
Abstract
Interleukin-1 (IL-1) is a major “alarm” upstream pro-inflammatory cytokine that mainly acts by inducing cascades of cytokine and inflammation-promoting mediators. In the tumor arena, IL-1 is produced by both malignant and microenvironmental cells. IL-1α and IL-1β are the major agonists of IL-1, while IL-1Ra is a physiological inhibitor of pre-formed IL-1. IL-1α and IL-1β differ in their compartmentalization and in the producing cells. IL-1β is only active in its inflammasome dependent processed and secreted form and has been considered as the major mediator of inflammation. On the other hand, IL-1α is mainly cell-associated in tissue resident cells, being also active in its precursor form. The role of the IL-1 molecules in the unique microenvironment in the colon is largely unknown. Here, we described the role of IL-1α and IL-1β in colon homeostasis, colon inflammation, colon carcinogenesis and invasiveness of colorectal cancer. Understanding of the integrative role of IL-1α and IL-1β in these processes will facilitate the application of novel IL-1 modulating approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Instruction
Colon cancer is one of the most common malignancies observed in clinical practice. More than 1.2 million cases of colorectal cancer (CRC) are diagnosed each year worldwide. CRC remains the fourth most common cause of cancer-related death in western countries, in spite of early screening and improved treatment, including advances in surgery, chemotherapy and biological therapy [reviewed in [1]]. Survival of CRC patients is highly dependent on the tumor stage at the time of diagnosis. Over one-third of patients die within five years of the initial diagnosis, mainly from liver metastasis [reviewed in [1]]. Colon carcinogenesis is a multistep process, in which normal colon epithelial cells (IECs) are transformed to malignant cells. Approximately 20 % of CRC cases have a familial background, however, the majority have been linked to environmental causes rather than heritable genetic changes. Environmental risk factors for CRC include food-borne mutagens, specific intestinal commensals and chronic intestinal inflammation [reviewed in [2]]. Fifteen percent of all epithelial tumors develop in organs in which chronic inflammation occurs. As in all solid tumors, the microenvironment of colon tumors consist of recruited inflammatory cells from the bone marrow (BM) and stromal cells. In most instances, the microenvironment supports invasiveness and tumor progression [(reviewed in [3–5]]. Correlation between prolonged inflammation and susceptibility to CRC is documented in patients with chronic inflammatory bowel disease (IBD), such as ulcerative colitis (UC) and Crohn’s disease (CD) [reviewed in [3–11]]. Patients suffering from UC for more than 35 years show an increased risk for CRC development from 18 to 35 %, while 8 % of CD patients are at risk of carcinogenesis [reviewed in [12, 13]]. The CRC subtype that is associated with IBD is difficult to treat and has a high mortality rate.
The Microenvironment of the Colon
Intestinal mononuclear phagocytes, which normally reside in the intestinal lamina propria (LP), such as macrophages and DCs, are important in preventing harmful responses to food constituents and commensal bacteria [14]. However, in the presence of pathogenic bacteria, the immune system can elicit robust inflammatory and protective host-defensive responses in the intestine [15–17]. A central question is how the immune system discriminates between commensal and pathogenic bacteria. This issue is particularly important in the intestine, where a large number of commensal microorganisms challenge the immune system without eliciting pro-inflammatory responses [18]. The signals that program mononuclear phagocytes to become anergic to Toll like receptor (TLR) ligands of commensals and thus maintain intestinal homeostasis have not yet been elucidated. Induction of anti-inflammatory cytokines following the engulfment and degradation of commensal bacteria in intestinal macrophages are among the possible mechanisms conferring tolerance. For example, mononuclear phagocytes of the LP do not produce TNFα and IL-6 when stimulated with TLR ligands or commensal bacteria [19, 20]. Another possible mechanism for tolerance is the low expression of TLRs or key components of patterns recognition receptors (PRRs) signaling in intestinal mononuclear cells [21]. Production of IL-10 by mononuclear cells and TGFβ by stromal cells were also shown to be involved in suppression of colon immunity to commensals [20, 22]. IL-10 is secreted by Treg cells, which are abundant in the intestine and play a significant role in colon homeostasis. Treg cells also suppress the induction and function of effector T cells, especially pro-inflammatory Th17, and inhibit the activation/function of mononuclear cells. Given that resident macrophages and dendritic cells are anergic to TLR ligands and commensal bacteria, it is unclear if they can actively promote host defenses against pathogenic bacteria. Alternatively, infiltrating myeloid cells, such as neutrophils, myeloid derived suppressor cells (MDSCs), inflammatory monocytes and macrophages differentiated from them could be responsible for eliminating pathogenic bacteria.
