
Abstract
Epidemiological and clinical studies have identified many physiological traits and biomarkers that are statistically associated with coronary artery disease (CAD). For some of these traits and biomarkers it is well established that they represent true causal risk factors for CAD. For other biomarkers, however, the distinct character of association is still a matter of debate. Randomized controlled trials (RCT) had a pivotal role in establishing causal associations between risk factors and biomarkers and CAD in some settings by demonstrating that therapeutic intervention targeting risk factors/biomarkers also affect the risk for clinical outcomes, such as CAD. In other scenarios, however, RCTs did not demonstrate clear benefits associated with lowering biomarker levels and therefore suggest that the association between these biomarkers (like C reactive protein) and CAD was driven by confounding or reverse causation. Even accurately conducted RCTs are not immune against incorrect causal inference. Moreover, the extensive costs and efforts required to conduct RCTs asked for alternative study designs to elucidate potential causal associations. Mendelian Randomization studies represent one such alternative by using genetic variants as proxies for specific biomarkers to investigate potential causal relations between biomarkers and clinical outcomes. In this review, we briefly describe the principles of MR studies and summarize recent MR studies in the context of CAD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epidemiological and clinical studies have identified many physiological traits and biomarkers that are statistically associated with coronary artery disease (CAD). For some of these traits and biomarkers it is well established that they represent true causal risk factors for CAD. For other biomarkers, however, the distinct character of association is still a matter of debate [1, 2]. Randomized controlled trials (RCT) had a pivotal role in establishing causal associations between risk factors and biomarkers and CAD in some settings by demonstrating that therapeutic intervention targeting risk factors/biomarkers also affect the risk for clinical outcomes, such as CAD [3]. In other scenarios, however, RCTs did not demonstrate clear benefits associated with lowering biomarker levels and therefore suggest that the association between these biomarkers (like C reactive protein) and CAD was driven by confounding or reverse causation [4, 5].
Even accurately conducted RCTs are not immune against incorrect causal inference.
Moreover, the extensive costs and efforts required to conduct RCTs asked for alternative study designs to elucidate potential causal associations. Mendelian Randomization studies represent one such alternative by using genetic variants as proxies for specific biomarkers to investigate potential causal relations between biomarkers and clinical outcomes [6]. In this review, we briefly describe the principles of MR studies and summarize recent MR studies in the context of CAD.
Principles of Mendelian Randomisation Studies
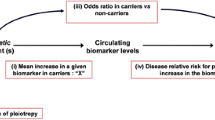
The aim of MR studies is to assess whether the associations between a biomarker (exposure) and a disease phenotype (outcome, e.g. CAD) are causal. One key feature of MR studies is that a genetic variant is used as a proxy for the biomarker of interest (Fig. 1). The ideal scenario is that the genetic variant causally effects the biomarker level. Due to the fact that the alleles of a genetic variant are distributed randomly at meiosis (Mendel’s second law of independent assortment), individuals are then “randomized” by nature to higher or lower biomarker levels. In this regard, MR studies and RCTs share an essential feature as individuals are being randomized in both scenarios – either by nature or by a standardised study protocol to either lower or higher levels of a biomarker. It is unlikely that the random distribution of alleles is influenced by other factors that could also affect the outcome, making MR studies, therefore, not susceptible to confounding, i.e. indirect modulation by factors associated with the biomarker and the outcome. Hence, both groups represented by the respective genotypes are comparable except for the biomarker which is influenced by the genetic variant [7]. A further relevant assumption for MR studies is the absence of pleiotropic effects. That means that the genetic variant affects the outcome only via the biomarker of interest. It is essential that the chosen SNP does not have any other significant effects on intermediate disease traits or biomarkers, which might also influence the outcome measure [8].

Basic principles of MR. A genetic variant has be to substantially associated with the biomaker of interest. It is independent of any confounders (measured or unmeasured). Moreover, the association between a SNP and the disease outcome has to be mediated only through the biomaker of interest. Fufilling these requirements, the association between a biomaker and disease outcome can be assumed to be causal. Adapted from Dovey Smith G and Hemani G: Mendelian randomisation: genetic anchors for causal inference in epidemiological studies, Hum Mol Genet., 2014
Such pleiotropic effects could lead to wrong conclusions in many ways. First, an observed effect might be driven by another biomarker, which was not subject of the conducted MR analysis but is also associated significantly with the studied SNP and the outcome. Second, another biomarker (correlated with SNP and outcome) might abolish true positive associations between a first biomarker and the outcome by counteracting its effect. A true causal association between biomarker 1 and the outcome could then be easily missed [7].
