
Abstract
In this study, a new organocatalyst derived from proline was developed and shown to be an efficient catalyst for asymmetric Michael addition reactions of ketones and aldehydes to nitroolefins with high diastereo- and enantioselectivities. (syn;anti up to 99:1, ee. up to 98 %.). Furthermore, the catalyst is easily recovered and could be reused six times without significant loss of its ability to affect the outcome of the asymmetric reactions. In addition, computational studies at the B3LYP/6-311G(d,p)//6-311 + G(2dp,f) level was conducted on a model reaction, and confirmed the following hypotheses: first, the hydrogen bonding between carboxyl group and nitro group plays an important role in catalysis, and second, the energy barrier for re-face attack in reactions of ketones to form 2S, 3R products is lower than that for the si-face attack leading to 2S, 3R products.
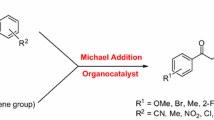
Graphical Abstract
Structural modification of a previously reported organocatalyst (lead compound in figure) was used to design an efficient and recyclable organocatalyst for asymmetric Michael addition. The introduced carboxyl group not only enhances the enantioselectivity but also brings convenience to the recovery of the catalyst.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Recently, organocatalyzed asymmetric carbon–carbon bond-forming reactions have received much attention [1–10].Footnote 1 Particularly, the organocatalytic asymmetric Michael reaction of aldehydes and ketones to nitroolefins is a key reaction in organic synthesis [11–22]. After the pioneering work of List and Barbas [23–25], many organo-catalysts derived from proline have been reported to exhibit high activities and excellent enantioselectivities for this reaction [15, 19, 26–47]. However, there are some major problems associated with these catalysts, including that a high catalyst loading (10–30 mol%) is generally required, and they are hard to recover. These problems limit the widespread application of the catalysts. The design and synthesis of highly active, easily recoverable and reusable catalysts have been proved to be a significant challenge. Recently, only limited success has been achieved [17, 41, 48–56]. Although initial strategies toward organocatalyst recycling using ionic liquid support, fluorous technologies and water-soluble salts have been developed, some of these catalysts still have some drawbacks. For example, the expensive fluorous solvents are required for phase separation [17, 53] or the addition of new protonic acid is necessary for reusing [54]. A catalyst overcoming these limitations would be advantageous. It is well known that structural modification is a valuable tool in the development of new catalysts. Recently, we have developed a series of proline-based reduced dipeptides, which produced by modifications of dipeptides, as efficient organocatalysts for asymmetric Michael addition [57]. In this paper, structural modification of a previously reported organocatalyst 1 [27, 28, 36] (Fig. 1, lead compound) was used to design and prepare an efficient and recyclable organocatlayst for asymmetric Michael addition.

New design of catalyst
2 Experimental Section
2.1 General Details
Commercial reagents were used without purification except for otherwise explanation. Analytical thin layer chromatography was performed on 0.20 mm silica gel plates and silica gel (200–300 mesh) was used for flash chromatography both purchased from Qingdao Haiyang Chem. Company, Ltd.
1H and 13C NMR were recorded on Varian-500 instruments. Chemical shifts were reported in ppm down field from internal Me4Si. All the multiplet patterns assigned the first-order splitting patterns. Mass spectra were recorded using electrospray ionization (ESI) on LCQ Advanced MAX Mass instruments. Optical rotations were tested on a WZZ-3 polarimeter using 10 mL cell with a 1 dm path length and Autopol II polarimeter using 1 mL cell with a 1 dm path length. HPLC analysis was measured using Chiralpak AS-H column.
