
Abstract
Human amniotic membrane (HAM) has useful properties as a dermal matrix substitute. The objective of our work was to obtain, using different enzymatic or chemical treatments to eliminate cells, a scaffold of acellular HAM for later use as a support for the development of a skin equivalent. The HAM was separated from the chorion, incubated and cryopreserved. The membrane underwent different enzymatic and chemical treatments to eliminate the cells. Fibroblasts and keratinocytes were separately obtained from skin biopsies of patients following a sequential double digestion with first collagenase and then trypsin–EDTA (T/E). A skin equivalent was then constructed by seeding keratinocytes on the epithelial side and fibroblasts on the chorionic side of the decellularizated HAM. Histological, immunohistochemical, inmunofluorescent and molecular biology studies were performed. Treatment with 1 % T/E at 37 °C for 30 min totally removed epithelial and mesenchymal cells. The HAM thus treated proved to be a good matrix to support adherence of cells and allowed the achievement of an integral and intact scaffold for development of a skin equivalent, which could be useful as a skin substitute for clinical use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autografts are currently the best treatment for cutaneous lesions and burns. However, autografts present problems with donor site morbidity and engraftment failure. In addition, when patients have 50 % or more of their body surface burned, donor sites are limited (Macri and Clark 2009). Tissue engineering has emerged as an interdisciplinary field with the aim of generating new biological materials for replacing diseased or damaged tissues or organs. For skin, the ultimate goal is to rapidly create a construct that effects complete regeneration of functional skin. For this to be achieved, there is a need for an appropriate source of cells and a scaffold (Theoret 2009). Therefore, an important component of tissue engineering is the supporting matrix upon which cells and tissue grow. This scaffold must easily integrate with host tissues and provide an appropriate environment for cell growth and differentiation (Niknejad et al. 2008).
Various biomaterials have been used for dermal matrix substitutes, including collagen–elastin membrane (Hafemann et al. 1999), acellular cadaveric dermis (Buinewicz and Rosen 2004), collagen–glycosaminoglycan (GAG) membrane (Orgill et al. 1999), xenogenic acellular dermal matrix (Yu et al. 2006), collagen/gelatin sponge scaffold (Takemoto et al. 2008), plasma and hyaluronic acid (Cervelli et al. 2010), human plasma based dermal equivalent (Meana et al. 1998; Llames et al. 2004; Mazlyzam et al. 2007).
The human amniotic membrane (HAM) is considered an important potential source as a scaffold because of its structure, biological properties, mechanical properties and low immunogenicity (Niknejad et al. 2008). The HAM is the innermost layer of the placenta and is composed of: (1) a single layer of columnar or cuboid epithelial cells; (2) a basal membrane, which resembles that of skin both morphologically and ultrastructurally, consisting of laminin 5 and types IV, VII, XVII collagen (Wilshaw et al. 2008); (3) an acellular compact layer; and (4) underlying fibroblast and spongy layers (Yang et al. 2006). The HAM contains no blood vessels or nerves (Niknejad et al. 2008).
In clinical use, the HAM has been used to cover clean partial-thickness wounds and donor sites and applied as a temporary dressing for freshly excised burns because it has good adherence to wounds, provides pain and inflammation relief, and promotes epithelialization, scar prevention, and prevention of infection. HAM also has other advantageous biological properties, such as being anti-inflammatory, anti-microbial, anti-fibrotic, anti-angiogenic, anti-microbial and anti-viral (Kesting et al. 2008). Additionally, HAM possesses good mechanical properties, including permeability, stability, elasticity, flexibility and plasticity (Niknejad et al. 2008).
Importantly, HAM shows little or no immunogenicity and an immune response against the graft, if any, is slight and ineffective; thus it does not present transplantation risks. In contrast, the chorion has high immunogenicity, and for this reason, is not used as a biomaterial for transplantation purposes (Díaz-Prado et al. 2011). Moreover, HAM can be easily obtained, processed, and transported (Niknejad et al. 2008), as well as sterilized and preserved at low cost for long periods without obvious architectural changes (Niknejad et al. 2008; Yang et al. 2009).
The objective of this work was to create a scaffold of decellularizated HAM and to develop an in vitro model of skin using this HAM as the scaffold for culture of keratinocytes on the basal membrane and the fibroblasts on the stromal side. The evaluation of the formation of this possible skin equivalent was done by histological, immunohistochemical and immunofluorescent techniques.