In the past decade, genome-wide association studies have identified numerous single nucleotide polymorphisms (SNPs) that display robust associations with cardiovascular biomarkers or disease traits, including diabetes, LDL or HDL cholesterol, triglycerides or various circulating proteins (e.g. C-reactive protein) [9, 10] as well as cardiovascular endpoints such as CAD, stroke or heart failure. The identification of genetic information associated with biomarker levels and the exploration how these variants associated with CAD is an important requirement for MR studies.
Cardiovascular Biomarkers and Disease Traits Tested in MR Studies
Fostered by enormous progress in cardiovascular genetics, MR studies have been conducted for more than ten years making important scientific contributions. Some of these contributions encouraged further drug developments like PCSK9 inhibitors [11, 12], but also reinforced disappointments from negative pharmacological trials on –for example- few CETP inhibitors [5, 13].
A systematic search in PubMed (using the terms “Mendelian randomization” and “coronary heart disease”) confirmed a recent rapid increase in the number of MR studies. Since our last review on MR studies for CAD in 2014 [7], the number of hits using the above search terms almost doubled up to 215. It is far beyond the scope of a single review to present all biomarkers and cardiovascular disease traits tested in MR settings in more detail. Therefore, we name several examples in Table 1 and focus on recent important discoveries.
Height
Many epidemiological studies observed an association between shorter adult height and the risk for CAD [14]. A meta-analysis reported that a decrease of 1 SD (approximately 6.5 cm) goes along with a relative risk increase of 8 % for CAD [15, 16]. Even after adjustment for known cardiovascular risk factors (e.g. hypertension, dyslipidemia, smoking habits, diabetes), which are also associated with shorter height, the effect remained. Hence, the distinct mechanisms linking height to CAD and whether this relation is causal remains unclear. A genomewide association study (GWAS) identified 180 genetic variants associated with height [17], which were tested for their association with CAD in the CARDIoGRAM and C4D-consortia involving more than 65,000 cases and 128,000 controls [16]. This analysis revealed that a 1-SD decrease of genetically determined height increases the risk of CAD by 13.5 % (95 % CI 5.4-22.1, p < 0.001). Some of these genetic variants also increase circulating levels of LDL-cholesterol and triglycerides. However, Nelson et al. estimated that less than one third of the observed association between height and CAD is driven by these established risk factors. Thus, other biological processes, determining height and CAD may explain the observed correlation.
Dysglycaemia and Type 2 Diabetes
Several observational studies reported that diabetes is a risk factor for CAD. Meta-analyses of clinical trials supported the strategy that long-term glucose lowering reduces adverse cardiovascular outcomes [18]. However, recent trials failed to reduce cardiovascular events by lowering blood glucose levels, thereby challenging the concept of a causal interference between diabetes and large vessel disease, such as CAD [19, 20]. Therefore, Ross and colleagues investigated the relation between dysglycemia and diabetes and CAD using an MR study [21]. By analysing 59 SNPs associated with type 2 diabetes (T2DM), they estimated the causal effect of diabetes on CAD as an OR of 1.63 (95 % CI 1.23-2.07), which is inline with individual observational studies and meta-analyses [21]. In a former study diabetes SNPs (n = 44) also proved a significant increase in CAD risk (the average increase in CAD risk per T2DM-SNP was 1.0076, p = 0.02), albeit to a lesser extent than expected given the SNP effects on T2DM and observational estimates derived from the Framingham Heart Study [22]. The different estimates of diabetes being a causal risk factor for CAD potentially arise from the varying number of SNPs studied in these two MR settings. Moreover, Ross and colleagues used the MR technique not only to assess the CAD risk increase per individual allele but also calculated the magnitude of diabetes as a true risk factor for CAD (OR 1.63).
Niemann-Pick C1-Like 1
LDL cholesterol is one of the most important modifiable risk factors for CAD. In addition to observational evidence and RCTs on the effect of statins [23], MR studies on PCSK9 provided support for a causal relationship between LDL and CAD [24]. This convincing evidence from observational data, RCTs and MR studies fostered promising drug developments [11, 12].
Beside statins, ezetimibe, which acts through the inhibition of Niemann-Pick C1-like 1 (NPC1L1), is frequently administered to lower LDL-cholesterol. However, the benefit of an NPC1L1-inhibition in terms of cardiovascular outcomes was not proven for a long time.