2.2 Preparation of Catalyst
2.2.1 (S)-1-(Pyrrolidin-2-ylmethyl) Pyrrolidine-2, 5-dione (3)
Succinimide (2.97 g, 30 mmol) and K2CO3 (4.97 g, 36 mmol) was added to a solution of tosylate 5 (3.89 g, 10 mmol) in 20 ml DMF. The reaction mixture was stirred at 50 °C for 24 h. The mixture was diluted with 60 mL of water, and the resulted mixture was extracted with CH2Cl2 (20 mL × 3). The combined organic layer was washed with brine (20 mL × 2), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was chromatographed to give the intermediate 6. The crude product was dissolved in EtOH (20 mL), and the 0.1 g Pd/C (10 %) was added. The reaction mixture was stirred at r.t under 1 atm H2 overnight. The Pd/C was filtrated and the solution was concentrated in vacuo. The residue was purified by flash chromatograph on silica gel to give the desired product 3 as yellowy solid. (0.94 g, 51.6 % yield). \( \left[ \alpha \right]^{\text{D}}_{\text{rt}} \) = −35.1° (1.01, MeOH), 1H NMR (500 MHz, CDCl3) δ: 1.32–1.38 (1H, m), 1.66–1.85 (3OH, m), 2.17 (1H, br), 2.69 (4H, s), 2.77–2.81(1H, m), 2.94–2.99 (1H, m), 3.32–3.35 (1H, m), 3.44–3.55 (2H, m). 13C NMR (125 MHz, CDCl3) δ: 25.70, 28.39, 30.08, 43.54, 46.75, 57.32, 177.85. MS (ESI, m/z) 183.1 (M+H+). HRMS (ESI) calcd for C9H15N2O2 (M+H+): 183.1128, found: 183.1131.
2.2.2 (S)-5-Oxo-1-((S)-pyrrolidin-2-ylmethyl) Pyrrolidine-2-carb-oxylic Acid (4a)
To a stirred solution of N-Boc-l-prolinol 7 (20.1 g, 0.1 mol) in dry CH2Cl2 (300 mL) were added pyridinium chlorochromate (30.0 g, 0.14 mol) and 4 Å molecular sieves (30.0 g). The mixture was stirred at room temperature over night, then diluted with 300 mL Et2O (300 mL). The mixture was filtered through a silica gel G pad and concentrated in vacuo. The residue was chromatographed to afford pure 8 as a light yellow oil (15.2 g, 76 %). \(\left[ \alpha \right]^{\text{D}}_{\text{rt}}\) = −101° (0.66, CHCl3) 1H NMR(500 MHz, CDCl3) δ: 1.37 (5H, s),1.42 (4H, s),1.76–2.15 (4H, m), 3.44 (2H, m), 4.00–4.15 (1H, m),9.41 (0.6H, d), 9.50 (0.4H, br). 13C NMR (125 MHz, CDCl3) δ: 23.89, 24.58, 27.91, 28.21, 28.33, 46.47, 46.80, 64.82, 64.98, 76.80, 77.12, 77.43, 80.11, 80.51, 154.86, 200.29, 200.55.
To a solution of 8 (2.0 g, 10 mmol) in 20 mL MeOH was added L-aspartic acid dimethyl ester(2.1 g, 12 mmol), 4 Å molecular sieves (2.0 g) and Pd/C (wet, 5 %, 0.2 g). The reaction mixture was stirred at room temperature under 1 atm H2 until the TLC (EtOAc: Petroleum ether = 1:1) showed that 8 had disappeared and intermediate 9 had generated. The mixture was filtered to remove the catalyst, then TsOH (0.34 g, 2 mmol) was added into the filtrate. The mixture refluxed for 24 h, then concentrated in vacuo. The residue was purified by chromatograph to afford product 10 as a light yellow oil. \(\left[ \alpha \right]^{\text{D}}_{\text{rt}}\) = 36.51° (0.1, MeOH) 1H NMR(500 MHz, CDCl3) δ: 1.44(9H, s), 1.80–1.97 (4H, m), 2.12 (1H, m), 2.36–2.50 (3H, m) 2.95–3.02 (1H, m), 3.30–3.37 (2H, m), 3.68–3.73 (1H, m), 3.77 (3H, s), 3.83–3.95 (1H, m), 4.26–4.35 (1H, m). 13C NMR (125 MHz, CDCl3) δ: 22.53, 23.36, 28.44, 29.20, 29.67, 45.58, 46.20, 52.42, 55.12, 59.42, 76.71, 77.02, 77.34, 79.58, 154.71, 172.24, 175.62.