Materials and methods
Harvest and preparation of HAMs
Human placentas (n = 5) from healthy donor mothers were obtained from selected caesarean sections at the Hospital Materno Infantil-Teresa Herrera A Coruña, Spain. All mothers provided written informed consent before collection. This study was approved by the Ethics Committee of Clinical Investigation of Galicia (Spain). Under stringent sterile conditions, the harvested placentas were placed in 199 medium (Invitrogen, Spain) with antibiotics during 24 h: cotrimoxazol (50 μg/ml; Almirall, Spain), vancomycin (50 μg/m; Hospira, Spain), amykacin (50 μg/ml; Normon, Spain), and B amphotericin (5 μg/ml; Bristol-Myers Squibb, Spain). The HAM was carefully separated from the chorion, which was discarded, and immediately washed three times with 0.9 % saline solution (NaCl) solution (Braun, Spain) to remove blood and mucus. The HAM was incubated again in 199 medium with the antibiotics listed above for 6–20 h at 4 °C and then cryopreserved.
Cryopreservation of HAMs
The HAM was cut into 4 × 4 cm patches and placed on a supportive sterile nitrocellulose filter in 20 ml of a 10 % cryoprotectant [dimethyl sulfoxide (DMSO) (Sigma, Spain)] in 199 medium. Each patch of HAM was cryopreserved following a protocol of controlled freezing using a CM 2000 (Carburos Metálicos, Spain). Freezing rates were −1 °C/min to a temperature of −40 °C, −2 °C/min to −60 °C, and −5 °C/min to −150 °C. All HAMs were stored in the gas phase of liquid nitrogen at −150 °C.
Thawing of HAMs
Thawing of HAMs was carried out for 5 min at room temperature followed by incubation at 37 °C until thawing was complete. To reduce cell damage due to osmotic changes, the DMSO was removed by sequential washing and progressive dilution with 0.9 % NaCl at 4 °C.
Decellularization of HAMs
To determine which treatment was best of those found in the bibliography for elimination of epithelial and mesenchymal cells, HAMs were incubated with one of the following treatments: (a) 0.25 % trypsin–ethylenediaminetetraacetic acid (EDTA) (T/E) (Sigma) at 37 °C for 10 or 30 min; (b) 1 % T/E at 37 °C for 10 or 30 min; (c) 0.1 % EDTA at 37 °C for 30 min or 1 h; (d) 0.1 % EDTA at 4 °C for 16 h; (e) 0.02 % EDTA at 37 °C for 1 or 2 h; (f) dispase (PAA Laboratories GmbH, Germany) 1.2 U/ml at 37 °C for 30 min or 1 h; and (g) accutase (PAA Laboratories GmbH, Germany) at 37 °C for 45 min or 1 h.
From each sample, one part was fixed in 4 % formol and embedded in paraffin for histologic (hematoxylin and eosin (H–E), Masson’s trichrome, alcian blue and Safranin O), and immunohistochemical staining for laminin, fibronectin and vimentin. Unpreserved parts of samples were utilized for analysis by contrast phase microscopy and DNA quantification.
DNA extraction and quantification from HAMs
For each fresh HAM (n = 5), 10 mg of the non-treated positive control and of those treated with the different procedures to remove the cells described above, was ground with a mortar and pestle and DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Germany) according to manufacturer’s instructions. The quality and concentration of DNA extracts were determined using an Infinite® 200 PRO NanoQuant (Tecan, Germany).
Skin biopsy collection
Skin samples of sizes between 2 and 4 cm, originating in different zones, were obtained from consenting patients (n = 10; 12–50 years-old) from the Burns Unit of the Complejo Hospitalario Universitario A Coruña, Spain.
Fibroblast isolation and culture
Each skin biopsy with no previous dermal–epidermal separation was minced using surgical scissors. Human fibroblasts were isolated by enzymatic digestion with 2 mg/ml collagenase type I (Sigma) containing 1 % antibiotic–antimycotic solution (Life Technologies, USA) with agitation for 3 h at 37 °C. Following incubation, the collagenase type I solution was removed and inactivated with Dulbecco’s minimal essential medium (DMEM) (Gibco, USA) and 10 % foetal bovine serum (FBS) (Gibco, USA) and centrifuged. The cell pellet was re-suspended in fibroblast medium (FM): DMEM, 10 % FBS and 1 % penicillin–streptomycin (P/S) (Gibco, USA). The cells were counted and 2 × 106 cells were seeded in a 25 cm2 flask (Greiner Bio One, USA) with FM and cultured at 37 °C in 5 % CO2. The medium was changed every 2–3 days. The fibroblasts were plated until the second passage.
Keratinocyte isolation and culture
The skin fragments remaining after collagenase treatment were enzymatically digested with 1 % T/E (Sigma), for 60 min at 37 °C. Following incubation, the T/E solution was removed and inactivated with DMEM and 10 % FBS and centrifuged. The cell pellet was re-suspended in human epidermal keratinocyte basal medium (EpiGRO; Millipore, Germany). The cells were counted and 2 × 106 cells were seeded in a 25 cm2 flask and cultured at 37 °C in 5 % CO2. The medium was changed every 2–3 days. The keratinocytes were plated until the second passage.
