
Abstract
A gene encoding halohydrin dehalogenase from an alphaproteobacterium (AbHHDH) was identified, cloned and over-expressed in Escherichia coli. AbHHDH was able to catalyze the stereoselective dehalogenation of prochiral and racemic halohydrins. It showed the highest enantioselectivity in the dehalogenation of 20 mM (R,S)-2-bromo-1-phenylethanol, which yielded (S)-2-bromo-1-phenylethanol with 99% ee and 34.5% yield. Moreover, AbHHDH catalyzed the azidolysis of epoxides with low to moderate (S)-enantioselectivity. The highest enantioselectivity (E = 18.6) was observed when (R,S)-benzyl glycidyl ether was used as the substrate. A sequential kinetic resolution catalyzed by HHDH was employed for the synthesis of chiral 1-chloro-3-phenoxy-2-propanol. We prepared enantiopure (S)-isomer with a high enantiopurity of ee > 99% and a yield of 30.7% (E-value: 21.3) by kinetic resolution of 20 mM substrate. The (S)-isomer with 99% ee readily obtained from 40 to 150 mM (R,S)-1-chloro-3-phenoxy-2-propanol. Taken together, the results of this study demonstrate the applicability of this HHDH for the production of optically active compounds.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Halohydrin dehalogenases (HHDHs) are employed by microbes for the biodegradation of halogenated organic compounds [1]. Biochemical and structural studies have shown that HHDHs and the enzymes of the short-chain dehydrogenases/reductases family (SDR) are not only evolutionarily but also mechanistically related, catalyzing the dehalogenation of ortho-halogenated alcohols into their corresponding epoxides [1, 2]. In the reverse reaction, they can accept a variety of alternative anionic nucleophiles such as CN−, NO2−, and N3−, to open the epoxide ring, which provides a novel biocatalytic strategy for the preparation of fine chemicals and pharmaceutic intermediates [2, 3].
Recent investigations focus on the biotechnological applications of HHDHs, especially regarding the stereoselective biotransformations [1]. At the early stage, HHDHs were utilized in the kinetic resolution of racemic halohydrins to obtain an optically active enantiomer [4,5,6,7]. Chiral epoxides are valuable and extremely important building blocks for the synthesis of enantiomerically pure pharmaceuticals and agrochemicals. In addition, the kinetic resolution of racemic epoxides by HHDHs is another way to obtain the optically pure isomers [3, 8,9,10]. The HHDH-catalyzed stereoselective ring opening of epoxides is widely used in the asymmetric synthesis of enantiopure β-substituted alcohols, which are important building blocks for a number of pharmaceuticals and biologically active compounds [11,12,13,14]. The most successful example is the synthesis of the statin side-chain precursor ethyl (R)-4-cyano-3-hydroxybutyrate by coupling highly engineered variant of HheC with stereoselective ketoreductase [15, 16].
Despite these diversity of reported biocatalytic applications, it is particularly surprising to remember that all the reactions studied in the past 20 years have involved only a very small number of HHDHs [2]. Before 2014, only six different HHDH sequences had been cloned, characterized and classified into three different phylogenetic subtypes A, B, and C by sequence homology and substrate specificity [1]. To explore novel promising and attractive HHDHs, many new techniques and approaches have been adopted. Among them, database mining is an effective strategy to discover new HHDHs. Using a database mining approach based on the two sequence motifs representing the nucleophile binding pocket (TX4[FY]XG) and catalytic triad (SX12YX3R), the number of available HHDH sequences was greatly increased [1, 17]. Four new HHDH family subtypes, D through G, could be identified, with percentage identities below 30% for protein sequence alignments between them [1, 17]. Of these HHDHs, 21 representative proteins from six phylogenetic subtypes were characterized in detail, several of which showed broad substrate spectra [13, 18,19,20,21,22]. Among all the HHDHs mentioned above, HheC still seems to be the only HHDH enzyme with relatively high enantioselectivity in the conversion of various substrates, whereas most other HHDHs are either non-selective or display only low to moderate enantioselectivity toward a few halohydrins and epoxides [1, 18]. Thus, there is still a lack of enantioselective HHDHs with high enantioselectivity for various halohydrins and epoxides. Therefore, it is necessary to identify and clone more enantioselective HHDH for further characterization of their biochemical and structural properties and for further applications in organic synthesis.
