfide, potentially alter the redox state of proteins, leading to their inactivation (Aksenov and Markesbery 2001). Furthermore, protein carbonylation, oxidation of thiol group and alterations of protein structure by oxidants may affect the function of different proteins, resulting in a partial or complete loss of its functionality (Halliwell and Gutteridge 2007; Levine et al. 1990).
DNA subjected to attack by ROS especially by hydroxyl radical generates a huge range of base and sugar modification products (Dizdaroglu et al. 2002). This attack to purine/pyrimidine bases and or deoxyribose sugar result in strand breaks, oxidized bases and formation of DNA adducts, as 8-hydroxy-2′-deoxyguanosine. Therefore, DNA oxidation can lead to mutations that may disturb DNA replication (Halliwell and Gutteridge 2007).
Healthy aerobic organisms had developed antioxidant defense mechanisms aiming to avoid or to reduce the cellular damage provoked by free radical production (or overproduction) maintaining the balance of ROS production (Halliwell and Gutteridge 2007). Antioxidant defense mechanisms involve both enzymatic and non-enzymatic strategies. The enzymatic antioxidant system is composed by superoxide dismutase (SOD), Catalase (CAT), and Glutathione Peroxidase (GPx). SOD dismutates the O •−2 to H2O2, which is less reactive and can be degraded by other enzymes, such as CAT or GPx. Both GPx and CAT detoxify H2O2 by reducing it to water (H2O) and oxygen (O2) (Fridovich 1999; Halliwell and Gutteridge 2007). GPx belong to selenoperoxidases’ family, which contain selenocysteine at their active site (Kühn and Borchert 2002). Besides of H2O2 removal, GPx also reduces organic peroxides into their corresponding alcohols. GPx uses reduced glutathione (GSH) as a hydrogen donor whereby GSH is oxidized to glutathione disulfide (GSSG). The regeneration of GSH is catalyzed by glutathione reductase (GSHRd) (Halliwell and Gutteridge 2007; Kühn and Borchert 2002).
The non-enzymatic antioxidant defenses comprise endogenous substances, such as metal-binding proteins (transferrin, ferritin), uric acid, GSH, ceruloplasmin, albumin, and dietary antioxidants, such as vitamins (A, C, E, B1, B2, B6, and B12), α-lipoic acid, mixed carotenoids, several bioflavonoids, antioxidant minerals (copper, zinc, manganese, and selenium), and folic acid (Halliwell and Gutteridge 2007; Davies 2000). GSH, the major thiol antioxidant and redox buffer of the cell, can be obtained from dietary sources or by endogenous synthesis from glutamate, cysteine, and glycine (Fang et al. 2002).
Redox homeostasis is preserved in vivo through equilibrium between production of oxidant species and antioxidant response. Nevertheless, under certain conditions, if there is a significant increase in RS generation or a decrease in radical elimination from the cell by low antioxidant activities, oxidative cellular stress occurs. The term oxidative stress is currently defined as an imbalance between pro- and antioxidants in favor of the former, which implicates a loss of redox signaling (Halliwell and Gutteridge 2007).
Recently, the role of oxidative stress in neurodegenerative diseases has received much attention, since brain and nervous system are more vulnerable to oxidative stress. Brain processes a large amount of O2 in a relatively small tissue mass. Additionally, some brain areas are rich in iron content that is able to catalyze the generation of ROS. Neuronal membranes are abundant in polyunsaturated fatty acids (PUFAs), which are particularly prone to RS attack and lipid oxidative damage due to their possession of unsaturated double bonds. Other aspects that give to the brain sensibility to oxidative stress include high calcium (Ca2+) traffic across neuronal membranes, the presence of excitotoxic amino acids, and high concentrations of autoxidizable neurotransmitters (dopamine and noradrenaline) (Halliwell 2006). Lastly, the brain contains low to moderate activity of SOD, CAT, and GPx compared to kidney or liver (Halliwell and Gutteridge 2007).
Regarding to important role of oxidative stress in neurodegenerative diseases, various findings have shown that oxidative stress participates in the pathophysiology of some IEM such as aminoacidopathies, organic acidemia, lysosomal, mitochondrial, and peroxisomal diseases (Wajner et al. 2004; Vargas et al. 2004; Barschak et al. 2006; Sirtori et al. 2005; Ribas et al. 2010; Negretto et al. 2014). Although the relation between oxidative stress and IEM pathophysiology is not well elucidated, the accumulation of toxic metabolites is appointed as the main reason for the increase of free radicals.
Oxidative Biomarkers and Antioxidant Defenses in Biological Samples from X-ALD Patients
In recent years, the role of oxidative stress has been highlighted in diverse neurodegenerative diseases, and the imbalance between oxidative stress measures and antioxidant levels in X-ALD is present because of the generation of ROS/RNS during lipid and protein oxidation (Table 1).
Lipid peroxidation is a critical process that form a large diversity of products in variable number (Halliwell and Gutteridge 2007) and is probably the most investigated area of research when it comes to ROS and its important role in cell biology and human health (Ayala et al. 2014). Consequently, mechanisms of lipid hydroperoxides and biologically active metabolites formation, together with their effect on cellular structure and function, are getting more importance in X-ALD pathogenesis study.
Di Biase et al. (2000) have observed in X-ALD patients a slight alteration of oxidative status and an increased susceptibility to LDL oxidation, what was protected by simvastatin therapy. Oxidized lipids can affect cell function by accumulating in the cell membrane causing leakage of the plasmalemma and interfering with the function of membrane bound receptors (Cai and Harrison 2000). Moreover, products of lipid peroxidation (e.g., unsaturated aldehydes and other metabolites) have cytotoxic and mutagenic properties, and LDL oxidation itself has a specific role in the pathogenesis of atherosclerosis, diabetes, and Alzheimer disease (Halliwell and Gutteridge 2007; Seifried et al. 2007).