Development of an in vitro skin equivalent model
A concentration of 5 × 105 fibroblasts was seeded in each well of a six-well culture plate in FM. One day later, when the fibroblasts reached confluency, a decellularizated 4 × 4 cm patch of HAM was put on the fibroblasts, with the stromal side in contact with the fibroblasts, and stabilized with a CellCrown (Scaffdex, Finland). Then, 1 × 106 keratinocytes were seeded on the epithelial side with keratinocyte culture medium (3:1) mixture of DMEM/HAM-F12 (Gibco, USA) supplemented with 10 % FBS, insulin (5 μl/ml) (Sigma), hydrocortisone (0.4 μl/ml) (Sigma), triiode-thyronine (1.3 ng/ml) (Sigma), adenine (24 μl/ml) (Sigma) and 1 % P/S (Gibco, USA). The medium was changed every 2–3 days; at the end of the first change and until the end of the culture, 10 ng/ml epidermal growth factor (EGF) was added (Austral Biologicals, USA). Cultures were incubated in a humidified 5 % CO2 atmosphere at 37 °C using the submerged method throughout culture (Peña et al. 2010). The skin equivalent was cultured for 7 days.
At harvest, part of the skin equivalent was fixed in 4 % formol and embedded in paraffin for histological and immunohistochemical analyses; another part was frozen in optimal cutting temperature (OCT) compound for immunofluorescent staining.
Histological, immunohistochemical and immunofluorescent analyses
For general histological analyses, 4 μm-thick paraffin sections were deparaffinized in xylol, rehydrated in a graded series of ethanol, and stained with H–E, Masson’s trichrome, Verhoeff hematoxylin, alcian blue and Safranin O using standard protocols.
For immunohistochemical analyses, 4 μm-thick paraffin sections, which had been deparaffinized and hydrated, were incubated with monoclonal antibodies to detect the presence of laminin (LAM-89; Sigma), fibronectin (SPM246; Santa Cruz Biotechnology, USA), vimentin (V9; Abcam, UK), cytokeratin (AE1/AE3; Dako, Denmark), cytokeratin-14 (Mob-186; Diagnostic BioSystems, USA), keratin 10 (DE-K10; Neomarkers, USA), p63 (4A4; Thermo Scientific, USA) and Ki67 (MM1; Leica Biosystems, UK). The peroxidase/DAB ChemMateTM DAKO EnVisionTM detection kit (Dako) was used to determine antigen–antibody interaction.
For immunofluorescent analyses, cryosections (4 μm) were fixed with cold acetone for 10 min. They were then washed and incubated with a monoclonal antibody to detect the presence of laminin (LAM-89; Sigma). Sections were incubated with goat anti-mouse secondary antibody conjugated with fluorescein isothiocyanate (FITC) (Dako). Finally, the nuclei were counterstained with DAPI (Sigma).
Results
Treatment of HAM with 1 % T/E at 37 °C for 30 min totally removed epithelial and mesenchymal cells
HAMs were treated with different enzymatic and chemical procedures to remove epithelial and mesenchymal cells and facilitate the adhesion of primary culture skin cells. The degree of removal of epithelial and mesenchymal cells following each treatment was analyzed by H–E staining and DNA quantification.
The results of the removal of epithelial and mesenchymal cells from HAMs assessed using H–E staining of histological deparaffinized sections (Fig. 1a) demonstrated that the best treatment for removing these cells was 1 % T/E at 37 °C for 30 min. With this treatment, all of the epithelial and mesenchymal cells were eliminated. However, the basal membrane and the extracellular matrix of the HAM remained intact; these latter two components of the HAM are necessary because they act as a support for other cells, such as skin cells, to establish models for tissue engineering.

a Hematoxylin–eosin (H–E) staining following different enzymatic and chemical treatments used to eliminate cells from human amniotic membranes (n = 5) (representative photographs, ×400 magnification). b Concentration of DNA obtained following different treatments enzymatic and chemical treatments used to eliminate cells from human amniotic membranes (n = 5). T/E = trypsin–ethylenediaminetetraacetic acid (EDTA); HAM = human amniotic membrane
The results of the other T/E or EDTA treatments showed that after digestion epithelial cells and mesenchymal cells were preserved. With these treatments, it was necessary to use a mechanical rake to completely eliminate these cells and this mechanical treatment damaged the HAM. Accutase treatment also removed epithelial cells but did not eliminate mesenchymal cells in the stroma of the HAM, while treatment with dispase eliminated epithelial and mesenchymal cells, but considerably damaged the matrix and basal membrane of the HAM.