Herein, a series of predicted putative HHDHs were selected as the candidates on the basis of NCBI database searches with the reported HheC and HheB as query sequences. Eight genes bearing 36–46% amino acid sequence identity with the probe HHDHs were cloned and expressed in Escherichia coli BL21(DE3). After one round of testing and screening, the HHDH from an alphaproteobacterium (isolate 46_93_T64), was identified as the best candidate based on its high enantioselectivity toward (R,S)-phenyl glycidyl ether. The substrate scope of this novel enzyme was investigated in detail. Furthermore, sequential kinetic resolution of (R,S)-1-chloro-3-phenoxy-2-propanol catalyzed by the halohydrin dehalogenase in conjunction with azide was performed to verify the practical applicability of HHDH.
2 Materials and Methods
2.1 Materials
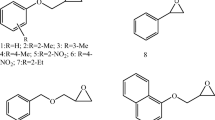
The reagents 1,3-dichloro-2-propanol (1), 1,3-dibromo-2-propanol (2), (R,S)-2,3-dichloro-propanol (3), (R,S)-2,3-dibromo-2-propanol (4), (R,S)-2-chloro-1-phenylethanol (5), (R,S)-2-bromo-1-phenylethanol (6), (R,S)-1-chloro-3-phenoxy-2-propanol (7), (R,S)-epichlorohydrin (8), (R,S)-styrene oxide (9), (R,S)-phenyl glycidyl ether (10), (R,S)-benzyl glycidyl ether (16), (R,S)-naphthyl glycidyl ether (17) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). All reagents were of analytical grade. The racemic epoxides 11–15 were prepared according to a previously described synthesis method [23]. Isopropyl-β-d-thiogalactopyranoside (IPTG) and kanamycin were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Yeast extract, tryptone and agar were purchased from Oxoid (Basingstoke, England). All other organic compounds were obtained from local commercial suppliers and used without further purification (Fig. 1).

Substrates used in the dehalogenation and epoxide ring opening reactions with AbHHDH [1: 1,3-dichloro-2-propanol, 2: 1,3-dibromo-2-propanol, 3: (R,S)-2,3-dichloro-propanol, 4: (R,S)-2,3-dibromo-2-propanol, 5: (R,S)-2-chloro-1-phenylethanol, 6: (R,S)-2-bromo-1-phentlethanol, 7: (R,S)-1-chloro-3-phenoxy-2-propanol, 8: (R,S)-epichlorohydrin, 9: (R,S)-styrene oxide, 10: (R,S)-phenyl glycidyl ether, 11: (R, S)-1,2-Epoxy-3-(2-methylphenoxy)propane, 12: (R, S)-1,2-epoxy-3-(3-methylphenoxy)propane, 13: (R, S)-1,2-epoxy-3-(4-methylphenoxy)propane, 14: (R, S)-1,2-epoxy-3-(2-nitrophenoxy)propane, 15: (R, S)-l-(2′-ethylphenoxy)-2,3-epoxy-propane, 16: (R,S)-benzyl glycidyl ether, 17: (R,S)-naphthyl glycidyl ether]
2.2 Strains and Plasmid
Escherichia coli BL21 (DE3) (Novagen, Darmstadt, Germany) was used as host strains for the heterologous expression of recombinant HHDH. pET28a (+) was used as vector for the expression of HHDH gene in E. coli BL21 (DE3). E. coli transformants were grown in Luria–Bertani (LB) medium.
2.3 Database mining for HHDHs
A toolbox of HHDHs was collected by database mining. The candidates were further confirmed as putative HHDHs based on the presence of the conserved motifs [1]. A total of 8 putative HHDHs were selected and confirmed by multiple sequence alignment with other reported HHDHs using ClustalW2 program (Table 1). The HHDH nucleotide sequences were synthesized by Suzhou Hongxun Biotechnological Development (China) using the PCR assembly method after condons optimization for E. coli. The HHDH genes were cloned individually into the pET28a vector between the NcoI and XhoI restriction endonuclease sites. The ligated plasmid pET28a-HHDHs were transformed into E. coli BL21 (DE3). After heterogeneous overexpression in E. coli BL21 (DE3), the activity and enantioselectivity toward (R,S)-phenyl glycidyl ether were determined, as listed in Table 1.