Recently, the NPC1L1 exome was sequenced in more 20,000 individuals from different ethnities and 15 distinct NPC1L1 inactivating mutations were identified [25]. Heterozygous carriers of these mutations (about 1 in every 650 persons) had on average 12 mg/dl lower LDL cholesterol levels compared to non-carriers, and a reduced CAD risk. These findings suggest that a lifelong inhibition of NPC1L1 decreases the risk of CAD. The authors speculated that such effect could also be achieved by a pharmacological inhibition. Indeed, IMPROVE-IT investigators recently reported that ezetimibe, when added to simvastatin, was able to further decrease the absolute risk difference of the primary endpoint (cardiovascular death, myocardial infarction, unstable angina, coronary revascularization or stroke) by 2 % (towards 32.7 %) over 7 years of treatment. Such results provide evidence that this agent not only decreases LDL cholesterol but also prevents future cardiovascular events [26].
APOC3
Recently, a large-scale MR-analysis suggested a causal role of triglycerides in CAD by studying 185 independent genetic variants associating with lipid traits [27]. Subsequently, the same group asked to which extent rare mutations contribute to the variation of triglyceride level and alter the risk of CAD [28]. The authors made great efforts and sequenced almost 20,000 genes in 3734 participants to identify such genetic variants, which were then tested for their association with CAD (34,002 cases and 76,968 controls). They found a combination of four loss-of-function mutations within the APOC3 gene associating strongly with triglyceride levels. About 1 in 150 persons carried any of these rare mutations, which were associated with a 39 % decrease in triglyceride levels compared to non-carriers. Also, HDL cholesterol (22 % increase) and LDL cholesterol (16 % decrease) were affected by these variations. Interestingly, heterozygous carriers of any of these mutations had a 40 % lower risk of CAD than noncarriers (OR 0.6, 95 % CI 0.47-0.75, p = 4 × 10−06) [28]. These results indicate that a loss-of-function of APOC3 contributes to the risk of CAD, making APOC3 an interesting potential target for future interventions to reduce the risk of CAD [29].
25-hydroxyvitamin D
Based on three large study samples from the Copenhagen area, Danish researchers assessed a potential causal association between 25-hydroxyvitamin D and CAD [30]. Numerous observational studies reported that low concentrations of 25-hydromyvitamin D (p-25(OH)D) were associated with a greater risk for CAD and myocardial infarction [31, 32]. A meta-analysis of 18 observational prospective studies found a 39 % risk increase for CAD when comparing individuals in the lowest vs. the highest quartile of p-25(OH)D concentrations [33].
RCTs investigating the health benefit of p-25(OH)D did not sufficiently focus on cardioprotective effects nor studied solely the effect of p-25(OH)D [33, 34]. Hence, an MR analysis would be suitable to investigate this association. In total, four genetic variants which reduce p-25(OH)D concentrations were tested in an MR setting using the above mentioned Danish cohorts with the following key results. Individuals in the lowest vs. the highest quartile of p-25(OH)D concentrations displayed an increased risk of ischemic heart disease (HR: 1.82 (95 % CI 1.42-2.32). Second, each allele increase associated with a 1.9 nmol/decrease in p-25(OH)D in an aggregated genetic risk score consisting of such four genetic variants within the CYP2R1 and DCHR7 loci. Third, the authors found no evidence that the analysed variants associated with risk for CAD (OR 0.98, 95 % CI 0.76-1.26). In sum, these data argue against a causal relationship between p-25(OH)D and CAD.
Reverse causation or confounding have to be taken into consideration to explain the positive associations observed in epidemiological settings. Since p-25(OH)D concentrations can also mirror socioeconomic factors, malnutrition and an unhealthy lifestyle, it is conceivable that the association might be driven by such confounders [33].
Vitamin C
Elevated vitamin C concentrations as a consequence of high intake of fruits and vegetables are commonly paralleled by multiple health benefits and reduced all-cause mortality [35]. Moreover, prospective studies reported that vitamin C might also have beneficial effects on CAD [36]. However, RCTs for vitamin C supplementation provided inconsistent results so far [37, 38]. In this regard, the Nordestgaard group conducted a MR analysis with samples from the general Danish community [38]. The authors found a strong association between a genetic variant (rs33972313) within the SLC23A1 gene -encoding for a sodium-dependent vitamin C transporter- and higher plasma vitamin C levels (11 % per allele). Comparing groups with the highest vs. the lowest intake of fruits and vegetables, the authors observed multivariable adjusted hazard ratios of 0.87 (95 % CI 0.78-0.97, p = 0.01) for CAD. Albeit these facts, the results obtained from the MR analysis drew an ambiguous picture: a genetically determined 25 % elevation of plasma vitamin C produced an OR for CAD of 0.90 with a 95 % CI including 1.0 (0.75–1.08). Since the OR-range is spanning 1.0, it makes it difficult to jump to a clear statement about causal interrelation. Nevertheless, given comparable effect sizes to those of fruit and vegetable intake, a certain effect of genetically elevated vitamin C concentrations on CAD seems likely.