DNA quantification of HAM incubated with different treatments confirmed the results from histological staining
DNA quantification results (Fig. 1b) indicated that the best treatment to remove cells was 1 % T/E at 37 °C for 30 min (remaining DNA: 1.38 ± 0.53 ng/µl), which, as also seen in the histological studies, completely eliminated epithelial and mesenchymal cells. The treatment with 1 % T/E at 37 °C for 10 min (19.13 ± 0.21 ng/µl) was insufficient to eliminate the cells, probably because, although mesenchymal cells have been totally eliminated, some zones with endothelium are present. The other treatments, 0.25 % T/E at 37 °C for 30 min (45.33 ± 10.35 ng/µl), 0.1 % EDTA at 4 °C for 16 h (54.95 ± 6.12 ng/µl), 0.1 % EDTA at 37 °C for 30 min (77.23 ± 0.38 ng/µl), 0.02 % EDTA at 37 °C for 2 h (39.7 ± 2.26 ng/µl), and 0.02 % EDTA at 37 °C for 1 h (46.7 ± 14.73 ng/µl) did not effectively eliminate all cell types The worst of these was 0.1 % EDTA at 37 °C for 30 min with the largest quantity of remaining DNA. After treatment with accutase at 37 °C for 1 h, which removed epithelial cells, the concentration of remaining DNA may be due to mesenchymal cells in the HAM stroma (10.79 ± 2.68 ng/µl), as observed in the histological study. Finally, treatment with dispase at 1.2 U/ml at 37 °C for 1 h (6.53 ± 4.2 ng/µl) eliminated all cells, as did T/E at 37 °C for 30 min, but damaged the basal membrane and the components of the extracellular matrix to a greater degree.
Treatment of HAM with 1 % T/E at 37 °C for 30 min eliminated all cells, but did not damage other HAM components
Inverted phase contrast microscopy demonstrated the condition of the HAM pre- and post-treatment. Non-treated HAM showed an epithelium of polygonal cells (Fig. 2a). After treatment with 1 % T/E at 37 °C for 30 min these cells were eliminated, leaving an acellular scaffold (Fig. 2b). Masson’s trichrome staining showed that the major components of the HAM matrix, such as collagen, were retained after treatment with 1 % T/E at 37 °C for 30 min (Fig. 2c, d). Alcian blue and safranin O staining showed that mucopolysaccharides (Fig. 2e, f) and proteoglycans (Fig. 2g, h), respectively, were retained after this treatment.

Comparison of non-treated human amniotic membrane (HAM) (a, c, e and g) with HAM after treatment with 1 % T/E at 37 °C for 30 min (b, d, f and h). By inverted phase contrast microscopy, epithelial cells can be observed anchored to the basement membrane in the non-treated HAM (a), while in the treated HAM, epithelial cells can no longer be observed (b). Masson’s trichrome shows that collagen fibers are present in both non-treated and treated HAM (c, d). Staining with alcian blue (mucopolysaccharides) and safranin O (proteoglycans) show few differences between non-treated and treated HAM (e, h). (a ×100 magnification) (c–h ×400 magnification)
To determine whether basal membrane proteins remain after decellularization, immunohistochemistry was performed to detect the presence of laminin and fibronectin. Both proteins were present after treatment with 1 % T/E at 37 °C for 30 min (Fig. 3b, d) at the same levels as in the non-treated HAM (Fig. 3a, c).

Immunohistochemistry to detect the proteins, laminin, fibronectin and vimentin, in non-treated human amniotic membrane (HAM) (a, c and e) with HAM after treatment with 1 % T/E at 37 °C for 30 min (b, d and f). Basal membrane proteins, laminin (a) and fibronectin (c) are present in non-treated HAM and are retained in the de-epithelialized HAM (b, d), respectively. Staining for vimentin is observed in non-treated HAM (e), but is negative in the de-epithelialized HAM (f) because of the elimination of mesenchymal cells. (×400 magnification)
Finally, to determine if the treatment for the decellularization also affected mesenchymal cells, immunohistochemistry was carried out to determine the expression of vimentin (an antigen present on those cells; Fig. 3e). The absence of staining for this antigen after treatment with 1 % T/E at 37 °C for 30 min demonstrated that most mesenchymal cells were eliminated with this treatment (Fig. 3f).