2.4 Protein Purification
Recombinant E. coli cells harboring the pET28a-AbHHDH were cultured in 2 L of LB with kanamycin (50 µg/mL) at 37 °C. When the optical density at 600 nm reached 0.6, the cells were induced by addition of 0.1 mM IPTG and growth was carried out at 28 °C for extra 10 h. The cells were harvested by centrifugation at 10,000×g for 10 min. The purification of recombinant HHDH was carried out at 0–4 °C. For preparation of the crude enzyme, 4.0 g of recombinant E. coli (wet cell weight) was resuspended in 40 mL Tris–SO4 buffer (20 mM, pH 8.0). After sonification for 15 min in an ice bath, crude enzyme was prepared by centrifugation of the lysate at 15,000×g for 20 min at 4 °C. The supernatant was fractionated by the addition of ammonium sulfate between 30 and 70% saturation. The active precipitate was collected by centrifugation at 12,000×g for 20 min and dissolved in the buffer. The as-prepared solution was loaded onto a HiTrap Phenyl Sepharose column, which had been equilibrated with 1.0 M (NH4)2SO4/20 mM Tris–SO4 buffer (pH 8.0). After washing the column with the same buffer, the adsorbed proteins were eluted with Tris–SO4 buffer (20 mM, pH 8.0) supplemented with a linear gradient of ammonium sulfate (from 1.0 to 0 M). The active fractions were combined and dialyzed against the Tris–SO4 buffer (20 mM, pH 8.0). Finally, the fractions with HHDH activity were loaded onto a Superdex 75 column, pre-equilibrated with Tris–SO4 buffer (20 mM, pH 8.0), and the enzyme was eluted with the same buffer at a flow rate of 0.5 mL/min. Aliquots containing pure HHDH were collected and used for subsequent studies. SDS-PAGE was applied to check the purity of the protein [24], and the protein concentration was determined using a Bradford protein assay kit with bovine serum albumin as the standard.
2.5 Substrate Specificity and Enantioselectivity of the AbHHDH
The substrate specificity of recombinant AbHHDH was determined by measuring the dehalogenation rates of seven different halohydrins as well as monitoring the ring-opening reaction of nine different epoxides in the presence of azide. Analytical-scale reactions were carried out in 2 mL Eppendorf tubes with a total reaction volume of 400 µL. Stock solutions comprising halohydrins 1–7 in ethanol were added to a final concentration of 20.0 mM into Tris–SO4 buffer (100 mM, pH 8.0). The epoxide ring-opening reactions were carried out in 100 mM Tris–SO4 buffer (pH 7.5) containing 20 mM epoxides 9–16 and 40 mM NaN3. The reactions were started by the addition of enzyme in Tris–SO4 buffer (pH 8.0 or 7.5) and incubated at 30 °C and 150 rpm. Samples were collected at regular time intervals, extracted twice with 1 mL ethyl acetate and dried over with anhydrous Na2SO4. The conversion, activity and enantiomeric excess (ee) values were determined using HPLC or GC analysis. One unit of enzyme activity was defined as the amount of enzyme that catalyzed the formation of 1 µmol of epoxide per min under the described assay conditions. The specific activities were expressed in U/mg, and all experiments were conducted in triplicate. Activity assay of the control contained cell-free extracts obtained using the pET28a vector without the HHDH gene in E. coli BL21(DE3), cultured in the same way.
2.6 Analytical Methods
The ee and yields of halohydrins, epoxides and azido alcohol were determined by chiral GC analysis on a capillary BGB-175 column, or HPLC analysis on Chiralcel AS-H and Chiralcel OD-H columns (250 × 4.6 mm, particle size 5 µm, Daicel Chemical Industries, Tokyo, Japan) under the conditions described in the Supplementary information. The enantiomers of the epoxides was assigned by comparison of their elution order on the GC or HPLC columns with published data [21, 25,26,27,28,29]. The enantiomers of the azido alcohols were identified by analogy according to the reduced epoxides. The ees and eep were derived from the remaining epoxide and formed azido alcohols of the two enantiomers as [ee(%) = (S − R)/(S + R) × 100]. R and S are the concentrations of (R) and (S)-enantiomer, respectively. E values were calculated by formula E = ln[(1 − c)(1 − ees)]/ln[(1 − c)(1 + ees)] (c, conversion) [30].