Major pathological features of colon chronic inflammation, such as IBD, include intestinal epithelial cells (IEC) injury and disruption of the mucosa barrier that lead to invasion of bacteria into the LP and subsequent colon inflammation. Carcinogenesis in the colon is promoted by aberrant interactions of the microflora with IECs and inflammatory cells through PRRs, which include cell surface TLRs or receptors recognizing bacterial or tissue damage ligands in the cytosol (Nod-like receptors, NLR). Diverse innate and adaptive immune cells and cytokines have been shown to be involved in chronic colon inflammation that may lead to CRC development. These include bone marrow (BM)-derived infiltrating myeloid cells, colon resident and stromal cells, such as IECs and fibroblasts (especially myofibroblasts), as well as T cell subsets, mainly Th17 and to some extent Th1 cells. However, the relative importance of specific cytokines in the overall cytokine network of IBD and CRC has not yet been elucidated [reviewed in [15, 23–32]].
In this review, we will discuss the multifaceted role of upstream, pro-inflammatory cytokine molecules from the IL-1 family in CRC development, based on our results showing that these molecules can be expressed in the tumor microenvironment by cancer, immune and tissue resident cells.
Characteristics of IL-1α and IL-1β- the Major IL-1 Agonistic Molecules
The IL-1 family consists of 11 agonist and antagonist molecules that are centrally involved in regulating inflammatory responses. These include IL-1α, IL-1β, IL-1Ra, IL-18, IL-33, IL-1Ra, IL-36α, IL-36β, IL36γ and IL-38 [reviewed in [33–39]]. Here, we will mainly focus on the two major IL-1 agonistic molecules i.e., IL-1α and IL-1β, and IL-1 receptor antagonist (IL-Ra), which is a physiological inhibitor of IL-1 signaling. IL-1 differs from most other cytokines by lack of a signal sequence, thus not passing through the endoplasmic reticulum-Golgi pathway. IL-1Ra, which has a signal peptide, is secreted in the ER-Golgi exocytic pathway. Generally, IL-1 is produced and secreted by various cell types upon inflammatory or stress conditions, predominantly by myeloid cells, which display the strongest capacity to produce and secrete IL-1. Stimulation of IL-1 production occurs through signaling of TLRs, which recognize conserved microbial molecules of pathogens, [reviewed in [40–42]] as well as endogenous molecules, which are products of damaged cells [reviewed in [43–45]]. The activation of TLR signaling via the NF-κB pathway leads to the generation of many cytokines; IL-1 is a central cytokine produced by this pathway. Signaling through surface IL-1Rs and most of the TLRs is common and converges from MyD88 to NF-κB activation and induction of an inflammatory response, including expression of IL-1.
Processing of the IL-1 Agonistic Molecules
IL-1α and IL-1β are synthesized as precursors of 31 kD that are further processed by proteases to their mature secreted 17 kD forms. The IL-1β-converting enzyme (ICE), or caspase-1, is a cysteine protease, which is activated in the cytosol on the inflammasome platform, and subsequently cleaves the inactive precursors of IL-1β and IL-18 into their mature secreted forms [reviewed in [34, 35, 46–52]. Thus, caspase-1 is synthesized as an inactive zymogen and becomes active after controlled dimerization and activation of the inflammasome. Here, we restrict our discussion mostly to functional aspects of the inflammasome [reviewed in [6, 8, 10, 11, 53]]. Inflammasomes are large cytosolic complex proteins that sense cytosolic perturbations [52, 54–56]. Inflammasome formation is triggered by diverse microorganisms and their products or by stress-associated signals that support the autocatalytic cleavage of pro-caspase-1, which activates acute inflammation by cleavage of pro-IL-1β and pro-IL-18, and secretion of the mature forms of these pro-inflammatory cytokines. Caspase-1 also induces an inflammatory form of cell death called pyroptosis, which limits the multiplication of intracellular pathogens, such as Salmonella, in the macrophage [57]. IL-1β and IL-18 secretion is also induced during pyroptosis, which leads to recruitment of immune cells to inactivate the released pathogens [58]. Inflammasomes are assembled around a set of core components that include a sensor protein (a member of the NLR and PYRIN families), an adaptor protein containing the apoptosis-associated speck-like protein, ASC that also contains a caspase activation and recruitment domain (CARD), and an inflammatory caspase (mainly Caspase-1). The most studied inflammasomes until now are those containing NLR sensors, such as NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRC4 and potentially NLRP12 [52]. Each sensor protein has its own ligands and at present such ligands are being extensively characterized. For inflammasome assembly and activation, all components must exist in the same cell. Caspase-1 and ASC are ubiquitously expressed in cells and tissues, while inflammasome sensors exhibit a restricted expression, suggesting that cell-type specific mechanisms sense tissue perturbations. It has recently become evident that inflammasomes in different cell types may induce different, but often complimentary responses.