Milk Intake
Some preliminary evidence indicates that higher milk intake goes along with increased blood cholesterol and higher risks for CAD and myocardial infarction [39], whereas some observational analyses did not support this concept [40]. RCTs are difficult to conduct when it comes to food intake because of the required longterm adherence to randomization [41]. Bergholdt et al. evaluated rs4988235 within the MCM6 locus (a genetic variant associated with lactase persistence/non-persistence) as a suitable proxy to assess the potential causal association between milk intake and CAD in an MR study [41]. Carriers of the TC and TT genotype are known to have a regular enzyme function while individuals with the CC genotype suffer from difficulties to digest milk products – many of them develop symptoms of lactose intolerance when continuing milk consumption [41]. Using datasets from almost 100,000 individuals from the general population, the authors found 1) an observational hazard ratio for a 1 glass/week of 1.0 (95 % CI 1.00-1.01) for both CAD and MI; 2) a median milk intake of 3 glasses/week in lactase CC non-persistent individuals compared with 5 glasses/week in individuals carrying the TC and TT genotype (p = 3 × 10−60) and; 3) no association with CAD nor MI when comparing lactase TC/TT persistent individuals with lactase CC non-persistent individuals (OR 1.0, 95 % CI 0.92-1.09 for CAD and OR 0.96, 95 % CI 0.84-1.09 for MI) genotype. These findings indicate neither an observational nor a genetic association between milk intake and CAD/MI.
Circulating Brain-Derived Neurotrophic Factor
Brain-Derived Neurotrophic Factor (BDNF) is a peptide playing an important role for the development of obesity by influencing behaviours like food intake and physical activity [42]. Animal models revealed inverse relations between circulating BDNF and unfavorable outcome measures such as obesity and an increased myocardial infarct size after experimental infarction [43–45]. Kaess and colleagues investigated the association between circulating BDNF levels and cardiovascular events and mortality within the Framingham Heart Study (FHS) cohort and found an inverse association between serum BDNF and CAD risk (HR per 1-SD increase 0.88, 95 % CI 0.80-0.97, p = 0.01) and mortality (HR 0.87, 95 % CI 0.80-0.93, p = 0.0002) [43]. Next, Kaess et al. performed a MR analysis using a nonsynonomous SNP (rs6365) within the BDNF gene. This SNP was associated with BDNF levels in the FHS cohort (0.772 ng/ml increase per minor allele copy) and with CAD in the CARDIoGRAM consortium (OR 0.957, 95 % CI 0.923-0.992). These data suggest that BDNF might have a causal and protective effect on the development of CAD.
Celiac Disease
In epidemiological settings patients with celiac disease are at increased risk for CAD [46, 47]. Whether this observation is due to a modified risk profile (e.g. an unfavourable lipid constellation) or because of a per se causal association between celiac disease and CAD remained obscure. A set of 41 genetic variants which was robustly associated with celiac disease in prior studies, were tested for association with CAD in CARDIoGRAM (please see above) [48]. Only 24 SNPs (58.5 %) produced ORs greater than 1 (CAD-OR range 1.001–1.081), while the remaining 17 variants displayed ORs of either 1.0 or below (ORs range 0.951–1.0). This proportion (58.5 %) of risk increasing alleles with consistent effects on celiac disease and CAD did not differ significantly (p = 0.069) from the proportion expected just by chance (50 %). Hence, a causal association between these two diseases is rather unlikely given the results from this genetically based analysis. Shared non-genetic factors like dietary or metabolic alterations in celiac disease are more likely to explain the observed association.
Leukocyte Type-1 Interferon Production
Cytokines play a pivotal role in chronic inflammatory diseases like atherosclerosis and CAD [49]. Type-1-interferons (IFN-I) as part of the antiviral response in inflammatory conditions have been intensively studied. The production of a subgroup (INF-α) is markedly elevated in autoimmune diseases like systemic lupus erythemathodes (SLE), which in turn displays a risk factor for CAD [50, 51].