2.7 Sequential Kinetic Resolution of (R,S)-1-Chloro-3-Phenoxy-2-Propanol
Freshly prepared E. coli cells expressing AbHHDH were resuspended to a density of 5 g/L cell dry weight (cdw) in 0.2 M Tris–SO4 buffer (pH 7.0, 7.5 or 8.0) containing (R,S)-1-chloro-3-phenoxy-2-propanol (20–150 mM) and 1 equiv of NaN3 to a 10 mL system in a shake-flask. Reactions were carried out at 20 °C and 150 rpm. Reaction progress was periodically monitored by taking samples (0.5 mL) from the reaction mixture. The samples were extracted with ethyl acetate (1 mL), dried over Na2SO4 and analyzed by HPLC.
2.8 Nucleotide Sequence Accession Number
The nucleotide sequence of AbHHDH has been deposited in the GenBank database under the accession number MH782148.
3 Results and discussion
3.1 Identification and screening of HHDHs
A genome mining approach was adopted to search for robust HHDHs that might be able to catalyze the azide-mediated ring opening reaction of epoxides to corresponding azido alcohols with high enantioselectivity. In total, 8 HHDHs with 36–46% identity with HheC or HheB were selected from the NCBI database and overexpressed in E. coli BL21 (DE3) to form a HHDH toolbox. After testing their activities and enantioselectivities toward (R,S)-phenyl glycidyl ether as a representative substrate (Table 1), HHDH from an alphaproteobacterium 46_93_T64 exhibited the high specific activity [550 U/g cell wet weight (cww)] and enantioselectivity (99% ee). Therefore, the AbHHDH was chosen for further studies.
A protein BLAST search was conducted with AbHHDH as the search sequence, which showed that it has moderate sequence similarity to known HHDH, reaching 41% identity with Hhe C from Agrobacterium radiobacter AD1 [31], 36% identity with HheA from Corynebacterium sp. N-1074 [32], 42% identity with TmHHDH from Tistrella mobilis ZJB1405 [21], 56% identity with PlHHDH from Parvibaculum lavamentivorans DS-1 [19]. Sequence analysis indicated that the cloned HHDH gene contained an open reading frame with 732 bp encoding a 243 amino-acid protein. Multiple alignments of AbHHDH and other known HHDHs showed that it shares the N-terminal motif TX4[FY]XG for nucleophile binding and the catalytic triad of Ser135-Tyr148-Arg152 (Fig. S1) [1]. The expression of HHDH was clearly visible in the SDS-PAGE of the soluble fraction of the E. coli cells taken at 10 h. When a cleavable His-tag was fused to the N- or C-terminus of AbHHDH, it was almost completely inactivated. Therefore, it was purified to apparent homogeneity by salting out with ammonium sulfate, hydrophobic chromatography and gel chromatography which yielded 52 mg/L purified AbHHDH. The purified enzyme had a molecular weight of 27 kDa (Fig. S2), which was in good agreement with the theoretical value (26.2 kDa) calculated using ExPASy. The specific activity of AbHHDH for the azidolysis of (R,S)-phenyl glycidyl ether was determined to be 27.5 U/mg protein.
3.2 Substrate specificity and enantioselectivity
Substrate specificity and enantioselectivity are among the most important enzyme characteristics for biocatalysis. To map the substrate spectrum of this newly mined HHDH, various halohydrins and epoxides of diverse structure were employed. The activity assay of the control, using a cell-free extracts obtained by expression of the pET28a vector without the HHDH gene in E. coli BL21(DE3), did not show any activity toward any of the tested substrates. The activity and enantioselectivity of AbHHDH in the dehalogenation of different halohydrins (1–7) are summarized in Table 2. Almost all the tested substrates were accepted by AbHHDH. The highest activity was measure for the dehalogenation of 1,3-dibromo-2-propanol (17.2 U/mg), while 1,3-dichloro-2-propanol, (R,S)-2-bromo-1-phenylethanol and (R,S)-2,3-dibromo-2-propanol were converted with good specific activities, reaching 48.8%, 12.2% and 6.9% relative activity, respectively. With the substrate (R,S)-2-chloro-1-phenylethanol, only a low activity could be detected, and there was no detectable activity were observed with (R,S)-2,3-dichloro-propanol. (R,S)-3 is efficiently dehalogenated by HheC but was a poor substrate for all other currently characterized HHDH. These results indicate that a halogen in the α position relative to the hydroxyl group seems to be beneficial with respect to the specific activity of the HHDHs. For instance, the activity of AbHDH toward (R,S)-compound 4 was less than 6.9% of that toward the prochiral halohydrin 2 (Table 2). Furthermore, the activity with bromine-substituted compounds were higher than with equivalent chlorine-substituted compounds. The reason why AbHHDH prefers brominated alcohols as substrates is that chlorine has a higher electronegativity than bromine, so that the strength of the carbon-bromine bond is much weaker than that of the carbon-chlorine bond [2, 18]. The specific activities toward the aromatic substrates (R,S)-2-chloro-1-phenylethanol and (R,S)-1-chloro-3-phenoxy-2-propanol were generally low. One of the reasons could be steric hindrance, which has been observed to affect the reactivity substantially.