Lipid peroxidation also leads to the production of conjugated diene hydroperoxides and unstable substances that disintegrate into various aldehydes like malondialdehyde (MDA), 4-hydroxynonenal (HNE), and thiobarbituric acid reactive species (TBA-RS). Considering that light emitted in spontaneous chemiluminescence assay usually arises from peroxidizing lipids due to an increase in ROS/RNS production and that TBA-RS assay reflects the amount of MDA formation (Halliwell and Gutteridge 2007), lipid peroxidation was verified by chemiluminescence and TBA-RS in plasma of X-ALD patients. Chemiluminescence was significantly increased in plasma of CCER patients, and TBA-RS measurement was significantly increased in plasma from AMN, CCER, and asymptomatic patients, as well as in plasma of HTZ for X-ALD (Vargas et al. 2004; Deon et al. 2006, 2007, 2008). In addition, it was observed a marked increase of MDA, measured by high-performance liquid phase chromatography (HPLC), in plasma from CCER and Addison-only patients before BMT (Rockenbach et al. 2012). Besides, it was verified that both human adrenal cortex and brain from X-ALD patients (juvenile and adult cerebral X-ALD and AMN) show evidence of oxidative damage, particularly from lipid peroxidation (HNE and MDA) immunoreactivity in astrocytes and microglia (Powers et al. 2005). Importantly, these alterations on oxidative biomarkers were mainly related to demyelinating cerebral and cerebellar lesions but they were also detected beyond the demyelinating limit (Powers et al. 2005). Khan et al. (2008) have observed, in CCER patient’s brain white matter, an increment of reactive lipid aldehydes (HNE and acrolein). These results strongly indicate that lipid peroxidation is stimulated in X-ALD.
Lipid peroxidation-mediated membrane defects have also been implicated in the decreased reactivity of thiol group membrane proteins (Ohyashiki et al. 1994). It is widely established that various reactive oxygen species react with membrane lipids and proteins, altering cell membrane fluidity, and giving rise to carbonyl group formation into side chains or reduction of sulfhydryl groups of susceptible amino acids (Levine and Stadman 2001). Thiols and carbonyls content are considered two biomarkers of protein damage.
Alterations of protein structure may affect the function of receptors, enzymes, and transport proteins, resulting in a partial or complete loss of protein functionality (Levine et al. 1990). Increased formation of carbonyl groups can also cause an autoimmune response due to the recognition of altered proteins as exogenous (Halliwell and Gutteridge 2007) which, in turn, can trigger and/or intensify an inflammatory process. On the other hand, sulfhydryl groups in proteins may have an antioxidant function since they may scavenge oxidants, thus sparing antioxidants and/or cellular constituents from attack. In this context, Khan et al. (2008) have reported increased levels of carbonyl proteins in all CCER brain areas. In addition, it has been verified a reduced plasma thiols in CCER, AMN and Addison-only patients and a higher plasma carbonyls in AMN and CCER patients compared with controls (Rockenbach et al. 2012; Petrillo et al. 2013). By contrast, plasma carbonyl content was not altered in plasma of Addison-only patients. A significant negative correlation between thiols content and C26:0 was observed in plasma of X-ALD individuals, which suggest a potential link between C26:0 accumulation and protein oxidative damage in pathogenesis of X-ALD (Rockenbach et al. 2012).
Furthermore, carbonyl compounds derived from the oxidation of carbohydrates and lipids can react with proteins, leading to the formation of advanced glycation and lipoxidation end products as malondialdehyde–lysine (MDAL), carboxylmethyl-lysine (CML), and carboxyethyl-lysine (CEL). Moreover, glutamic semialdehyde (GSA—derived from metal-catalyzed oxidation of proline and arginine) and aminoadipic semialdehyde (AASA—resulted from lysine oxidation) represent direct oxidation of carbonyl residues (Autrup et al. 1999; Boyd-Kimball et al. 2005). Therefore, it has shown that the concentration of selected protein oxidative biomarkers (GSA and AASA for carbonylation, CML and CEL for glycoxidation/lipoxidation, and MDAL for lipoxidation), analyzed by CG/MS, were increased by twofold in fibroblasts and peripheral mononuclear cells derived from X-ALD patients (Fourcade et al. 2008; Fourcade et al. 2010).
Oxide nitric (NO•) is a diffusible free radical that performs many roles in diverse physiological and pathological circumstances. The direct toxicity of NO• is modest, involving oxidation reactions, since itself is only a weak oxidant. However, in the presence of superoxide, the overproduction of NO• can be transformed to its most reactive and harmful derivative, peroxynitrite (ONOO−). Therefore, NO-mediated toxicity via peroxynitrite may cause the inhibition of mitochondrial respiration, formation of iron–NO complexes in iron containing enzyme systems, inhibition of antioxidant enzyme systems, induction of lipid peroxidation, elevation of toxic VLCFA, oxidation of protein sulfhydryl groups, nitration of proteins, and nitrosylation of nucleic acids, as well as deamination of DNA bases leading to strand break (Radi et al. 1991; Kahn et al. 1998; Halliwell and Gutteridge 2007). The most studied reaction of RNS with proteins is the conversion of tyrosine to 3-nitrotyrosine which has been related to inflammatory conditions in neurodegenerative diseases, like Alzheimer, Parkinson, and Amyotrophic Lateral Sclerosis (Halliwell and Gutteridge 2007). Nitrotyrosine itself could be toxic by undergoing to redox cycling, by disturbing with signal transduction or by being incorporated into the microtubule protein tubulin and distorting cytoskeleton, leading eventually to cell death (Halliwell and Gutteridge 2007). Gilg et al. (2000) verified the presence of inducible nitric oxide synthase (iNOS) in the astrocytes and microglia/macrophages of ALD inflammatory demyelinative lesions. They also demonstrated the abnormal nitration of proteins with anti-tyrosine antibodies in the center of the ALD brain lesions. The abnormal nitration of proteins is irreversible and prevents phosphorylation, which interferes with their normal metabolic functions. Nitrotyrosylation of proteins is considered to be strong circumstantial evidence for the presence of the highly toxic peroxynitrite molecule and free radicals. Powers et al. (2005) have found the increased iNOS in astrocytes and macrophages along with nitrotyrosylated proteins in the demyelinating lesions, which corroborated with findings of Gilg et al. (2000).