10 (2.3 g, 6 mmol) was dissolved in MeOH 20 mL. NaOH aqueous solution (1 M, 12 mL) was added into the solution. After stirring for 2 h, the mixture was acidified with 1 M HCl (pH = 4–5) and the solvent was removed in vacuo. The residue was added into 20 mL CH2Cl2 and the slurry chilled to 5 °C, then was added 10 mL TFA. The mixture stirred until TLC showed the starting material disappeared. After removed the solvent in vacuo, the residue was dissolved in 30 mL H2O. The aqueous solution was neutralized with NaHCO3, and then poured into a column with cation exchange resin (30 g). The target compound 4a was washed off by 2 N aqueous ammonia then purified by chromatograph. (0.95 g, 74 %). \(\left[ \alpha \right]^{\text{D}}_{\text{rt}}\) = −40.1° (1.0, MeOH) 1H NMR (500 MHz,CD3OD) δ: 1.71–1.79(1H, m), 1.98–2.18 (4H, m), 2.31–2.42 (2H, m), 2.44–2.51 (1H, m), 3.23–3.28 (1H, m), 3.33–3.38 (1H, m), 3.48 (1H, dd, J1 = 14.5 Hz, J2 = 3.5 Hz), 3.72–3.77 (1H, m), 3.82 (1H, m), 4.07–4.09 (1H, m). 13C NMR (125 MHz, CD3OD) δ: 24.13, 25.59, 28.67, 30.84, 45.42, 45.98, 61.00, 65.98, 178.16, 179.10. MS (ESI m/z): 213.2 (M+H+). HRMS (ESI) calcd for: C10H17N2O3 (M+H+), m/z 213.1328, found 213.1323.
2.2.3 (R)-5-Oxo-1-((S)-pyrrolidin-2-ylmethyl) Pyrrolidine-2-carb-oxylic Acid (4b)
The catalyst 4b was synthesized from D-aspartic acid dimethyl ester utilizing the similar procedure of 4a. \(\left[ \alpha \right]^{\text{D}}_{\text{rt}}\) = +91.4° (1.0, MeOH) 1H NMR(500 MHz, CD3OD) δ: 1.68–1.75(1H, m), 1.97–2.09 (3H, m), 2.15–2.22 (1H, m), 2.32–2.49 (3H, m), 3.24 (1H, dd, J1 = 9.0 Hz, J2 = 6.5 Hz), 3.27–3.33 (2H, m), 3.57 (1H, dd, J1 = 14.5 Hz, J2 = 4.0 Hz), 4.12 (1H, t, J = 8.0 Hz), 4.35–4.40 (1H, m). 13C NMR (125 MHz, CD3OD) δ: 24.22, 24.39, 28.24, 30.20, 44.99, 46.34, 48.84, 56.46, 64.50, 178.19, 178.97. MS (ESI m/z): 213.2 (M+H+). HRMS (ESI) calcd for: C10H17N2O3 (M+H+), m/z 213.1328, found 213.1325.
2.3 General Experimental Procedure for the Michael Addition
2.3.1 General Experimental Procedure for the Michael Addition of Cyclohexanone to Nitroalkene by Chiral Catalyst 2 and 3
To a solution of the amide catalyst (0.1 mmol), TFA (0.1 mmol) and the nitroalkene (0.5 mmol) in solvent (1 mL) was added cyclohexanone (5 mmol), and the solution was stirred at ambient temperature until TLC showed the nitroalkene disappeared. Ethyl acetate (10 volumes) was added. The solution was washed with water and 1 N HCl, dried (Na2SO4) and concentrated to give the crude product which was purified by flash chromatography on silica gel. Relative and absolute configurations of the products were determined by comparison with the known 1H NMR, chiral HPLC analysis, and optical rotation values.
2.3.2 General Experimental Procedure for the Michael Addition of Cyclohexanone to Nitroalkene by Chiral Catalyst 4a and 4b
To a solution of the catalyst (0.075 mmol) in MeOH (0.5 mL) was added cyclohexanone (2.5 mmol) and the nitroalkene (0.5 mmol). The solution was stirred at ambient temperature until TLC showed the nitroalkene disappeared. The mixture was concentrated and the residue diluted by 1 mL mixture solvent (ethyl acetate: petroleum ether = 1:1) to precipitate the catalyst. The catalyst was recovered by filter and washed with petroleum ether. The filtrate was concentrated to give crude product which was purified by chromatography on silica gel. Compounds 13a–i reported in Table 2 (entries 1–9) are known in literature and our spectroscopic data are in agreement with published data.