Our interest was focused on the enantioselectivity of AbHHDH in the conversion of various halohydrins. AbHHDH catalyzed the transformation of prochiral 1,3-dichloro-2-propanol and 1,3-dibromo-2-propanol into (S)-epichlorohydrin (ee = 78.3%) and (S)-epibromohydrin (ee = 62.9%). Asymmetric synthesis using prochiral halohydrins is particularly attractive because the theoretical yield of one enantiomer in this case is 100% [33]. Compared with other HHDHs, the enantioselectivity of AbHHDH toward prochiral halohydrins was similar to that of HHDH from T. mobilis ZJB1405 and different from that of the HheB and HheB8 from Corynebacterium sp. N-1074 and Bradyrhizobium erythrophlei [21, 22]. The epoxides produced by HheC and HheA were almost racemic, suggesting that enantioselective dehalogenation of prochiral halohydrins is not a general property of all HHDHs [34,35,36]. Furthermore, the enantioselectivity of AbHHDH in the conversion of racemic haloalcohols 3–7 was studied by chiral GC and HPLC. AbHHDH was able to convert (R,S)-2,3-dibromo-2-propanol into (S)-epibromohydrin. This indicated that the (R)-2,3-dibromo-2-propanol was converted preferentially, followed by a slower conversion of the (S)-enantiomer. Similar to the (R)-selective HheC, AbHHDH also preferentially converted the (R)-enantiomer of (R,S)-2-chloro-1-phenylethanol [37]. So far, HheC still seems to be the only enzyme exhibiting a comparably high R-enantioselectivity in the conversion of (R,S)-2-chloro-1-phenylethanol (E = 65), while other reported HHDHs are either non-selective or display only low enantioselectivity toward haloalcohol 5 (E ≤ 4.7) [18, 37]. In the dehalogenation reaction of (R,S)-2-chloro-1-phenylethanol, AbHHDH showed a moderate enantioselectivity with an E value of 6.43. We also investigated the enantioselectivity of AbHHDH in the dehalogenation of (R,S)-2-bromo-1-phenylethanol. The (R)-enantiomer was converted preferentially, followed by a much slower conversion of the (S)-enantiomer. (S)-2-bromo-1-phenylethanol was obtained with > 99% ee and 34.5% yield (E = 27.7). Thus, AbHHDH exhibited a relatively high enantioselectivity among the reported HHDHs, with the 2nd highest R-enantioselectivity of all known HHDHs in literature [1, 2]. From this point of view, AbHHDH may be of considerable interest in biocatalysis.
In the reverse reaction, which is epoxide-ring opening, different terminal epoxides can be accepted. However, there were significant differences in the activity and enantioselectivity among the accepted substrates. The substituents at the phenyl ring had an effect on the activity, but no general trend was observed. The position of the substituent at the phenyl ring also affected the enzyme activity, which increased from 3′<4′<2′ for the CH3 group. AbHHDH showed high activity in the kinetic resolution of (R,S)-phenyl glycidyl ether (27.2 U/mg) but only low activity toward (R,S)-1-chloro-3-phenoxy-2-propanol (0.37 U/mg). This could imply that AbHHDH preferentially catalyzes epoxide ring opening of (R,S)-10 using azide as a nucleophile, compared to the elimination of a chloride ion from the corresponding (R,S)-7. Although, the methyl- substituted phenyl glycidyl ether derivatives were well accepted by the AbHHDH, their nitro- and methoxy- derivatives were transformed at a slower rate. Moreover, the enzyme did not perform well with (R,S)-naphthyl glycidyl ether (2.6 U/mg) and benzyl glycidyl ether (1.7 U/mg), which might be due to steric hindrance. Moreover, AbHHDH displayed only poor activity on styrene oxide.