Fourcade et al. (2008) have reported that excess of C26:0 generates intracellular ROS levels in X-ALD human fibroblasts, analyzed by ROS-sensitive 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) probe. Uto et al. (2008) have observed that X-ALD lymphoblasts accumulate VLCFA, synthesize higher levels of free radicals and present higher levels of the membrane-anchored NADPH oxidase subunit gp91 PHOX. Therefore, they have suggested that accumulation of VLCFA in X-ALD lymphoblasts may cause the increased levels of the catalytic subunit of NADPH oxidase gp91 PHOX in membrane fractions and total cell homogenate. These data support previous reports, showing that the accumulation of VLCFA induces changes in cell membrane properties and results in cell signaling alteration. It could be implied that the higher synthesis of free radicals observed in X-ALD lymphoblasts may be a consequence of the higher levels of the catalytic subunit gp91 PHOX, probably playing an important role as modulators of the development/progression of neuroinflammation in X-ALD disease. NADPH oxidases have become the focus of attention due to the discovery that they may play a role in the development/progression of inflammation in disorders affecting the CNS (Uto et al. 2008). Moreover, their results also indicate that X-ALD lymphoblasts produce higher levels of NO• and cytokines like tumoral necrosis factor alpha (TNF-α) and interleukin 1β (IL-1β), which propose additional alterations of other membrane proteins, and support previous studies reporting that cells derived from X-ALD patients display an increased proinflammatory response, through interferon gamma (IFN-γ) and interleukin 12 (IL-12) (Powers et al. 2005).
Most important source of ROS production in many aerobic cells is usually related to the mitochondrial electron transport chain, and this ROS production could contribute to damage of mitochondrial proteins, lipids and DNA (Halliwell and Gutteridge 2007). Comparing nuclear DNA and mitochondrial DNA (mtDNA), the last one seems to be more sensitive to oxidative stress because of the proximity between mtDNA and ROS generated during electron transport and the lack of protective histones (Halliwell and Gutteridge 2007; Lee and Wei 2005). López-Erauskin et al. (2013) have observed, in X-ALD patients’ fibroblasts, which excess of C26:0 generates mtDNA oxidation and specifically impairs oxidative phosphorylation (OXPHOS) triggering mitochondrial ROS production from electron transport chain complexes. They also have detected increased mtDNA oxidation ratios in the white matter zones from AMN patients, which show active demyelinating plaques.
The increment of ROS/RNS formation has been speculated to cause antioxidant consumption and thus, a decline in antioxidant levels or generation of oxidation products from them (e.g., ascorbyl radical and GSSG) can be measured as an index of oxidative stress (Halliwell and Gutteridge 2007). Antioxidant defense mechanisms involve both enzymatic and non-enzymatic strategies. Common non-enzymatic antioxidants include the vitamins A, C, and E, GSH, mixed carotenoids, coenzyme Q10 (CoQ10), several bioflavonoids, minerals (copper, zinc, manganese, and selenium), and cofactors like folic acid, uric acid, albumin, and vitamins B1, B2, B6, and B12 (Halliwell and Gutteridge 2007; Davies 2000).
It is known that albumin, urate, and ascorbate make the major contributions to the total antioxidant capacity of human plasma, largely because of their high concentrations relative to other blood antioxidants such as bilirubin, alpha-tocopherol, and beta-carotene (Shofield and Braganza 1996). Several techniques (colorimetry, fluorescence, and chemiluminescence) have been developed to assess the total antioxidant capacity (Lissi et al. 1995; Kampa et al. 2002; Halliwell and Gutteridge 2007).
Vargas et al. (2004) have observed that total radical-trapping antioxidant potential (TRAP) measurement, which is indicative of the tissue non-enzymatic antioxidant defenses, was not altered in plasma of CCER patients. Since the major contributors of TRAP value in plasma are urate, plasmatic proteins, ascorbic acid, and vitamin E, it is feasible that the concentrations of these substances are not modified in plasma from X-ALD patients (Vargas et al. 2004). However, the known antioxidants do not account for all the measures of TRAP, since in human plasma it has been found that unidentified antioxidants contribute up to 35 % to the experimentally determined TRAP value (Halliwell and Gutteridge 2007).
On the other hand, total antioxidant reactivity (TAR) measurement, which is a measure of the tissue capacity to react with free radicals, was markedly reduced in plasma from CCER patients (Vargas et al. 2004). Also, it was observed a significant decrease of TAR measurement in plasma from HTZ patients (Deon et al. 2008). TAR parameter, a simple method based on luminol-enhanced chemiluminescence, reflects the capacity of the additive to engage in the electron transfer processes to luminol-derived radicals. So, it should be noted that TAR corresponds to an useful index of the capacity of a given tissue to modulate the damage associated with an increased production of free radicals and reflects not only the quantity of antioxidants (given by TRAP), but also, and particularly, its quality (given by its reactivity) (Lissi et al. 1995). Deon et al. (2006) have reported that plasma TAR measurement was not altered in X-ALD patients not receiving LO treatment. In this respect, it should be noticed that three out of four patients were asymptomatic, differently from Vargas’ study that utilized only symptomatic CCER patients and where a significant decrease in plasma TAR was observed (Vargas et al. 2004). Therefore, it appears that TAR values were reduced in plasma of symptomatic but not of asymptomatic X-ALD individuals (Deon et al. 2006).