Development of an in vitro skin equivalent model
Once we had concluded that treatment of the HAM with 1 % T/E at 37 °C for 30 min was the best procedure for decellularization of the membrane, the suitability of HAM as a scaffold for an in vitro skin equivalent mode was assessed as follows:
-
(a) Fibroblast and keratinocyte primary cultures
The seeded fibroblasts reached confluency within 5–6 days. The cells had high proliferative capability and showed spindle cytoplasms with thin and long projections, typical fibroblast morphology. Primary keratinocytes cultures on the first day showed a short and wide spindle shape, and proliferated cell colonies could be seen at days 3–4. Those cells were polygonal in shape, like pavement stones, with round, centered nuclei; and cultures reached confluency between 10 and 12 days (data not shown).
-
(b) In vitro skin equivalent model
Fibroblasts were seeded on the stromal side and keratinocytes on the epithelial side of HAM decellularizated with 1 % T/E at 37 °C for 30 min; these constructs were cultivated for a week in the presence of keratinocyte culture medium, as described in “Materials and methods”. The use of the submerged method obtained a skin equivalent in less time, considerably reducing the culture time compared with other previously used air-lift methods. By the end of the culture period, the fibroblasts acquired their typical spindle-like shape and started to proliferate (Fig. 4a) and the keratinocytes appeared more compactly arranged with a stone pavement-like morphology (Fig. 4b).

Cell morphology of the in vitro skin equivalent model. a Morphology of fibroblasts on the stromal side of de-epithelialized human amniotic membrane (HAM) 5 days after seeding. b Morphology of keratinocytes on the epidermal side of de-epithelialized HAM 5 days after seeding. (×100 magnification)
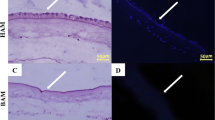
After 1 week in submerged culture to maintain undifferentiated cells, in vitro skin equivalent models exhibited good morphology and good mechanical properties, showing good flexibility, so it was possible to manipulate the skin equivalent easily during the culture without separation of the epidermis from the dermis. These properties were demonstrated by histological and immunological analysis of the skin equivalent created. H–E staining showed a top sheet of keratinocytes and a bottom with fibroblasts (Fig. 5a). Immunohistochemistry confirmed the presence of keratins in the epidermis (AE1/AE3) (Fig. 5b). In the epidermis, positive K14 basal cells could be observed, which indicated the cells were in proliferative state (Fig. 5c). No suprabasal stratum existed as is shown by the K10 antibody, a marker for the late differentiation of keratinocytes (Fig. 5d). There was also some evidence of p63, indicating the presence of epidermal stem cells (Fig. 5e). But, there were not positive Ki67 cells (Fig. 5f).

Morphological and phenotypical characterization of the in vitro skin equivalent cultured for 7 days. a Staining with hematoxylin–eosin (H–E). Immunohistochemistry for cytokeratin (b), keratin 14 (c), keratin 10 (d), p63 (e), Ki67 (f), and vimentin (g). Immunofluorescence for laminin (h). (a–g, ×400 magnification) (h, ×200 magnification)
Fibroblasts infiltrating the HAM and the synthesized stromal collagen matrix, were, as demonstrated by immunohistochemistry for vimentin (Fig. 5g). The basement membrane remained intact, as demonstrated by the expression of laminin seen with immunofluorescence (Fig. 5h). The basement membrane is important for anchoring keratinocytes on the HAM. The absence of DAPI nuclei fluorescence again demonstrated the complete elimination of unwanted cells.
Discussion
Tissue engineering techniques applied to the repair of damaged skin in patients with different pathologies generally include reconstruction of the skin in vitro by isolating epidermal keratinocytes and dermal fibroblasts from the patient and their expansion through specific cultivation techniques, including construction of a matrix or support for the final cultured dermal cells. For this purpose, various alternatives have been followed that provide a sheet of epithelium with a dermal base. One of the most novel alternatives has been the formation of a artificial skin structure that includes both epithelial culture and culture of the dermal component, the latter based on the creation of an extracellular matrix in human serum using a gel which provides a three-dimensional structure with human fibroblasts inside (Llames et al. 2004).
Following development of this model, several groups have investigated the use of other supports in skin tissue engineering. HAM has shown great potential because of its structure, biological properties, mechanical properties and low immunogenicity (Niknejad et al. 2008). For use as scaffold, HAM must first be decellularized; many methods have been used with varying degrees of success. Some of these methods are too complex for practical use and others fail because they not only eliminate all cells but also the cellular components of the tissue matrix (Wilshaw et al. 2006). The methods most commonly used to remove the HAM epithelium are chemical agents, such as EDTA, enzymatic proteolytic enzymes, such as dispase (Lim et al. 2009), or enzymes in chemical combination, such as T/E at a concentration range from 0.1 to 0.25 % at 37 °C for 30 min, followed by a mechanical treatment (Hopkinson et al. 2008; Riau et al. 2010).