For the secretion of IL-1β and IL-18, two signals are essential. The first is supplied by products of microorganisms or tissue damage that signal through activated NF-κB to induce transcription of ProIL-1β or ProIL-18. The second signal activates the inflammasome and leads to secretion of the mature cytokines. The second signal can be supplied by various stimuli. For example, extracellular ATP released from dying cells, signals through the P2X7 adrenergic receptor to form pores in the membrane, which stimulate potassium efflux from the cell. This efflux leads to emplacement of large-pored pannexin-1 channels into the membrane, through which products of microorganisms and tissue damage can access the cytosol and directly activate NLRP3 or other NLRs. Larger molecules, such as crystalline or particulate ligands, enter the cell by phagocytosis and following rupture of the phagolysosome, can activate the inflammasome directly or through ROS, which are generated in the process of lysosome damage. The process of secretion of the inflammasome-dependent cytokines is not yet clear; secretion occurs immediately following processing, without the accumulation of these cytokines in the cell. The role of the inflammasome in cancer development and progression has been studied [reviewed in [46]].
The precursor of IL-1α (ProIL-1α) is processed by the Ca2 +-dependent protease calpain into the mature 17 kD form and the 16 kD N-terminal cleavage product- the propiece of IL-1α, also termed IL-1α N-terminal peptide (IL-1NTP). The latent form of calpain is activated in cells under inflammatory conditions and especially upon loss of plasma membrane integrity, which occurs during necrosis [59]. However, intracellular ProIL-1α, which is biologically active, is present in many cells because they contain calpain inhibitors and are thus unable to process and secrete IL-1α. Recently, some involvement of the inflammasome in IL-1α secretion has also been demonstrated [60–62]. A biologically active membrane-associated form of IL-1α (23 kD), which is anchored to the membrane via a mannose-like receptor, has been found in activated cells that express the cytokine.
IL-1 Receptors
Exogenous IL-1α and IL-1β signal through the same IL-1Rs that belong to the immunoglobulin (Ig) supergene family, and are abundantly expressed on many cell types, suggesting that both IL-1 agonists are synonymous in their function. IL-1R of type I (IL-1R1) (80 kD) is a signaling receptor, whereas the IL-1R of type II (IL-1R2) (68 kD) serves as a decoy target, acting to reduce excessive amounts of IL-1 [reviewed in [33, 34, 36–39, 63]]. Following the binding of IL-1 to IL-1R1, the IL-1R acceptor protein (IL-1RAcP) is recruited. This heterodimeric complex triggers IL-1 signaling by activating the IL-1 receptor-associated kinase (IRAK) and ultimately leads to activation of NF-κB and its target genes. IL-1R2 and the IL-1Ra do not form this heterodimeric complex with the IL-1RAcP and therefore do not recruit IRAK. Signaling through surface IL-1R1 represents an evolutionary conserved mechanism homologous to the TLR pathway.