The non-enzymatic antioxidant defense system has many components, comprising endogenous substances and dietary antioxidants (Halliwell and Gutteridge 2007). A deficiency in any of these components can cause a reduction in the overall antioxidant status and it has been implicated in several disease states, such as atherosclerosis, cancer, diabetes mellitus, and arterial hypertension (Kampa et al. 2002). Total antioxidant status assay (TAS) enables assessment of the integrated antioxidant system, which encompasses all biological components with antioxidant activity like bilirubin, vitamin C, uric acid, polyphenols, and proteins (Kampa et al. 2002). TAS measurement, which represents the quantity of the tissue antioxidants, was significantly reduced in plasma of CCER and AMN patients but not in plasma of asymptomatic and HTZ subjects, when compared to controls (Deon et al. 2007, 2008). Besides, it was found that TAS was significantly reduced in AMN patients when compared to the CCER group (Deon et al. 2007). The authors have verified that the non-enzymatic antioxidant defenses are reduced in plasma of symptomatic but not in plasma of asymptomatic X-ALD individuals, and they suggested that these defenses are possibly preserved in asymptomatic patients because the amount of non-enzymatic antioxidant defenses is probably sufficient to counterbalance the free radical generated.
Plasmalogens comprise all plasmenyl phospholipids containing ethanolamine or choline in the head group of the glycerol backbone and its biosynthesis are allocated in peroxisomes. In mammals, the distribution and composition of plasmalogens varies among different tissues: nervous tissues, kidney, and testis have relatively high levels of plasmalogens and liver has very low amounts of this compound. Plasmalogens have been implicated in several biological processes where they can affect membrane fluidity, mediate signal transduction, and protect against oxidative stress (Wanders and Waterham 2006). Nervous tissues, kidney, and testis are also characterized by high levels of plasmenylethanolamine (PlsEtn), whereas heart and skeletal muscle have high levels of plasmenyl choline (Braverman and Moser 2012). PlsEtn in white matter myelin contains mainly PUFA, including linoleic, arachidonic, and docosahexacosanoic acids (Norton 1984). A drastic reduction in PlsEtn leads PUFA oxidation, and membrane lipid dysregulation in myelin/oligodendrocytes may not only cause demyelination/destabilization but also lead to membrane fragmentation, lipid peroxidation, and produce chemotactic molecules for vascular immune cells (Latorre et al. 2003; Gorgas et al. 2006). Kahn et al. (2008) have observed, in brain white matter of CCER patients, not only increased levels of VLCFA but also reduced levels of PlsEtn. They have reported that the loss of PlsEtn was greatest in the plaque area and lesser, but significant, in histologically normal-looking areas and that its reduction was related to oxidative stress. These authors have suggested that peroxisomal dysfunction during a secondary insult might also contribute to reduced levels of PlsEtn in CCER brain.
The major thiol antioxidant and redox buffer of the cell, the tripeptide GSH, in addition to being a cofactor for the GPx enzyme, is involved in many others metabolic processes, including protective role against oxidative stress. Because GSH blood concentration may reflect GSH status in other less accessible tissues, measurement of both GSH and GSSG in blood has been considered essential as an index of whole-body GSH status (Halliwell and Gutteridge 2007). Fourcade et al. (2008) have observed that the addition of C26:0 (10 mM) to the medium produced a marked decrease of GSH in X-ALD fibroblasts. Petrillo et al. (2013) have proposed to analyze the dysregulation of the redox homeostasis in the blood of X-ALD patients, with particular focus on the GSH system. The lymphocyte concentration of the GSH was significantly decreased and all the oxidized glutathione forms (GSSG + GS-Pro) were significantly increased in AMN phenotype. Confirming the imbalance of the redox status in these patients, the GSSG/GSH was significantly increased. No significant differences between CCER and controls were observed in lymphocytes; however, in erythrocytes, GSH content was significantly decreased.
Activity of GPx, SOD, and CAT in X-ALD patients is also challenging, since not all three antioxidant enzymes are affected in all studies or present a similar standard of variation. In addition, antioxidant enzymes that vary on condition of oxidative stress in X-ALD remain uncertain, since antioxidant enzymes like GPx and SOD may be induced by oxidative stress (to increase their level or activity) or consumed (to decrease their level or activity).
A moderate but significant increase of GPx activity was detected in erythrocytes from CCER patients. CAT and SOD activities were much greater in fibroblasts from these patients. The significant increase of these antioxidant enzyme activities may have been a response to high sustained levels of RS. The increase of CAT and GPx activity suggests that H2O2 is formed in excess, whereas the increase of SOD suggests that O •−2 is formed in excess (Vargas et al. 2004).
Deon et al. (2006) have also evaluated the CAT, SOD and GPx activities in erythrocytes from asymptomatic patients before LO treatment and no significant difference were observed when compared to controls. So far, they have also suggested that the enzymatic and non-enzymatic antioxidant defenses are not significantly altered in X-ALD asymptomatic patients. Corroborating with the above, GPx and SOD activity in erythrocytes was similar between patients and controls in the study of Petrillo et al. (2013).
On the other hand, Powers et al. (2005) have shown an increased manganese-SOD (MnSOD) activity in astrocytes from juvenile and AMN patients. Fourcade et al. (2008) have observed an induction of SOD2 and no induction of SOD1, CAT, or GPx in fibroblasts from X-ALD patients. Brose et al. (2012) have reported that the reduced function variant C47T and the GTAC haplotype in the SOD2 gene are significantly associated with cerebral demyelination in adolescent cerebral X-ALD, adult cerebral X-ALD, and AMN cerebral patients. Considering that X-ALD patients with cerebral demyelination have reduced SOD2 activity, SOD2 genotyping, or enzymatic assays may be used as a predictive measure of cerebral disease development and be used to determine which patients may benefit from antioxidant therapy.