To further confirm the enantiopreference of the enzyme, its selectivity in the ring-opening reaction of epoxides was also investigated, as shown in Table 3. AbHHDH showed moderate enantioselectivity for substrates 10, 15, and 16, while poor enantioselectivity was observed for substrates 8, 9 and 17. In contrast to (R)-selective HheA, AbHHDH preferentially converted the (S)-enantiomer of epoxide 10 [10]. The substituent and its position at the phenyl ring also affected the enantioselectivity of HHDH in the biocatalytic azidolysis of epoxides. Compared to the epoxides with a substitution at the para- or ortho position (11, 13), the epoxide with a substitution at the meta position (12) was more selectively converted (higher ee). It is worth noting that a nitro group at the 2′-position greatly increased the enantioselectivity to 99% ee, while the ee values were 32.4% with 2′-CH2CH3 substituents and 56.1% ee with 2′-CH3 substituents. It was also found that a larger side-chain (17) could decrease the enantioselectivity to some extent, which is in agreement with earlier reports [25].
Thus AbHHDH was able to catalyze the enantioselective conversion of various prochiral and racemic halohydrins, as well as epoxides, yielding enantiomerically enriched (S)-epoxides, (S)-halohydrins and (R)-epoxides. This preference is significantly different from other well-known HHDHs such as HheC and HheA, demonstrating the unique substrate specificity and special synthetic applications of AbHHDH.
3.3 Sequential kinetic resolution catalyzed by halohydrin dehalogenase
Compound 7, (R,S)-1-chloro-3-phenoxy-2-propanol was chosen as the substrate to investigate the sequential kinetic resolution process. The reactions that can occur starting from a halohydrin and N3− are depicted in Fig. 2. The ring closure of (R,S)-7 yields (R,S)-phenyl glycidyl ether (10), which can again act as a substrate for the enzyme. Ring opening of (R,S)-10 by N3− results in the formation of the corresponding azido alcohol.

Sequential kinetic resolution of (R,S)-1-chloro-3-phenoxy-2-propanol
Firstly, kinetic resolution of (R,S)-10 catalyzed by AbHHDH was tested in the presence of NaN3. When the kinetic resolution was conducted at 20 mM (R,S)-10, a moderate enantioselectivity (E = 9.98) was obtained, leading to the formation of 16.7% (R)-10 with an ee > 99%, and the enantiopurity of the product was very low (data not shown). The azidolysis of (R,S)-10 leads to 1-azido-3-phenoxy-2-propanol, which was established through a comparison with the 1H NMR spectrum reported in the literature [38]. The enantiomers of 1-azido-3-phenoxy-2-propanol were assigned by analogy according to the reduced epoxides configuration and comparing the retention times with reference materials [28]. To date, limited numbers of HHDHs were reported to catalyze azidolysis of (R,S)-10. HHDH from Arthrobacter sp. AD2 was reported to catalyze the ring-opening of (R,S)-10 with low (S)-enantioselectivity (E = 2) [10]. In contrast to the (S)-selective AbHHDH, the recombinant HHDH from Bradyrhizobium erythrophlei catalyzed the azidolysis of (R,S)-10, yielding (S)-10 with 99% ee [22].
Bioconversions of (R,S)-7 in the absence of azide were subsequently carried out. At the initial stage of the reaction, AbHHDH converted (R)-7 to the corresponding epoxide in preference to the other enantiomer. However, as the enzyme reaction is reversible, the released chlorine opens the epoxides, resulting in an equilibrium within an hour, producing racemic epoxides. This issue can be solved by removing the produced epoxide or the chloride. We tested the ring-closure reaction of AbHHDH in the presence of NaN3, allowing the AbHHDH itself to eliminate the (R,S)-compound 10 by catalyzing a successive step. The advantage of the sequential reaction is that the enantioselectivity of the first step can be improved. The accumulation of (R,S)-epoxide 10 can be avoided, and (S)-7 is obtained in the presence of azide in a one-pot reaction starting from (R,S)-7. When this reaction was carried out, the conversion rate of (R,S)-7 increased markedly, resulting in the formation of 1-azido-3-phenoxy-2-propanol. Only a small amount of (R,S)-10 transiently accumulated. Since the intermediate epoxide (S)-10 was efficiently and irreversibly converted by the AbHHDH to (R)-1-azido-3-phenoxy-2-propanol, the conversion of (R)-7 was drawn to completion. The reaction rate of the (S)-enantiomer was slower, and this resulted in a high optical purity of the remaining halohydrin (> 99% ee). As shown in Fig. 3, the sequential kinetic resolution of 20 mM (R,S)-7 by using resting E. coli cells reached an ee of > 99% within 13 min, while the final yield of enantiopure (S)-7 and the enantiomeric ratio (E) were 30.7% (theoretical yield = 50%) and 21.3, respectively.