2.4 Computational Details
DFT calculations were carried out with the Gaussian 09 package [58]. The transition structure are fully optimized by B3LYP [59–61] method using 6-311G(d,p) basis set. Frequency calculations were performed at the same level on all optimized geometries to ensure only positive eigenvalues for minima and one negative imaginary frequency for transition states. The transition state was further verified by the connectivity between the reactant and transition sate confirmed by intrinsic reaction coordinate (IRC) [62] calculation. The single point energy is recalculated by B3LYP method using 6-311 + G(2dp,f) basis set on the optimized geometries. All the energies discussed in the paper are Gibbs free energies unless otherwise specified.
3 Result and Discussion
In previous studies, the hydrogen bond was used as a general strategy for designing the new catalyst [17, 63–66]. The common transition state TS1 (Fig. 1) shows that hydrogen bonding is an important factor in forming a favorable transition state. Inspired by this, derivatives of the lead compound 2–4b, which contain more hydrogen acceptor or donor groups were prepared [67]Footnote 2 as shown in Scheme 1, and evaluated for the model Michael addition reaction of cyclohexanone 11 to nitrostyrene 12. The representative results are summarized in Table 1 text (although the first paragraph and paragraphs that follow headings should not be indented).

Preparation of the catalysts

Derivative 2 was designed because the oxygen atom of the carbonyl group adjacent to the chiral center might provide an extra hydrogen bond acceptor. Unfortunately, it yielded poorer enantioselectivity (36 % ee, Table 1, entry 2) than that of the lead compound (84 % ee) [36]. Thus, the carbonyl group has a negative impact on the enantioselectivity. Two possible reasons are: the carbonyl group decreases the electron density on the N atom and thus weakens the hydrogen bond, or the steric effect of the ortho-carbonyl may hinder the N atom from moving to the appropriate position for forming the hydrogen bond.
In order to understand the effect of the carbonyl, amide 3 (Fig. 1) was prepared and employed to catalyze the model reaction. Interestingly, catalyst 3 afforded better enantio-selectivity (93 % ee, Table 1, entry 6) than the lead compound 1. This encouraging result implies that the strategy of introducing more potential hydrogen bond acceptors works. A comparison of 2 and 3 indicates that it is the steric effect of the ortho-carbonyl that is responsible for hindering the enantioselectivity.
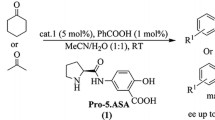
Although amide 3 afforded good yield and a high level of diastereo- and enantioselection, it is very difficult to recover this catalyst because the added acid makes the reaction system much more complicated. When amide 2 or 3 was used to catalyze the reaction without a protonic acid, the Michael product was not obtained (Table 1, entries 1, 3, 5, 7), because a large amount of polymerized 12 was quickly formed under these conditions. Other studies have indicated that amines behave as polymerization initiators [68], and Barbas’s group has reported that the addition of Brønsted acids can promote the formation of enamines and thus inhibit polymerization [69]. Therefore the use of catalysts containing carboxyl group might be a way to simplify the reaction system [57]. The introduced carboxyl groups could take the place of the added Brønsted acid as a hydrogen donor. In addition, the carboxyl group and the secondary amine can form an inner salt, which should enhance the aqueous solubility of the catalyst and thus provide easy recovery of the catalysts. To test this hypothesis, derivatives 4a–b were prepared and their catalytic performance in the model reaction was examined. The model reaction proceeded effectively in methanol in the presence of 20 mol% 4a without any additives, with a high yield (93 %), an excellent diastereoselectivity of 99:1 (syn:anti) and an enantio-selectivity of 97 % ee (Table 1, entry 9). The catalyst loading could be reduced to 15 or 5 mol% to get similar results, but a longer reaction time was needed (Table 1, entries 10 and 11). When the loading was further reduced to 1 mol%, the reaction was too slow to be detected (Table 1, entry 12). Interestingly, the diastereo-isomer 4b led to lower yield (67 %) and slightly decreased enantioselectivity (93 % ee), even with a prolonged reaction time of 168 h. (Table 1, entry 13).