In our study, the first objective was to analyze which of all the methods described in the literature was the most effective to eliminate not only the epithelium, but also stromal cells (Yang et al. 2009; Lim et al. 2009; Hopkinson et al. 2008; Barreto de Melo et al. 2007; Kim et al. 2009). We were able to establish that digestion with 1 % T/E for 30 min at 37 °C completely eliminated not only the epithelium, but also the cells of the stroma. This method excluded the need for the mechanical treatment described in other methods that damages the basal membrane or the matrix, while maintaining the content of proteins, such as laminin, collagen fibers, mucopolysaccharides and proteoglycans.
It has been stated that complete elimination of the epithelial cells of the HAM before seeding keratinocytes increases their percentage of confluency. The elimination of the epithelium of the HAM facilitates the migration of keratinocytes (Yang et al. 2009) and other cells, such as limbal cells (Shortt et al. 2009) and neurons (Davis et al. 1987).
In the other treatments that we analyzed, 0.25 % T/E, EDTA or accutase, did not eliminate the epithelium completely, making the use of a rake necessary, which resulted in the mechanical corruption of the membrane, as shown by absence of expression of laminin. While the use of dispase completely eliminated the epithelium, the basal membrane was also enzymatically damaged, as shown again by the lack of expression of laminin.
These results agree with those of other authors who, after using treatment with EDTA, only managed to remove the epithelium in some areas while leaving it intact in others (Barreto de Melo et al. 2007), or after using a solution of accutase containing proteolytic enzymes, only the cells of the epithelium were removed, not the mesenchymal cells of the stroma.
To confirm the total elimination of the cells of the epithelium and stroma, as an indirect measurement of the presence of cells, we also examined the content of residual DNA following the different treatments. Our results confirmed that treatment with 1 % T/E for 30 min at 37 °C was the most effective because it resulted in the lowest content of residual DNA. This analysis was used by Wilshaw et al. (2006) to assess the DNA content after the use of sodium dodecyl sulphate (SDS) to eliminate the epithelium.
Although complete elimination of epithelial cells of the HAM is necessary for its use as a “scaffold”, it is also vital that the extracellular matrix remains intact for cellular repopulation, because the extracellular matrix contains the essential components to anchor cell proliferation and remodel the tissue (Liang et al. 2009). The main components of the extracellular matrix are the proteins, collagen, fibronectin and laminin, which promote in vitro adherence of cells. Our results demonstrate the presence of all those components after treatment with 1 % T/E for 30 min at 37 °C and, therefore, confirm that the treatment does not damage the extracellular matrix.
Previous studies have shown that other treatments, including the use of dispase, digest several molecules of the extracellular matrix, particularly those of the basal membrane, including collagen VI, fibronectin and laminin. Transmission electron microscopy confirmed breakage of the ultrastructure of the HAM, starting with alteration of the basal lamina and continuing with loss of the stromal collagen network and its molecular composition (Hopkinson et al. 2008). Also, prolonged incubation with dispase caused significant disruption of the structure of the denuded HAM, which can affect cell growth in cultured explants (Lim et al. 2009).
Once we determined the most effective treatment for decellularization of the HAM, our next objective became analysis of the HAM obtained for its suitability as a support in an in vitro skin equivalent model. With the concept of providing more consistency and better cosmetic results from a dermo-epidermic union (Velásquez et al. 2008), we generated an in vitro model that had keratinocytes and fibroblasts cultivated on a HAM support.
For creation of in vitro skin models, various approaches have been used. As it was described previously (Llames et al. 2004), we used collagenase to optimize the extraction of fibroblasts from the biopsies and subsequently isolated the keratinocytes from skin biopsies by digestion with T/E. Our in vitro skin model was cultured using a submerged method, and after only 7 days we had already obtained a skin equivalent. This method considerably reduced the time needed for culture by other investigators who used the air-lift method (Yang et al. 2009). Animal model studies have shown that airlift cultures produced degeneration of the grafted epithelium. This does not occur when a monolayer is cultured using the submerged method (Peña et al. 2010), producing an equivalent structure with several layers in which keratinocytes proliferate to give the stratified appearance.
In previous studies, our group found that keratinocytes grow and thrive best on the basal lamina because of laminin, an essential element for the anchoring, polarization and migration of keratinocytes. In our in vitro skin model, that protein remains in the HAM after the enzymatic treatment, promoting anchoring of the keratinocytes. The fibroblasts infiltrate, grow and thrive best on the stromal side of the HAM. These results agree with those of other authors (Yang et al. 2009), who showed that the existence of the components of the basal membrane in the dermal matrix help to improve the morphology of the epidermis and are necessary for the formation of the hemidesmosomes and the development of dense lamina (Yang et al. 2006).