To date, more than 50 % of genetic variants, which have been identified to associate with SLE are involved in the INF-I pathway. Further evidence for a causal role of IFN-I in SLE comes from epidemiological reflections: an OR of ≈7.5 for the association of CAD in SLE patients cannot be attributed to traditional CAD risk factors. Hence, Nelson and colleagues had good reasons to choose SLE associated genetic variants as proxies to study the relation of INF-I-production and CAD since GWAS on INF-I production were missing to date [51]. First, they calculated a genetic risk score consisting of 3 SNPs, which correlated significantly with INF-α production in cell culture experiments but did not associated with CAD in the CARDIoGRAM consortium (OR 1.0, 95 % CI 0.98-1.02). In addition, the authors tested a set of SLE-associated SNPs (n = 11) and –again- found no association with CAD. These MR based analyses raise doubt about INF production being an indispensable and causal step in CAD development. The utility of drugs targeting INF-I production has to be critically evaluated [51].
Chronic Kidney Disease and Marker of Renal Function
Several adverse outcomes are closely related to chronic kidney disease (CKD), and CAD represents the most common cause of death in patients with CKD [52]. Since traditional and novel risk factors are not able to fully explain the association between CKD and CAD, Olden and colleagues asked whether a genetic link might close the remaining gap [52]. A total of 19 SNPs associated with kidney function in GWAS, was tested for its association with CAD using data from more than 100,000 individuals. Only one SNP (rs653178) near SH2B3 produced a p-value <0.05 with a direction-consistent OR of 1.08 (95 % CI 1.04-1.11) for association with CAD, indicating only limited evidence for a common genetic architecture of CKD and CAD.
Svensson-Färbom and his group took a close look on cystatin C, which represents an optimal marker of impaired renal function [53]. Not only CKD but also higher levels of cystatin C itself increase the risk for CAD and mortality as compared to individuals with lower cystatin C levels [54]. This association holds true even in patients with normal kidney function [55]. Different explanations for this phenomenon including toxic effects of cystatin C and progression of dysmetabolic states in patients with elevated cystatin C have been intensively discussed [53]. Based on these pathophysiological considerations one might speculate about a causal relationship between cystatin C and CAD. A recent GWAS brought a genetic variant (rs13038305) to light, which associates with cystatin C independently of creatinine based measures of kidney function [56]. The authors tested this SNP for its association with cystatin C levels in the Malmö Diet and Cancer study (MDC, 4743 subjects) and found a 0.34 SD increase in cystatin C per minor allele. Next, they confirmed results from previous observations by showing a clear relation between cystatin C and CAD. Finally, using the above mentioned SNP as a proxy for cystatin C levels, the authors conducted an MR analysis and found no evidence for an association between the genetic variant and CAD, neither in the MDC study (OR 1.0, 95 % CI 0.94-1.07, p = 0.92), nor within CARDIoGRAM (OR 0.99, 95 % CI 0.96-1.03, p = 0.84), suggesting no causal association between cystatin C and CAD. Rather, the epidemiological observations have to be seen in the light of impaired kidney function and connected pathophysiological changes [53].
Summary
Mendelian randomization studies are now being conducted for more than 10 years, contributing important evidence regarding the possible causal association of cardiovascular biomarkers with clinical CAD. While some disputable biomarkers provided no evidence for causality in MR studies [57], other MR results paved the way for promising drug developments [58]. As intensively discussed by others, it is essential to adhere strictly to the principles of MR studies to minimize the risk of misinterpretations coming e.g. from pleiotropy. Ruling out pleiotropy is particularly difficult because not all biological mechanisms of a genetic variant of interest might be known. Therefore, an advanced knowledge about genes, their interactions and their effect on (patho-) physiological traits is mandatory to allow a precise selection of genetic variants for MR studies. On the other hand, statistical methods are rapidly improving to pinpoint causal associations. For example, many authors used multiple genetic variants [16] to increase statistical power and decrease the likelihood of relevant pleiotropic effects. Other (statistical) developments to further improve conclusions from MR studies and facilitate the conduction of such studies include 1) two-sample MR studies to allow testing the biomarker and e.g. CAD in different cohorts 2) bidirectional MR for testing the causal direction between a biomarker and CAD, and 3) multi-phenotype MR studies, where regression methods are used to separate the effects from multiple phenotypes on e.g. CAD [59].