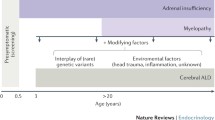
According with all exposed, oxidative stress is an important mediator of neurodegeneration since brain has relatively low levels of antioxidant defenses, high-lipid content, specially unsaturated fatty acids and catecholamines, which are highly susceptible to reactive oxygen species attack (Halliwell and Gutteridge 2007). Evidences of oxidative stress have been found in blood, fibroblasts samples and post-mortem brains from X-ALD patients (Table 1). Also, it was observed signs of oxidative damage/oxidative stress occurring at pre-symptomatic stages (asymptomatic X-ALD patients and HTZ) as well as in symptomatic patients (AMN and CCER). Studies described here, using different areas of brain from CCER patients, report that loss of PlsEtn and alterations in cellular redox are early events that might be involved in the transitions from metabolic to neurologic disease in X-ALD patients. These results suggest that oxidative stress is a generalized phenomenon in different cell types linked to the loss of function of the ABCD1 transporter/ALD protein and the accumulation of fatty acids (Fig. 1). Another interesting issue, that provides proofs of contribution of oxidative damage to X-ALD pathogenesis, was the in vivo and in vitro effect of antioxidants in reversing oxidative damage.

The role of oxidative stress and inflammation in X-ALD
Therapeutics and Oxidative Stress in X-ALD
The absence of curative treatment for X-ALD is well documented. However, it was demonstrated that dietary treatment with a 4:1 mixture of glycerol trioleate and glycerol trierucate (Lorenzo’s Oil, LO) lowers and almost normalizes plasma VLCFA concentrations (particularly the C26:0 levels), but did not ameliorate or alter the rate of the rapid progression of neurological symptoms in the cerebral variants of X-ALD. In addition, favorable responses occur only in patients beginning treatment before the appearance of neurological symptoms (Moser et al. 2005b). Deon et al. (2006) have investigated and evaluated whether oxidative stress parameters were altered in plasma and erythrocytes from asymptomatic and symptomatic X-ALD patients and whether LO treatment might alter these parameters. This study reinforces the hypothesis that lipid peroxidation is induced in plasma from asymptomatic and symptomatic X-ALD patients. Also, it has demonstrated that LO treatment was not capable to reduce this pathogenic process.
Lovastatin and simvastatin were tested as therapeutic drugs for normalizing VLCFA levels in plasma and skin fibroblasts of X-ALD patients (Pai et al. 2000; Singh et al. 1998). Therefore, statins were postulated to be a promissory therapy for X-ALD not only by decreasing VLCFA in X-ALD patients, but also by inhibiting the neuroinflammatory process (Khan et al. 2008). Simvastatin therapy seemed to protect LDL oxidation (Di Biase et al. 2000) and lovastatin decreased the synthesis of NO (Uto et al. 2008) in X-ALD.
Considering the treatment, BMT or hematopoietic stem cell transplantation (HSCT) has been the only known method to halt cerebral demyelination of X-ALD in boys. The mechanism by which BMT is able to halt the demyelinating process remains unclear (Moser 2006; Berger and Gärtner 2006; Peters et al. 2004). Tolar et al. (2007) showed that administration of the antioxidant N-acetyl-l-cysteine (NAC) before and after HSCT in three boys with advanced X-ALD, whose neurologic status and brain radiographic findings were stabilized, resulted in survival of a disease process that would be expected to be fatal. They concluded that NAC merits investigation as therapeutic strategy for patients with advanced X-ALD and has potential to change the lethal disease to a condition amendable to treatment with BMT (Tolar et al. 2007). Rockenbach et al. (2012) have observed that BMT was capable to reduce lipid peroxidation and protein damage in plasma from X-ALD patients (Rockenbach et al. 2012). Since BMT for X-ALD has been proven to be successful, the authors have speculated that the administration of antioxidants should be considered as a potential adjuvant therapy for patients affected by X-ALD which undergone to BMT.
Furthermore, it has been reported that vitamin E (alfa-tocopherol) reverses the oxidative damage in fibroblasts from X-ALD patients (Fourcade et al. 2008). Besides, Fourcade et al. (2010) verified that the ABCD2 mRNA levels in peripheral blood mononuclear cells increased 1.2–2.1-fold above baseline levels in patients X-ALD treated with valproic acid (VPA), a widely used anti-epileptic drug. It was also measured the oxidative biomarkers in the mononuclear cells from control individuals and X-ALD patients, before and after VPA treatment. In X-ALD patients, before VPA treatment, the levels GSA, AASA, and MDAL were 1.3–2.4-fold higher, and 6 months of treatment normalized the values AASA, CML, and CEL, and markedly decreased the levels of GSA and MDAL. Besides, alfa-tocopherol, NAC, or lipoic acid (LA), individually, normalized C26:0 acid-dependent ROS generation in vitro in X-ALD human fibroblasts or all antioxidants combining in a synergistically effects, at low doses, were capable to prevent fully this ROS accumulation (López-Erauskin et al. 2011). López-Erauskin et al. (2012) have provided evidence that oxidative stress induced under galactose conditions leads to mitochondrial damage in the form of mitochondrial inner membrane potential dissipation, ATP drop and necrotic cell death, together with increased levels of oxidative modifications in cyclophilin D protein in fibroblasts from X-ALD patients. NAC in vitro treatment rescues mitochondrial damage markers in fibroblasts from X-ALD patients, including cyclophilin D oxidative modifications, and reverses cyclophilin D induction. These results, demonstrating that antioxidant therapy prevents or rescues axonal degeneration (Fourcade et al. 2008, 2010; López-Erauskin et al. 2011, 2012), have led to phase II clinical trial with a cocktail of antioxidants (NCT01495260) in AMN patients (Galea et al. 2012; Fourcade et al. 2014). The efficacy of this clinical trial will be monitored by longitudinal assessment of oxidative lesions in blood cells and by clinical symptoms (Galea et al. 2012).