Biotransformation of (R,S)-3-chloro-1-phenoxy-2-propanol with recombinant E. coli expressing AbHHDH in the presence of NaN3. The reactions were performed with (R,S)-3-chloro-1-phenoxy-2-propanol (20 mM), 50 mM NaN3 and resting cells (5 g cdw/L) in Tris–SO4 buffer (200 mM, pH 7.5) at 30 °C
The reaction steps shown in Fig. 2 can be influenced by several factors such as pH and the concentration of the organic substrates. The pH of the reaction medium influences the rate of enzyme-catalyzed ring-closure and ring-opening reactions and the position of the equilibrium between these reactions. To identify suitable reaction conditions, the influence of pH on all aspects of the sequential kinetic resolution process was investigated. Table 4 shows that the conversion rate of the (R,S)-7 increased as the pH of the reaction mixture was increased from 7.0 to 8.0. The enantiomeric excess was not affected by the pH over the range 7.0–8.0, but the yield of (S)-7 decreased.
To further test the feasibility of HHDH catalyzing enantioselective resolution of (R,S)-7, resting cells of E. coli expressing AbHHDH were incubated at pH 7.0 with (R,S)-7 at various substrate concentrations ranging from 40 to 150 mM. Enantiopure (S)-7 with a high optical purity (ee > 99%) was readily obtained from 40 to 150 mM (R,S)-7 within 120 min. As shown in Table 5, the final yield of (S)-7 decreased as more (R,S)-7 was used initially, while the reaction time required for > 99% ee was prolonged. A yield of 27.8% enantiopure (S)-7 could be obtained from 40 mM (R,S)-7 after 22 min incubation, while the optical yield from the conversion of 60 mM (R,S)-7 was 26.4% after 40 min incubation. When the initial concentration was further increased up to 150 mM, (S)-7 with 99% ee was prepared with 22.1% yield. These results suggest that HHDH-catalyzed sequential kinetic resolution can be used to synthesize useful chiral building blocks.
4 Conclusions
In summary, a toolbox comprising several novel HHDHs was developed by data mining approaches. A novel HHDH, which exhibited a high activity and good enantioselectivity toward (R,S)-phenyl glycidyl ether, was successfully discovered from an alphaproteobacterium. AbHHDH exhibits varied activities (0.02–27.6 U/mg) toward the tested halohydrins and epoxides. AbHHDH catalyzed the conversion of prochiral halohydrins 1 and 2 to (S)-epichlorohydrin and (S)-epibromohydrin with 78.3% ee and 62.9% ee, demonstrating its potential for the direct synthesis of chiral epoxides. Among all the test racemic halohydrins, HHDH exhibited the highest enantioselectivity toward (R,S)-2-bromo-1-phenylethanol, producing (S)-2-bromo-1-phenylethanol with 99% ee and 34.5% yield. To the best of our knowledge, AbHHDH has the second-highest R-enantioselectivity toward halohydrins of all the HHDHs reported to date, and thus may be of considerable interest in biocatalysis. In addition, AbHHDH catalyzed the ring opening of epoxides with low to moderate enantioselectivity. The best result was obtained in the preparation of (S)-benzyl glycidyl ether, reaching 99% ee with 28.5% yield. Furthermore, this is also the first study to our knowledge to describe a sequential kinetic resolution process using AbHHDH, which provides a green and efficient synthesis route for (S)-1-chloro-3-phenoxy-2-propanol, an intermediate for the preparation of β-adrenergic blockers, in highly enantioenriched form (> 99% ee). These features make this a very attractive enzyme for potential applications in the biocatalytic preparation of enantiopure halohydrins, epoxides or azido alcohols.