Future MR studies will profit from these -currently evaluated- methods and will potentially bring even more biomarkers to light. However, new discoveries have to be interpreted using other evidences –like RCTs- to strengthen causal assumptions. By now, MR studies were already able to identify important drug targets and to catalyze promising pharmacological developments.
References
Sabater-Lleal M, Huang J, Chasman D, Naitza S, Dehghan A, Johnson AD, et al. Multiethnic meta-analysis of genome-wide association studies in >100 000 subjects identifies 23 fibrinogen-associated loci but no strong evidence of a causal association between circulating fibrinogen and cardiovascular disease. Circulation. 2013;128:1310–24.
IL6R Genetics Consortium Emerging Risk Factors Collaboration, N S, AS B, DF F, J G, P W, et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet Lond Engl. 2012;379:1205–13.
ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The antihypertensive and lipid-lowering treatment to prevent heart attack trial. major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA. 2002;288:2981–97.
Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis–chicken or egg? N Engl J Med. 2008;359:1953–5.
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–63.
Jansen H, Samani NJ, Schunkert H. Mendelian randomization studies in coronary artery disease. Eur Heart J. 2014;35:1917–24.
Smith GD, Timpson N, Ebrahim S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann Med. 2008;40:524–41.
DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium, A M, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–44.
Benjamin EJ, Dupuis J, Larson MG, Lunetta KL, Booth SL, Govindaraju DR, et al. Genome-wide association with select biomarker traits in the Framingham Heart Study. BMC Med Genet 2007; 8 Suppl 1:S11.
Roberts R A genetic basis for coronary artery disease. Trends Cardiovasc Med. 2015;25:171–8.
Giugliano RP, Sabatine MS. Are PCSK9 inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol. 2015;65:2638–51.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99.
Paajanen TA, Oksala NKJ, Kuukasjärvi P, Karhunen PJ. Short stature is associated with coronary heart disease: a systematic review of the literature and a meta-analysis. Eur Heart J. 2010;31:1802–9.
Emerging Risk Factors Collaboration. Adult height and the risk of cause-specific death and vascular morbidity in 1 million people: individual participant meta-analysis. Int J Epidemiol. 2012;41:1419–33.
Nelson CP, Hamby SE, Saleheen D, Hopewell JC, Zeng L, Assimes TL, et al. Genetically determined height and coronary artery disease. N Engl J Med. 2015;372:1608–18.
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8.
Ray KK, Seshasai SRK, Wijesuriya S, Sivakumaran R, Nethercott S, Preiss D, et al. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet Lond Engl. 2009;373:1765–72.
ORIGIN Trial Investigators, Gerstein HC, Bosch J, Dagenais GR, Díaz R, Jung H, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N. Engl. J. Med. 2012; 367:319–28.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–26.
Ross S, Gerstein HC, Eikelboom J, Anand SS, Yusuf S, Paré G. Mendelian randomization analysis supports the causal role of dysglycaemia and diabetes in the risk of coronary artery disease. Eur Heart J. 2015;36:1454–62.
Jansen H, Loley C, Lieb W, Pencina MJ, Nelson CP, Kathiresan S, et al. Genetic variants primarily associated with type 2 diabetes are related to coronary artery disease risk. Atherosclerosis. 2015;241:419–26.
Cholesterol Treatment Trialists’ (CTT) Collaboration, C B, L B, J E, LE H, C R, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet Lond Engl. 2010;376:1670–81.
Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Myocardial Infarction Genetics Consortium Investigators, NO S, H-H W, AC M, GM P, R D, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82.
Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97.
Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52.
TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, J C, GM P, PL A, DR C, NO S, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
Graham MJ, Lee RG, Bell TA, Fu W, Mullick AE, Alexander VJ, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90.
Brøndum-Jacobsen P, Benn M, Afzal S, Nordestgaard BG. No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: a Mendelian randomization study. Int J Epidemiol. 2015;44:651–61.
Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117:503–11.
Kilkkinen A, Knekt P, Aro A, Rissanen H, Marniemi J, Heliövaara M, et al. Vitamin D status and the risk of cardiovascular disease death. Am J Epidemiol. 2009;170:1032–9.
Brøndum-Jacobsen P, Benn M, Jensen GB, Nordestgaard BG. 25-hydroxyvitamin d levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arterioscler Thromb Vasc Biol. 2012;32:2794–802.
Elamin MB, Abu Elnour NO, Elamin KB, Fatourechi MM, Alkatib AA, Almandoz JP, et al. Vitamin D and cardiovascular outcomes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96:1931–42.