Concluding Remarks
Brain has low antioxidant defenses compared with other tissues, a fact that makes this tissue more vulnerable to increased RS. Mounting evidence have shown that oxidative stress has been implicated in the pathophysiology of common neurodegenerative disorders, such as Parkinson´s disease, Alzheimer’s disease, multiple sclerosis as well as in epileptic seizures and demyelination. X-ALD is clinically characterized by central and peripheral demyelination and adrenal insufficiency; however, the mechanisms that underlying the brain damage in X-ALD are poorly known. X-ALD patients (all phenotypes) are susceptible to oxidative damage caused by an increase in free radical production and by depletion in antioxidant capacity. Findings from literature suggest that oxidative stress may contribute to the neurological disturbance in X-ALD and may act synergistically with other underlying mechanisms involved in the pathophysiology of this disease. Therefore, the administration of antioxidants should be considered an adjuvant therapy for X-ALD patients.
Abbreviations
- AASA:
-
Aminoadipic semialdehyde
- ABCD1:
-
ATP-binding cassette (ABC) transporter subfamily D member 1
- ABAP:
-
2,2′-Azo-bis-(2-aminidinopropane)
- AMN:
-
Adrenomyeloneuropathy
- ATP:
-
Adenosine triphosfate
- BMT:
-
Bone marrow transplant
- C22:0:
-
Docosanoic acid
- C24:0:
-
Tetracosanoic acid
- C26:0:
-
Hexacosanoic acid
- Ca2+ :
-
Calcium
- CAT:
-
Catalase
- CCER:
-
Cerebral childhood ALD
- CEL:
-
Carboxyethyl-lysine
- CML:
-
Carboxylmethyl-lysine
- CNS:
-
Central nervous system
- DNA:
-
Deoxyribonucleic acid
- GPx:
-
Glutathione peroxidase
- GSA:
-
Glutamic semialdehyde
- GSH:
-
Glutathione
- GSHRd:
-
Glutathione reductase
- GSSG:
-
Oxidized glutathione
- H2DCFDA:
-
6-Carboxy 2′,7′-dichlorodihydrofluorescein
- H2O2 :
-
Hydrogen peroxide
- HNE:
-
4-Hydroxynonenal
- HO-1:
-
Hemoxygenase-1
- HPLC:
-
High-performance liquid chromatography
- HSCT:
-
Hematopoietic stem cell transplantation
- HTZ:
-
Heterozygote or female carriers
- IEM:
-
Inborn errors of metabolism
- IFN-γ:
-
Interferon gamma
- IL-12:
-
Interleukin 12
- IL-1β:
-
Interleukin 1 beta
- iNOS:
-
Inducible nitric oxide synthase
- LA:
-
Lipoic acid
- LDL:
-
Low-density lipoprotein
- LO:
-
Lorenzo’s oil
- MDA:
-
Malondialdehyde
- MDAL:
-
Malondialdehyde–lysine
- MnSOD:
-
Manganese–superoxide dismutase
- MRI:
-
Magnetic resonance image
- mtDNA:
-
Mitochondrial DNA
- NAC:
-
N-acetyl-cystein
- NaPA:
-
Sodium phenylacetate
- NO:
-
Oxide nitric
- O2 :
-
Oxygen
- O •−2 :
-
Superoxide radical
- OH• :
-
Hydroxyl radical
- ONOO− :
-
Peroxynitrite
- OXPHOS:
-
Oxidative phosphorylation
- PlsEtn:
-
Plasmenylethanolamine
- PUFA:
-
Polyunsaturated fatty acid
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- RS:
-
Reactive species
- SOD:
-
Superoxide dismutase
- TAR:
-
Total antioxidant reactivity
- TAS:
-
Total antioxidant status
- TBA-RS:
-
Thiobarbituric acid reactive species
- TNF-α:
-
Tumor necrosis factor alpha
- TRAP:
-
Total radical-trapping antioxidant potential
- VLCFA:
-
Very long chain fatty acidy
- VPA:
-
Valproic acid
- X-ALD:
-
X-linked adrenoleukodystrophy
References
Aksenov M, Markesbery WR (2001) Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 302:141–145
Autrup H, Daneshvar B, Dragsted LO et al (1999) Biomarkers for exposure to ambient air pollution–comparison of carcinogen-DNA adduct levels with other exposure markers and markers for oxidative stress. Environ Health Perspect 107(3):233–238
Ayala A, Muñoz MF, Argüelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:360438. doi:10.1155/2014/360438
Barschak AG, Sitta A, Deon M et al (2006) Evidence that oxidative stress is increased in plasma from patients with maple syrup urine disease. Metab Brain Dis 21:279–286
Berger J, Gärtner J (2006) X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetics aspects. Biochim Biophys Acta 1763:1721–1732
Berger J, Pujol A, Aubourg P, Forss-Petter S (2010) Current and future pharmacological treatment strategies in X-linked adrenoleukodystrophy. Brain Pathol 20:845–856
Berger J, Forss-Petter S, Eichler FS (2014) Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 98:135–142
Bezman L, Moser AB, Raymond GV et al (2001) Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol 49:512–517
Boyd-Kimball D, Castegna A, Sultana R et al (2005) Proteomic identification of proteins oxidized by Abeta (1–42) in synaptosomes: implications for Alzheimer’s disease. Brain Res 1044:206–215
Braverman NE, Moser AB (2012) Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 1822(9):1442–1452
Brose RD, Avramopoulos D, Smith KD (2012) SOD2 as a potential modifier of X-linked adrenoleukodystrophy clinical phenotypes. J Neurol 259:1440–1447
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Davies KJ (2000) Oxidative stress, antioxidant defenses, and damage removal, repair and replacement systems. IUBMB Life 50(4–5):279–289
Deon M, Wajner M, Sirtori LR et al (2006) The effect of Lorenzo’s oil on oxidative stress in X-linked adrenoleukodistrophy. J Neurol Sci 247:157–164
Deon M, Sitta A, Barschak AG et al (2007) Introduction of lipid peroxidation and decrease of antioxidant defenses in symptomatic and asymptomatic patients with X-linked adrenoleukodystrophy. Int J Dev Neurosci 25:441–444