References
Schallmey A, Schallmey M (2016) Appl Microbiol Biotechnol 100:7827
You ZY, Liu ZQ, Zheng YG (2013) Appl Microbiol Biotechnol 97:9
Dijoux GH, Elenkov MM, Spelberg JHL et al (2008) ChemBioChem 9:1048
Kasai N, Tsujimura K, Unoura K et al (1992) J Ind Microbiol Biotechnol 10:37
Kasai N, Tsujimura K, Unoura K et al (1992) J Ind Microbiol Biotechnol 9:97
Kasai N, Suzuki T, Furukawa Y (1998) Chirality 10:682
Nakamura T, Nagasawa T, Yu F et al (1992) J Bacteriol 174:7613
Spelberg JHL, Vlieg JETV, Bosma T et al (1999) Tetrahedron 10:2863
Hasnaoui G, Spelberg JHL, Vries ED et al (2005) Tetrahedron 16:1685
Mikeušević A, Primožič I, Hrenar T et al (2016) Tetrahedron 27:930
Elenkov MM, Hauer B, Janssen DB (2006) Adv Synth Catal 348:579
Elenkov MM, Primozic I, Hrenar T et al (2012) Org Biomol Chem 10:5063
Koopmeiners J, Diederich C, Solarczek J et al (2017) ACS Catal 7:6877
Chen SY, Yang CX, Wu JP et al (2013) Adv Synth Catal 355:3179
Ma SK, Gruber J, Davis C et al (2010) Green Chem 12:81
Fox RJ, Davis SC, Mundorff EC et al (2007) Nat Biotechnol 25:338
SchallmeyM KoopmeinersJ, Wells E et al (2014) Appl Environ Microbiol 80:7303
Koopmeiners J, Halmschlag B, Schallmey M et al (2016) Appl Microbiol Biotechnol 100:7517
Wan NW, Liu ZQ, Huang K et al (2014) RSC Adv 4:64027
Xue F, Liu ZQ, Wan NW et al (2014) Appl Biochem Biotechnol 174:352
Xue F, Liu ZQ, Wang YJ et al (2015) J Mol Catal B-Enzym 115:105
Xue F, Gao J, Zhang L et al (2018) Catal Lett 148:1181
Shapenvoa DS, Belyatskii MK, Panicheva LP (2010) Russ J Org Chem 46:1017
Liu ZQ, Wu L, Zhang XJ et al (2017) J Agr Food Chem 65:3721
Zhao J, Chu YY, Li AT et al (2011) Adv Synth Catal 353:1510
Wu K, Wang HL, Sun HH et al (2015) Appl Microbiol Biotechnol 99:9511
Wu SK, Li AT, Chin YS et al (2013) ACS Catal 3:752
Ulrich A, Volker M, Manfred PS (1993) Chirality 5:554
Xu Y, Xu JH, Pan J et al (2004) J Mol Catal B 27:155
Chen CS, Fujimoto Y, Girdaukas G et al (1982) J Am Chem Soc 104:7294
de Jong RM, Rozeboom HJ, Kalk KH et al (2002) Acta Crystallogr D 58:176
Nagasawa T, Nakamura T, Yu F et al (1992) Appl Microbiol Biotechnol 36:478
Assis HMS, Bull AT, Hardman DJ (1998) Enzyme Microb Technol 22:545
Nakamura T, Nagasawa T, Yu FJ et al (1994) Appl Environ Microbiol 60:1297
Liu ZQ, Gao AC, Wang YJ et al (2014) J Ind Microbiol Biotechnol 41:1145
Xue F, Liu ZQ, Wang YJ et al (2015) Catal Commun 72:147
Guo C, Chen Y, Zheng Y et al (2015) Appl Environ Microbiol 81:2919
Schrittwieser JH, Lavandera I, Seisser B et al (2009) Eur J Org Chem 14:2293
Acknowledgements
This project was sponsored by the National Natural Science Foundation of China (No. 21606192), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 16KJB180029) and the China Postdoctoral Science Foundation (No. 2016M601795).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declared that they have no conflicts of interest to this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xue, F., Ya, X., Xiu, Y. et al. Exploring the Biocatalytic Scope of a Novel Enantioselective Halohydrin Dehalogenase from an Alphaproteobacterium. Catal Lett 149, 629–637 (2019). https://doi.org/10.1007/s10562-019-02659-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-019-02659-0