The addition of cyclohexanone to trans-β-nitrostyene was used as a model to examine the recyclability of the catalyst 4a (at 20 mol%). After the reaction was completed, the reaction mixture was concentrated and the residue was diluted with a mixture solvent (ethyl acetate: petroleum ether = 1:1) to precipitate the catalyst, which was easily recovered by filtration. The first time recovered catalyst was characterized by 1H NMR to confirm that the structure did not change after the reaction. The recovered catalyst was used for the next run of the reaction. As shown in Table 2, catalyst 4a can be recycled and reused for at least six times without a significant loss of stereoselectivity (ee > 95 %) although the catalytic activity decreased gradually.

The scope of the Michael reactions using catalyst 4a in MeOH was examined with a variety of carbonyl compounds and nitroolefins (Table 3).

As demonstrated in Table 3, catalyst 4a can be applied to several Michael reactions for a variety of carbonyl compounds and nitroolefins in MeOH. Cyclohexanone efficiently underwent Michael reactions with different aryl-substituted nitroolefins to give Michael adducts 13a–h in high yields with excellent enantio-(91–98 % ee) and diastereoselectivities (syn/anti ratio up to >20:1). The results in Table 3 also show that the nature of the substituents on aryl groups slightly influences the yields and enantio-selectivities. For nitroolefins with electron-rich groups (methyl and methoxyl), the reaction proceeded smoothly to afford Michael adduct 13b–c in excellent enantio- (91–98 % ee) and diastereoselectivities (syn/anti > 20:1) (Table 3, entries 2–3). For nitroolefins with electron-deficient groups, the Michael adducts 13d–g were also obtained in high yields (87–93 %) with excellent enantio- (95–98 % ee) and diastereoselectivities (syn/anti ratio up to >20:1) (Table 3, entries 4–7). In addition, 3-pentanone and n-butyraldehyde are also suitable Michael donor with high diastereoselectivity as well as enantios (Table 3, entries 8 and 9). We also tested the aliphatic nitroolefin as Michael reaction substrate but only give the moderate enantioselectivity (76 % ee, Table 3, entry 10).These results indicate that catalyst 4a should be broadly plicable to the synthesis of γ-nitro carbonyl compounds.
The stereochemical results may be explained by the transition state (Favorable TS) shown in Scheme 2. Similar to the protonated nitrogen atom in TS1, the carboxyl group hydrogen bonds with the nitroalkene to keep it in a favorable position for the approach of the nitroalkene from the re face of the anti-enamine.

The proposed mechanism and transition state
4 Computation Studies
To gain a more detailed understanding of the origin of the high enantio- and diastereoselectivity of the processes catalyzed by 4a, we have computationally studied the transition states by density functional theory (DFT) at the B3LYP/6-311G(d,p) level. The geometric and energetic parameters of possible transition states are listed in Table 3 and structures are displayed in Fig. 2.

Transition-state geometries for the reaction between 11 and 12 catalyzed by 4a, calculated at B3LYP/6-311G(d,p) level
As show in Scheme 2, the enamine intermediates can adopt anti and syn conformations, For each of them, two different transition states exist for the approach of the nitropropene to the diastereotopical Re and Si faces of the enamine, resulting in the formation of four different transition states, two for each enantiomer (Fig. 2). The two transition states arising from the anti enamine (TS A and TS B ) can benefit from hydrogen-bonding activation between the carboxyl group and the nitro group, and our initial hypothesis was that this interaction might contribute to a lowering in the energy barriers, resulting in faster reaction rates. Meanwhile, reaction through syn-enamine conformations would occur without the help of hydrogen-bonding activation (TS C and TS D ). As shown in Table 4, we found that the lowest in energy (9.8 kcal/mol) corresponds to TS A , the one that leads to the experimentally observed 2S, 3R enantiomer. According to our initial hypothesis, this result shows the COOH delivers the nitroalkene by hydrogen bonding thus favours the approach of nitroalkene from the re face of the anti-enamine. The minor enantiomer 2R, 3S is formed through a Si approach of the nitro alkene to the anti enamine (TSB), whose activation energy is 12.7 kcal/mol. The different activated barriers between TS A and TS B should be considered that TS A has the stronger hydrogen bond than TS B (1.59 Å vs 1.67 Å, Table 3). Meanwhile, reaction through syn-enamine conformations (TS C and TS D ) cannot benefit from hydrogen bonding (Fig. 1). Therefore, their activation energies are much higher than those of their activated counterparts (activation energy: TS C > TS A and TS D > TS B , Table 4).