Deon M, Sitta A, Barschak AG et al (2008) Oxidative stress is induced in female carriers of X-linked adrenoleukodystrophy. J Neurol Sci 266:79–83
Di Biase A, Salvati S, Varí R et al (2000) Susceptibility to oxidation of plasma low-density lipoprotein in X-linked adrenoleukodystrophy: effects of simvastatin treatment. Mol Genet Metab 71(4):651–655
Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H (2002) Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med 32(11):1102–1115
Engelen M, Kemp S, de Visser M et al (2012) X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis 7:51
Fang YZ, Yang S, Wu G (2002) Free radicals, antioxidants, and nutrition. Nutrition 18(10):872–879
Fourcade S, López-Erauskin J, Galino J et al (2008) Early oxidative damage underlying neurodegenaration in X-adrenoleukodystrophy. Hum Mol Genet 17:1762–1773
Fourcade S, Ruiz M, Guilera C et al (2010) Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Hum Mol Genet 19:2005–2014
Fourcade S, López-Erauskin J, Ruiz M et al (2014) Mitochondrial dysfunction and oxidative damage cooperatively fuel axonal degeneration in X-linked adrenoleukodystrophy. Biochimie 98:143–149
Fridovich I (1999) Fundamental aspects of reactive oxygen species, or what’s the matter with oxygen. Ann N Y Acad Sci 893:13–18
Galea E, Launay N, Portero-Otin M et al (2012) Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta 1822:1475–1488
Gilg GA, Singh AK, Singh I (2000) Inducible nitric oxide synthase in the central nervous system of patients with X-adrenoleukodystrophy. J Neuropathol Exp Neurol 59(12):1063–1069
Gorgas K, Teigler A, Komljenovic D, Just WW (2006) The ether lipid-deficient mouse: tracking down plasmalogen functions. Biochim Biophys Acta 1763(12):1511–1526
Halliwell B (1994) Free radicals, antioxidants, and human disease: curiosity, cause or consequence? Lancet 344:721–724
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Halliwell B, Gutteridge JMC (2007) Free radicals in biology and medicine. Oxford University Press, Oxford
Kampa M, Nistikaki A, Tsaousis V, Maliaraki N, Notas G, Castanas E (2002) A new automated method for the determination of the total antioxidant capacity (TAC) of human plasma, based on the crocin bleaching assay. BMC Clin Pathol 2(1):1–16
Kemp S, Wanders RJA (2010) Biochemical aspects of X-linked adrenoleukodystrophy. Brain Pathol 20(4):831–837
Kemp S, Berger J, Aubourg P (2012) X-linked adrenoleukodystrophy: clinical, metabolic, genetic andpathophysiological aspects. Biochim Biophys Acta 1822:1465–1474
Khan M, Pahan K, Singh AK, Singh I (1998) Cytokine-induced accumulation of very long-chain fatty acids in rat C6 glial cells: implication for X-adrenoleukodystrophy. J Neurochem 71(1):78–87
Khan M, Singh J, Singh I (2008) Plasmalogen deficiency in cerebral adrenoleukodystrophy and its modulation by lovastatin. J Neurochem 106(4):1766–1779
Kühn H, Borchert A (2002) Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic Biol Med 33(2):154–172
Latorre E, Collado MP, Fernández I, Aragonés MD, Catalán RE (2003) Signaling events mediating activation of brain ethanolamine plasmalogen hydrolysis by ceramide. Eur J Biochem 270(1):36–46
Lee H-C, Wei Y-H (2005) Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37:822–834
Levine RL, Stadtman ER (2001) Oxidative modification of proteins during aging. Exp Gerontol 36(9):1495–1502
Levine RL, Garland D, Oliver CN et al (1990) Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186:464–478
Lissi E, Salim-Hanna M, Pascual C, Del Castillo MD (1995) Evaluation of total antioxidant potential (TRAP) and total antioxidant reactivity from luminol-enhanced chemiluminescence measurements. Free Rad Biol Med 18:153–158
López-Erauskin J, Fourcade S, Galino J et al (2011) Antioxidants halt axonal neurodegeneration in a mouse model of X-adrenoleukopystrophy. Ann Neurol 70:84–92
López-Erauskin J, Galino J, Bianchi P, Fourcade S et al (2012) Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain 135(Pt12):3584–3598
López-Erauskin J, Galino J, Ruiz M et al (2013) Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum Mol Genet 22(16):3296–3305
Maier EM, Kammerer S, Muntau AC, Wichers M, Braun A, Roscher AA (2002) Symptoms in carriers of Adrenoleukodystrophy relate to skewed X inactivation. Ann Neurol 52(5):683–688
Miller DM, Buettner GR, Aust SD (1990) Transition metals as catalysts of “autoxidation” reactions. Free Radic Biol Med 8(1):95–108
Moser HW (2006) Therapy of X-linked adrenoleukodystrophy. NeuroRx 3:246–253
Moser HW, Moser AB (1991) Measurement of saturated very long chain fatty acid in plasma. In: Hommes FA (ed) Techniques in diagnostics human biochemical genetics. Wiley-Liss, New York, pp 177–191
Moser AB, Kreiter N, Bezman L et al (1999) Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol 45(1):100–110
Moser H, Smith KD, Watkins PA, Powers J, Moser AB (2001) X-linked adrenoleukodystrophy. In: Scriver CR, Sly WS (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New-York, pp 3257–3301
Moser HW, Raymond GV, Dubey P (2005a) Adrenoleukodystrophy- new approaches to a neurodegenerative disease. JAMA 294:3131–3134