Leenders M, Sluijs I, Ros MM, Boshuizen HC, Siersema PD, Ferrari P, et al. Fruit and vegetable consumption and mortality: European prospective investigation into cancer and nutrition. Am J Epidemiol. 2013;178:590–602.
Crowe FL, Roddam AW, Key TJ, Appleby PN, Overvad K, Jakobsen MU, et al. Fruit and vegetable intake and mortality from ischaemic heart disease: results from the European prospective investigation into cancer and nutrition (EPIC)-heart study. Eur Heart J. 2011;32:1235–43.
Myung S-K, Ju W, Cho B, Oh S-W, Park SM, Koo B-K, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;346:f10.
Kobylecki CJ, Afzal S, Davey Smith G, Nordestgaard BG. Genetically high plasma vitamin C, intake of fruit and vegetables, and risk of ischemic heart disease and all-cause mortality: a Mendelian randomization study. Am J Clin Nutr. 2015;101:1135–43.
Huth PJ, Park KM. Influence of dairy product and milk fat consumption on cardiovascular disease risk: a review of the evidence. Adv Nutr Bethesda Md. 2012;3:266–85.
Elwood PC, Givens DI, Beswick AD, Fehily AM, Pickering JE, Gallacher J. The survival advantage of milk and dairy consumption: an overview of evidence from cohort studies of vascular diseases, diabetes and cancer. J Am Coll Nutr. 2008;27:723S–34S.
Bergholdt HKM, Nordestgaard BG, Varbo A, Ellervik C. Milk intake is not associated with ischaemic heart disease in observational or Mendelian randomization analyses in 98,529 Danish adults. Int J Epidemiol. 2015;44:587–603.
Rosas-Vargas H, Martínez-Ezquerro JD, Bienvenu T. Brain-derived neurotrophic factor, food intake regulation, and obesity. Arch Med Res. 2011;42:482–94.
Kaess BM, Preis SR, Lieb W, Beiser AS, Yang Q, Chen TC, et al. Circulating brain-derived neurotrophic factor concentrations and the risk of cardiovascular disease in the community. J Am Heart Assoc. 2015;4:e001544.
Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–300.
Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, et al. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Dev Camb Engl. 2000;127:4531–40.
Ludvigsson JF, James S, Askling J, Stenestrand U, Ingelsson E. Nationwide cohort study of risk of ischemic heart disease in patients with celiac disease. Circulation. 2011;123:483–90.
Wei L, Spiers E, Reynolds N, Walsh S, Fahey T, MacDonald TM. The association between coeliac disease and cardiovascular disease. Aliment Pharmacol Ther. 2008;27:514–9.
Jansen H, Willenborg C, Schlesinger S, Ferrario PG, König IR, Erdmann J, et al. Genetic variants associated with celiac disease and the risk for coronary artery disease. Mol Genet Genomics MGG. 2015;290:1911–7.
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–79.
Kahlenberg JM, Kaplan MJ. Mechanisms of premature atherosclerosis in rheumatoid arthritis and lupus. Annu Rev Med. 2013;64:249–63.
Nelson CP, Schunkert H, Samani NJ, Erridge C. Genetic analysis of leukocyte type-I interferon production and risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2015;35:1456–62.
Olden M, Teumer A, Bochud M, Pattaro C, Köttgen A, Turner ST, et al. Overlap between common genetic polymorphisms underpinning kidney traits and cardiovascular disease phenotypes: the CKDGen consortium. Am. J. Kidney Dis. Off. J Natl Kidney Found. 2013;61:889–98.
Svensson-Färbom P, Almgren P, Hedblad B, Engström G, Persson M, Christensson A, et al. Cystatin C is not causally related to coronary artery disease. PLoS One. 2015;10:e0129269.
Shlipak MG, Sarnak MJ, Katz R, Fried LF, Seliger SL, Newman AB, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med. 2005;352:2049–60.
Ix JH, Shlipak MG, Chertow GM, Whooley MA. Association of cystatin C with mortality, cardiovascular events, and incident heart failure among persons with coronary heart disease: data from the heart and soul study. Circulation. 2007;115:173–9.
Kottgen A, Glazer NL, Dehghan A, Hwang SJ, Katz R, Li M, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nature genetics 2009,41:712–717.
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet Lond Engl. 2012;380:572–80.
Kathiresan S Developing medicines that mimic the natural successes of the human genome: lessons from NPC1L1, HMGCR, PCSK9, APOC3, and CETP. J Am Coll Cardiol. 2015;65:1562–6.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–98.