Histological and immunohistochemical results show that the equivalent of skin structure obtained in our model is organized into several layers of keratinocytes and fibroblasts with a matrix. The expression of cytokeratin and keratin 14 indicates formation of a structure with keratins in the foil of the epidermis with proliferative capacity of the keratinocyte, while keratin 10 is absent, indicating that there is no terminal differentiation of keratinocytes. Some of the keratinocytes are p63 positive indicating possible presence of epidermal stem cells (Kim et al. 2004). The expression of p63 is increased during the healing of the wounds as well as in the basal and suprabasal layers of normal skin. The location of the p63 positive cells is associated with the stratum of the proliferative epithelium. It is possible that p63 may play a role in maintenance of the proliferative potential of keratinocytes and prevent terminal differentiation (Coolen et al. 2007). The presence of vimentin-positive cells reveals the location of the fibroblasts in the stromal side of the HAM.
However, in our model we do not observe expression of the cell proliferation marker Ki-67. Other authors (Peña et al. 2010) also reported a negative result in their plasma model, offering the explanation that when the epithelium arrives at confluency, proliferative activity is reduced. Another explanation is that at the end of the G1 phase cell cycle and the beginning of S-phase, the quantity of the Ki-67 antigen is minimal. Therefore, a cell in G1 phase and S-phase could be negative for Ki-67, but be proliferating.
We conclude that the best treatment to eliminate the cells of the HAM is 1 % T/E for 30 min at 37 °C. The use of the HAM matrix thus created to develop a skin equivalent model synthesized from the HAM as a support with the patient’s own cells to develop a dermis and epidermis has great potential for use in clinical practice.
References
Barreto de Melo GB, Gomes JA, da Glória MA, Martins MC, Haapalainen EF (2007) Morphological assessment of different amniotic membrane epithelial denuding techniques. Arq Bras Oftalmol 70:407–411
Buinewicz B, Rosen B (2004) Acellular cadaveric dermis (AlloDerm): a new alternative for abdominal hernia repair. Ann Plast Surg 52:188–194
Cervelli V, Lucarini L, Spallone D, Brinci L, de Angelis B (2010) Use of platelet rich plasma and hyaluronic acid on exposed tendons of the foot and ankle. J Wound Care 19:188–190
Coolen NA, Verkerk M, Reijnen L, Vlig M, van den Bogaerdt AJ, Breetveld M, Gibbs S, Middelkoop E, Ulrich MM (2007) Culture of keratinocytes for transplantation without the need of feeder layer cells. Cell Transplant 16:649–661
Davis GE, Engvall E, Varon S, Manthorpe M (1987) Human amnion membrane as a substratum for cultured peripheral and central nervous system neurons. Brain Res 430:1–10
Díaz-Prado S, Muiños-López E, Hermida-Gómez T, Rendal-Vázquez ME, Fuentes-Boquete I, de Toro FJ, Blanco FJ (2011) Isolation and characterization of mesenchymal stem cells from human amniotic membrane. Tissue Eng Part C Methods 17:49–59
Hafemann B, Ensslen S, Erdmann C, Niedballa R, Zühlke A, Ghofrani K, Kirkpatrick CJ (1999) Use of a collagen/elastin-membrana for the tissue engineering of dermis. Burns 25:373–384
Hopkinson A, Shanmuganathan VA, Gray T, Yeung AM, Lowe J, James DK, Dua HS (2008) Optimization of amniotic membrane (AM) denuding for tissue engineering. Tissue Eng Part C Methods 14:371–381
Kesting MR, Wolff KD, Hohlweg-Majert B, Steinstraesser L (2008) The role of allogenic amniotic membrane in burn treatment. J Burn Care Res 29:907–916
Kim DS, Cho HJ, Choi HR, Kwon SB, Park KC (2004) Isolation of human epidermal stem cells by adherence and the reconstruction of skin equivalents. Cell Mol Life Sci 61:2774–2781
Kim SS, Song CK, Shon SK, Lee KY, Kim CH, Lee MJ, Wang L (2009) Effects of human amniotic membrane grafts combined with marrow mesenchymal stem cells on healing of full-thickness skin defects in rabbits. Cell Tissue Res 336:594–599
Liang HS, Liang P, Xu Y, Wu JN, Liang T, Xu XP, Liu EZ (2009) Denuded human amniotic membrane seeding bone marrow stromal cells as an effective composite matrix stimulates axonal outgrowth of rat neural cortical cells in vitro. Acta Neurochir (Wien) 151:1113–1120