Moser HW, Raymond GV, Lu SE et al (2005b) Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch Neurol 62(7):1073–1080
Negretto GW, Deon M, Burin M et al (2014) In vitro effect of genistein on DNA damage in leukocytes from mucopolysaccharidosis IVA patients. Mol Genet Metab 111(2):205–208
Niki E, Yoshida Y, Saito Y, Noguchi N (2005) Lipid peroxidation: mechanisms, inhibition, and biological effects. BBRC 338(1):668–676
Norton WT (1984) Some thoughts on the neurobiology of the leukodystrophies. Neuropediatrics 15:28–31
Ohyashiki T, Sakata N, Matsui K (1994) Changes in SH reactivity of the protein in porcine intestinal brush-border membranes associated with lipid peroxidation. J Biochem 115:224–229
Pai GS, Khan M, Barbosa E et al (2000) Lovastatin therapy for X-linked adrenoleukodystrophy: clinical and biochemical observations on 12 patients. Mol Genet Metab 69:312–322
Peters C, Charnas LR, Tan Y et al (2004) Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood 104(3):881–888
Petrillo S, Piemonte F, Pastore A et al (2013) Glutathione imbalance in patients with X-linked adrenoleukodystrophy. Mol Genet Metab 109:366–370
Powers JM, Pei Z, Heinzer AK et al (2005) Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 64:1067–1079
Radi R, Beckman JS, Bush KM, Freeman BA (1991) Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 288(2):481–487
Ribas GS, Manfredini V, De Mari J et al (2010) Reduction of lipid and protein damage in patients with disorders of propionate metabolism under treatment: a possible protective role of L-carnitine supplementation. Int J Dev Neurosci 28(2):127–132
Rockenbach FJ, Deon M, Marchese DP et al (2012) The effect of bone marrow transplantation on oxidative stress in X-linked adrenoleukodystrophy. Mol Genet Metab 106:231–236
Schofield D, Braganza JM (1996) Shortcomings of an automated assay for total antioxidant status in biological fluids. Clin Chem 42(10):1712–1714
Seifried HE, Anderson DE, Fisher EI, Milner JA (2007) A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem 18:567–579
Shichiri M (2014) The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr 54(3):151–160
Singh I, Pujol A (2010) Pathomecanisms underlying X-adrenoleukodystrophy: a three-hit hypothesis. Brain Pathol 20:838–844
Singh I, Pahan K, Khan M (1998) Lovastatin and sodium phenylacetate normalize the levels of very long chain fatty acids in skin fibroblasts of X-adrenoleukodystrophy. FEBS Lett 426:342–346
Sirtori LR, Dutra-Filho CS, Fitarelli D et al (2005) Oxidative stress in patients with phenylketonuria. Biochim Biophys Acta 1740:68–73
Smith KD, Kemp S, Braiterman LT et al (1999) X-linked adrenoleukodystrophy: genes, mutations, and phenotypes. Neurochem Res 24(4):521–535
Stadtman ER, Levine RL (2003) Free-radical mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25:207–218
Tarpey MM, Fridovich I (2001) Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res 89(3):224–236
Tolar J, Orchard PJ, Bjoraker KJ, Ziegler RS, Shapiro EG, Charnas L (2007) N-acetyl-l-cysteine improves outcome of advanced cerebral adrenoleukodystrophy. Bone Marrow Transplant 39(4):211–215
Triantafyllou P, Economou M, Vlachaki E et al (2014) Multiple endocrine disorders associated with adrenomyeloneuropathy and a novel mutation of the ABCD1 gene. Pediatr Neurol 50(6):622–624
Uto T, Contreras MA, Gilg AG, Singh I (2008) Oxidative imbalance in nonstimulated X-adrenoleukodystrophy-derived lymphoblasts. Dev Neurosci 30:410–418
Valko M, Leibfritz D, Moncola J, Cronin M, Mazura M, Telser I (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Vargas CR, Wajner M, Sirtori LR et al (2004) Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochim Biophys Acta 1688:26–32
Wajner M, Latini A, Wyse AT, Dutra-Filho CS (2004) The role of oxidative damage in the neuropathology of organic acidurias: insights from animal studies. J Inherit Metab Dis 27:427–448
Wanders RJ, Waterham HR (2006) Peroxisomal disorders: the single peroxisomal enzyme deficiencies. Biochim Biophys Acta 1763(12):1707–1720
Wanders RJA, Schutgens RBH, Barth PG, Tager JM, Van Den Bosch H (1993) Postnatal diagnosis of peroxisomal disorders: a biochemical approach. Biochemie 75:269–279
Zheng M, Storz G (2000) Redox sensing by prokaryotic transcription factors. Biochem Pharmacol 59(1):1–6
Acknowledgments
The authors were supported by grants from the Brazilian Foundation Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Grant No. 007481/2011-13), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundo de Incentivo à Pesquisa e Eventos (FIPE/HCPA) (Project Nos. 01-0432, 10-0482 and 13-0247).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors (Marion Deon, Desirèe P. Marchetti, Bruna Donida, Moacir Wajner and Carmen R. Vargas) declare that there are no financial interests and/or no conflict of interest disclosure associated with this manuscript.
Ethical Standards
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Rights and permissions
About this article

Cite this article
Deon, M., Marchetti, D.P., Donida, B. et al. Oxidative Stress in Patients with X-Linked Adrenoleukodystrophy. Cell Mol Neurobiol 36, 497–512 (2016). https://doi.org/10.1007/s10571-015-0234-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-015-0234-2