Differences Between IL-1α and IL-1β
IL-1β is not present under homeostatic conditions; it is induced and secreted only upon inflammatory signals and its secretion is tightly controlled at the levels of transcription, mRNA stability, translation, post-translational modifications and processing. On the other hand, IL-1α is present in the cytosol, nucleus or on the cell membrane in homeostatic steady-state conditions, as well as during inflammation, when its expression is upregulated. IL-1α is mainly active in its ProIL-1α form, which is expressed in lumen-lining epithelial cells, as well as in other tissue cells. Furthermore, IL-1α is only rarely secreted by living cells and in most cases is undetectable in body fluids. Previously, we demonstrated that in vivo, in steady-state homeostasis and in inflammation, IL-1α and IL-1β are differentially expressed in tissues, possibly pointing to their different physiological roles [64, 65]. IL-1α and IL-1β differ in the sub-cellular compartments in which they are active. IL-1β is solely active as an extracellular secreted product, while its precursor is inactive and there is no membrane-associated form of IL-1β. On the other hand, IL-1α is mainly present in its cell-associated forms (ProIL-1α, IL-1NTP and membrane-associated forms), but is only marginally secreted in its mature form, with the exception of activated myeloid cells [reviewed in [33–39]]. Very little is known about the biological activity of IL-1NTP. ProIL-1α was shown to translocate to the nucleus, due to a nuclear localization sequence (NLS) located within the structure of ProIL-1α and IL-1NTP, but lacking in the mature form of IL-1α. In cells that express ProIL-1α, but do not secrete it, the cytokine possibly acts in an intracrine manner from within the cell, via signaling pathways that are not yet fully characterized. We have hypothesized that intracellular forms of IL-1α evolved as intracellular effector molecules undertaking important homeostatic regulatory functions beyond the realm of immunity and inflammation. These include effects on gene expression, cell growth and differentiation, which have been demonstrated in tissue-resident cells, such as endothelial cells, fibroblasts, smooth muscle cells, keratinocytes, epithelial cells and brown fat cells [reviewed in [33–35]]. Thus, IL-1α belongs to a group of “dual function” cytokines (i.e., HMBG1 and IL-33) that are expressed in the cytosol and can enter the nucleus, where they perform homeostatic functions, but upon cell necrosis, they are released into the microenvironment and serve as alarmins by inducing inflammation [reviewed in [66, 67]].
Inflammatory Activities of IL-1
IL-1 together with TNFα are defined as “alarm cytokines” that are secreted by macrophages and initiate inflammatory responses, by inducing a cascade of other pro-inflammatory genes [reviewed in [33–39]]. Of major importance are cyclooxygenase type 2 (COX-2), inducible nitric oxide synthase (iNOS), chemokines/cytokines and matrix metalloproteinases (MMPs). The IL-1 molecules stimulate their own and each other’s production; this represents an important amplification loop of the inflammatory response. Also, IL-1 increases the expression of integrins on endothelial cells, stromal cells and leukocytes and thereby promotes cell infiltration into inflamed tissues. IL-1β induces inflammation in a classical manner, being secreted from myeloid cells immediately after processing of its precursor. Due to the limited secretion of IL-1α, its role in inflammation is not yet clear.
Recently, the unique alarmin function of ProIL-1α in sterile inflammation has been described by us and others [68–73]. In tissue cells, such as epithelial cells, endothelial cells and fibroblasts, ProIL-1α is located in the cytosol and nucleus. Upon stress, expression of ProIL-1α increases and it translocates into the nucleus, where it is bound to chromatin in a highly dynamic manner. Upon necrotic cell death, ProIL-1α is released from the chromatin and induces inflammation [74]. However, following apoptotic cell death, the mobility of chromatin-bound IL-1α is greatly reduced; it concentrates in dense nuclear foci and is not released into the environment [69]. This represents a novel mechanism that explains why inflammatory responses are not generated upon apoptosis. Also, this mechanism indicates that IL-1α released from necrotic tissue provides an alarm signal that initiates inflammation, which is manifested by recruitment of neutrophils, while subsequently, infiltrating cells, especially macrophages, secrete other pro-inflammatory cytokines, such as IL-1β that propagate and/or terminate the inflammatory response [69, 71]. Recently, a novel mechanism to control IL-1α activity in necrotic cells has been described by Zheng, et al. 2013, [75] and reviewed in [76]. Thus, under normal conditions, IL-1α is synthesized as a p33 precursor that is sequestered in the cytosol by IL-1R2, where it cannot be cleaved by proteases or activate IL-1R1 signaling. However, after inflammasome activation, IL-1R2 can be cleaved by caspase-1 and ProIL-1α can be released and further processed by calpain to the highly active p17 mature form of IL-1α. It is still debatable whether ProIL-1α and mature IL-1α are active to the same extent [77]. Moreover, necrosis-induced IL-1α activity is tightly controlled in a cell type-specific manner [75]. Thus, in cell types with a silent necrotic phenotype, IL-1R2 remains associated with ProIL-1α. In contrast, in cells with an inflammatory, necrotic phenotype, IL-1R2 is either absent or caspase-1 is activated before necrosis. Overall, the extent of inflammation in damaged tissues depends on the concentration of cleaved IL-1α, as well as the local expression of IL-1R1. This control mechanism evolved in order to prevent exacerbation of inflammation induced by necrotic cells in tissues with limited regenerative capacity, such as kidney, heart and brain [64, 75]. These findings suggest that sterile inflammation can occur even without activation of IL-1β. Other studies have also demonstrated inflammasome-dependent IL-1α release in sterile inflammation, which may further lead to ProIL-1β caspase-1-dependent processing and release [reviewed in [34, 76]].