Dastani Z, Johnson T, Kronenberg F, Nelson CP, Assimes TL, März W, et al. The shared allelic architecture of adiponectin levels and coronary artery disease. Atherosclerosis. 2013;229:145–8.
Holmes MV, Dale CE, Zuccolo L, Silverwood RJ, Guo Y, Ye Z, et al. Association between alcohol and cardiovascular disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2014;349:g4164.
Ross S, D’Mello M, Anand SS, Eikelboom J. CARDIoGRAMplusC4D consortium, Stewart AFR, et al. Effect of bile acid sequestrants on the risk of cardiovascular events: a Mendelian randomization analysis. Circ. Cardiovasc. Genet. 2015;8:618–27.
Stender S, Frikke-Schmidt R, Nordestgaard BG, Grande P, Tybjaerg-Hansen A. Genetically elevated bilirubin and risk of ischaemic heart disease: three Mendelian randomization studies and a meta-analysis. J Intern Med. 2013;273:59–68.
Lieb W, Jansen H, Loley C, Pencina MJ, Nelson CP, Newton-Cheh C, et al. Genetic predisposition to higher blood pressure increases coronary artery disease risk. Hypertension. 2013;61:995–1001.
C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC), Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, et al. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ 2011; 342:d548.
Laugsand LE, Ix JH, Bartz TM, Djousse L, Kizer JR, Tracy RP, et al. Fetuin-A and risk of coronary heart disease: a Mendelian randomization analysis and a pooled analysis of AHSG genetic variants in 7 prospective studies. Atherosclerosis. 2015;243:44–52.
van Meurs JBJ, Pare G, Schwartz SM, Hazra A, Tanaka T, Vermeulen SH, et al. Common genetic loci influencing plasma homocysteine concentrations and their effect on risk of coronary artery disease. Am J Clin Nutr. 2013;98:668–76.
Casas JP, Ninio E, Panayiotou A, Palmen J, Cooper JA, Ricketts SL, et al. PLA2G7 genotype, lipoprotein-associated phospholipase A2 activity, and coronary heart disease risk in 10 494 cases and 15 624 controls of European ancestry. Circulation. 2010;121:2284–93.
Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Lipoprotein(a) and risk of myocardial infarction–genetic epidemiologic evidence of causality. Scand J Clin Lab Invest. 2011;71:87–93.
Benn M, Tybjaerg-Hansen A, McCarthy MI, Jensen GB, Grande P, Nordestgaard BG. Nonfasting glucose, ischemic heart disease, and myocardial infarction: a Mendelian randomization study. J Am Coll Cardiol. 2012;59:2356–65.
Nordestgaard BG, Palmer TM, Benn M, Zacho J, Tybjaerg-Hansen A, Davey Smith G, et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 2012;9:e1001212.
Tang WHW, Hartiala J, Fan Y, Wu Y, Stewart AFR, Erdmann J, et al. Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arterioscler Thromb Vasc Biol. 2012;32:2803–12.
Barbati E, Specchia C, Villella M, Rossi ML, Barlera S, Bottazzi B, et al. Influence of pentraxin 3 (PTX3) genetic variants on myocardial infarction risk and PTX3 plasma levels. PLoS One. 2012;7:e53030.
Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–7 427e1–2.
Ahmad OS, Morris JA, Mujammami M, Forgetta V, Leong A, Li R, et al. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat Commun. 2015;6:7060.
Holmes MV, Simon T, Exeter HJ, Folkersen L, Asselbergs FW, Guardiola M, et al. Secretory phospholipase A(2)-IIA and cardiovascular disease: a mendelian randomization study. J Am Coll Cardiol. 2013;62:1966–76.
Palmer TM, Nordestgaard BG, Benn M, Tybjærg-Hansen A, Davey Smith G, Lawlor DA, et al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. BMJ. 2013;347:f4262.
Acknowledgments
Supported by the EU-funded Integrated Projects CVgenes@target and the BMBF-funded German National Genome Network (NGFN-Plus) Project Atherogenomics (FKZ: 01GS0831) and e:AtheroSysMed, with participation in the German Excellence Center of Cardiovascular Research (DZHK), partner site Munich Heart Alliance and the Leducq consortium CADgenomics.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jansen, H., Lieb, W. & Schunkert, H. Mendelian Randomization for the Identification of Causal Pathways in Atherosclerotic Vascular Disease. Cardiovasc Drugs Ther 30, 41–49 (2016). https://doi.org/10.1007/s10557-016-6640-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-016-6640-y