5 Conclusion
In summary, a novel organocatalyst 4a which can be used to promote highly efficient asymmetric Michael addition reactions of ketones and aldehydes to nitroolefins has been developed. The main advantages of this catalyst are ease of synthesis and low loading (5 mol%) for high steroselectivities (ee up to 98 %, syn/anti up to 99/1) at room temperature. Moreover, the catalyst is easily recovered and reused. These advantages make 4a a potential catalyst for industrial applications. Computational studies at the B3LYP/6-311G(d,p) level have been conducted on a model reaction, confirming the initial hypothesis that the hydrogen bonding between carboxyl group and nitro group plays an important role in the activation of the nitro alkene and helps to discriminate between the two diastereofacial approaches. The computational results, which are in good agreement with the experimental observations, are discussed in the context of the stereochemical course of these Michael addition reactions. These results provide valuable insight into the mechanisms of asymmetric organocatalysis and might help the design of new and more efficient organocatalysts.
References
Dalko PI (2004) Angew Chem Int Ed 43:5138
Guillena G, Ramon DJ (2006) Tetrahedron Asymmetry 17:1465
Dalko PI (2007) Enantioselective rganocatalysis. Wiley, Weinheim
List B (2007) Chem Rev 107:5413
Rongming W, Linhai J, Dabin Q (2015) Tetrahedron Lett 56:2867
Masanori Y, Yuki N, Ami K, Shoji H, Masahiro Y (2013) Tetrahedron 69:10003
Clarke ML, Fuentes JA (2007) Angew Chem Int Ed 46:930
Almaşi D, Alonso DA, Gómez-Bengoa E, Nagel Y, Nájera C (2007) Eur J Org Chem 2007:2328
Diez D, Gil MJ, Moro RF, Marcos IS, García P, Basabe P, Garrido NM, Broughton HB, Urones JG (2007) Tetrahedron 63:740
Lu A, Wu R, Wang Y, Zhou Z, Wu G, Fang J, Tang C (2011) Eur J Org Chem 122:3507
Berner OM, Tedeschi L, Enders D (2002) Eur J Org Chem 12:1877
Krause N, Hoffmann-Röder A (2001) Synthesis 1:171
Tsogoeva SB (2007) Eur J Org Chem 11:1701
Almasi D, Alonso DA, Nájera C (2007) Tetrahedron Asymmetry 18:299
Cao CL, Ye MC, Sun XL, Tang Y (2006) Org Lett 8:2901
Freund M, Schenker S, Tsogoeva SB (2009) Org Biomol Chem 7:4279
Zu L, Wang J, Li H, Wang W (2006) Org Lett 8:3077
Lu AD, Gao P, Wu Y, Wang YM, Zhou ZH, Tang CC (2009) Org Biomol Chem 7:3141
Wang J, Li H, Lou B, Zu L, Guo H, Wang W (2006) Chem Eur J 12:4321
Lu A, Wu R, Wang Y, Zhou Z, Wu G, Fang J, Tang C (2010) Eur J Org Chem 2010:2057
Chen JR, Lai YY, Lu HH, Wang XF (2009) Tetrahedron 65:9238
Cao YJ, Lu HH, Lai YY, Lu LQ, Xiao WJ (2006) Synthesis 22:3795
List B, Pojarliev P, Martin HJ (2001) Org Lett 3:2423
Betancort JM, Barbas CF III (2001) Org Lett 3:3737
Enders D, Seki A (2002) Synlett 1:26
Ishii T, Fujioka S, Sekiguchi Y, Kotsuki H (2004) J Am Chem Soc 126:9558
Mase N, Thayumanavan R, Tanaka F, Barbas CFIII (2004) Org Lett 6:2527
Betancort JM, Sakthivel K, Thayumanavan R, Tanaka F, Barbas CFIII (2004) Synthesis 9:1509
Alexakis A, Andrey O (2002) Org Lett 4:3611