Lim LS, Riau A, Poh R, Tan DT, Beuerman RW, Mehta JS (2009) Effect of dispase denudation on amniotic membrane. Mol Vis 15:1962–1970
Llames SG, Del Rio M, Larcher F, García E, García M, Escamez MJ, Jorcano JL, Holguín P, Meana A (2004) Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation 77:350–355
Macri L, Clark RA (2009) Tissue engineering for cutaneous wounds: selecting the proper time and space for growth factors, cells and the extracellular matrix. Skin Pharmacol Physiol 22:83–93
Mazlyzam AL, Aminuddin BS, Fuzina NH, Norhayati MM, Fauziah O, Isa MR, Saim L, Ruszymah BH (2007) Reconstruction of living bilayer human skin equivalent utilizing human fibrin as a scaffold. Burns 33:355–363
Meana A, Iglesias J, Del Rio M, Larcher F, Madrigal B, Fresno MF, Martin C, San Roman F, Tevar F (1998) Large surface of cultured human epithelium obtained on a dermal matrix based on live fibroblast containing fibrin gels. Burns 24:621–630
Niknejad H, Peirovi H, Jorjani M, Ahmadiani A, Ghanavi J, Seifalian AM (2008) Properties of the amniotic membrane for potential use in tissue engineering. Eur Cell Mater 15:88–99
Orgill DP, Straus FH, Lee RC (1999) The use of collagen-GAG membranes in reconstructive surgery. Ann N Y Acad Sci 888:233–248
Peña I, Junquera LM, Meana A, García E, García V, De Vicente JC (2010) In vitro engineering of complete autologous oral mucosa equivalents: characterization of a novel scaffold. J Periodontal Res 45:375–380
Riau AK, Beuerman RW, Lim LS, Mehta JS (2010) Preservation, sterilization and de-epithelialization of human amniotic membrane for use in ocular surface reconstruction. Biomaterials 31:216–225
Shortt AJ, Secker GA, Lomas RJ, Wilshaw SP, Kearney JN, Tuft SJ, Daniels JT (2009) The effect of amniotic membrane preparation method on its ability to serve as a substrate for the ex vivo expansion of limbal epithelial cells. Biomaterials 30:1056–1065
Takemoto S, Morimoto N, Kimura Y, Taira T, Kitagawa T, Tomihata K, Tabata Y, Suzuki S (2008) Preparation of collagen/gelatin sponge scaffold for sustained release of bFGF. Tissue Eng Part A 14:1629–1638
Theoret C (2009) Tissue engineering in wound repair: the three “R”s–repair, replace, regenerate. Vet Surg 38:905–913
Velásquez DA, Pineda C, Cardona ME, Gómez NE, Gartz GJ, Úsuga IC, Tróchez DF, Londoño C (2008) Soluciones terapéuticas para la reconstrucción de la dermis y la epidermis. Oportunidades en el medio antioqueño. Rev Ing Bioméd 2:77–83
Wilshaw SP, Kearney JN, Fisher J, Ingma E (2006) Production of an acellular amniotic membrane matrix for use in tissue engineering. Tissue Eng 12:2117–2129
Wilshaw SP, Kearney J, Fisher J, Ingma E (2008) Biocompatibility and potential of acellular human amniotic membrane to support the attachment and proliferation of allogeneic cells. Tissue Eng Part A 14:463–472
Yang L, Shirakata Y, Shudou M, Dai X, Tokumaru S, Hirakawa S, Sayama K, Hamuro J, Hashimoto K (2006) New skin-equivalent model from de-epithelialized amnion membrane. Cell Tissue Res 326:69–77
Yang L, Shirakata Y, Tokumaru S, Xiuju D, Tohyama M, Hanakawa Y, Hirakawa S, Sayama K, Hashimoto K (2009) Living skin equivalents constructed using human amnions as a matrix. J Dermatol Sci 56:188–195
Yu YR, Min DH, Liu SJ, Wang M, Guo GH, Li GH (2006) Experimental study on xenogenic acellular dermal matrix incorporated with silver. Zhonghua Shao Shang Za Zhi 22:296–300
Acknowledgments
This work was supported by a grant from “Consellería de Innovación e Industria Dirección Xeral de I+D+I (No. 08CSA065916PR)”. A. Verdes-Sanluis was supported by a grant from “Centro de Investigación Biomédica en Red en Bioingeniería, Biomateriales y Nanomedicina” (CIBER-BBN-CB06/01/0040).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Sanluis-Verdes, A., Yebra-Pimentel Vilar, M.T., García-Barreiro, J.J. et al. Production of an acellular matrix from amniotic membrane for the synthesis of a human skin equivalent. Cell Tissue Bank 16, 411–423 (2015). https://doi.org/10.1007/s10561-014-9485-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10561-014-9485-2