Andrey O, Alexakis A, Tomassini A, Bernardinelli G (2004) Adv Synth Catal 346:1147
Cobb AJA, Longbottom DA, Shaw DM, Ley SV (2004) Chem Commun 16:1808
Cobb AJA, Shaw DM, Longbottom DA, Gold JB, Ley SV (2005) Org Biomol Chem 3:84
Reyes E, Vicario JL, Badia D, Carrillo L (2006) Org Lett 8:6135
Wang W, Wang J, Li H (2005) Angew Chem Int Ed 44:1369
Pansare SV, Pandya K (2006) J Am Chem Soc 128:9624
Mase N, Watanabe K, Yoda H, Takabe K, Tanaka F, Barbas CFIII (2006) J Am Chem Soc 128:4966
Vishnumaya, Singh VK (2007) Org Lett 9:1117
Gua L, Zhao G (2007) Adv Synth Catal 349:1629
Ni B, Zhang Q, Headley AD (2007) Tetrahedron Asymmetry 18:1443
Xu DQ, Wang LP, Luo SP, Wang YF, Zhang S, Xu ZY (2008) Eur J Org Chem 6:1049
Bukuo N, Zhang QY, Kritanjali D, Allan DH (2009) Org Lett 11:1037
Diana A, Diego AA, Enrique GB, Yvonne N, Carmen N (2007) Eur J Org Chem 14:2328
Terakado D, Takano M, Oriyama T (2005) Chem Lett 34:962
Wang L, Liu J, Miao T, Zhou W, Li P, Ren K, Zhang X (2010) Adv Synth Catal 352:1629
Zh W, Lu CF, Ch Yang G, Chen ZX, Nie JQ (2015) Catal Commun 62:34
Han Y, Mouming L, Sheng H (2014) Tetrahedron 70:8380
Cao YJ, Lai YY, Wang X, Li YJ, Xiao WJ (2007) Tetrahedron Lett 48:21
Ni B, Zhang Q, Headley AD (2007) Green Chem 9:737
Zhang Q, Ni B, Headley AD (2008) Tetrahedron 64:5091
Wu LY, Yan ZY, Xie YX, Niu YN, Liang YM (2007) Tetradedron Asymmetry 18:2086
Luo S, Mi X, Zhang L, Liu S, Xu H, Cheng JP (2006) Angew Chem Int Ed 45:3093
Alza E, Cambeiro XC, Jimeno C, Pericàs MA (2007) Org Lett 9:3717
Wang B-G, Ma B-C, Wang Q, Wang W (2010) Adv Synth Catal 352:2923
Zheng Z, Perkins BL, Ni B (2010) J Am Chem Soc 132:50
Xin H, Wen-Bin Y, Danash A, Wei Z (2013) Tetrahedron Lett 54:6064
Xuefei Q, Jun T, Yang L, Ligong Ch, Xilong Y (2015) Catal Commun 71:70
Cao X, Wang G, Zhang R, Wei Y, Wang W, Sun H, Chen L (2011) Org Biomol Chem 9:6487
Gaussian 09, Revision D.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian, Inc., Wallingford CT
Becke AD (1993) J Chem Phys 98:1372
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Gonzalez C, Schlegel HB (1990) J Phys Chem 94:5523
Knudsen RK, Mitchell CET, Ley SV (2006) Chem Commun 1:66
Tang Z, Yang Z, Chen X, Cun L, Mi A, Jiang Y, Gong L (2005) J Am Chem Soc 127:9285
Martin HJ, List B (2003) Synlett 12:1901
Lu D, Gong Y, Wang W (2010) Adv Synth Catal 352:644
Asami M (1990) Bull Chem Sc Jpn 63:721
Carter ME, Nash JL Jr, Drueke JW Jr, Schwietert JW, Butler GB (1978) J Polym Sci Polym Chem Ed 16:937
Mase N, Tanaka F, Barbas CFIII (2003) Org Lett 5:4369
Acknowledgments
This work was supported by Supported by National Natural Science Foundation of China (No. 21306038) and Natural Science Foundation of Hebei Province (No. B2013202216).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, M., Zhang, Y., Zhao, J. et al. A Recyclable Organocatalyst for Asymmetric Michael Addition. Catal Lett 146, 587–595 (2016). https://doi.org/10.1007/s10562-016-1693-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-016-1693-x