Differential Activities of IL-1α and IL-1β in the Malignant Process
Here, we will summarize in brief our comparative studies on the role of IL-1α and IL-1β in various phases of the malignant process. We have hypothesized that the cell type that express IL-1 or the localization of the IL-1 molecules in the context of the producing cell and its microenvironment dictate the biological function of the IL-1 molecule in normal homeostasis and also in the malignant process [reviewed in [33, 78–80]]. Our results demonstrate that in many cases, IL-1α and IL-1β perform distinct functions in malignancy.
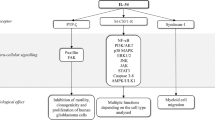
In the tumor arena, IL-1 is an abundant cytokine that can be secreted by malignant or microenvironment cells and affect local or even systemic inflammation and anti-tumor immunity. It is involved in all phases of the malignant process, such as tumorigenesis, tumor invasiveness and progression, as well as activation/suppression of anti-tumor immunity. In the malignant process, the target cells of IL-1 can include pre-malignant or malignant cells, as well as cells of the microenvironment that are activated by exogenous IL-1, usually to produce inflammatory mediators that promote invasiveness. In tumorigenesis, IL-1 of microenvironment origin can propagate the initial mutations by ROS or NO, produced by phagocytes, other microenvironment cells or mutated epithelial cells. IL-1 can then rescue initiated cells from apoptosis, enable their proliferation and further accumulation of mutations, ultimately leading to a malignant phenotype. IL-1 can then potentiate the invasiveness of malignant cells through stimulation of growth factors, angiogenesis and tumor cell motility, ultimately leading to metastasis. In some cases, IL-1 can also enhance the immunogenicity of malignant cells and consequently reduce tumor invasiveness. As IL-1 is an upstream cytokine, its effects on the malignant process may be direct or indirect, being mediated by cytokines/mediators that it induces. Thus, at tumor sites, IL-1 induces a local cytokine network that is determined by the array of expressed cytokines, their relative concentrations and the expression pattern of their receptors. This cytokine network dictates the dominant “net cytokine effect”, which is dependent on the local repertoire of cytokines and their receptors in malignant and tumor microenvironment cells and fluctuates at various phases of tumor development (Scheme 1).

IL-1 molecules in colon homeostasis, inflammation, carcinogenesis and CRC invasiveness. IL-1β can be produced and secreted during colon inflammation by infiltrating myeloid cells, as well as by IECs and other stromal cells. ProIL-1α is expressed in steady-state conditions in IECs. Upon tissue damage, ProIL-1α is released from necrotizing IECs and acts as an alarmin to induce inflammation. In addition, IL-1α can also be secreted by recruited infiltrating cells. In CRC, these patterns of IL-1 expression are also valid. However, malignant cells can secrete both Il-1α and/or IL-1β upon transformation or during tumor progression. The IL-1 molecules can act on IECs or transformed colon cells, as well as propagate local inflammation by activating microenvironment cells, which in the case of CRC increases tumor progression
Effects of IL-1 on Tumorigenesis
Tumorigenesis encompasses the in vivo induction of tumors cells by carcinogens or oncogenes, as well as in vitro transformation of normal or immortalized non-tumor forming cells into overt malignant cells that are capable of tumor formation in mice. We have demonstrated the role of host-derived IL-1 molecules on susceptibility to chemical carcinogenesis induced by 3-methylcholantrene (3-MCA), which acts both as an initiator and a tumor promoter, using a battery of IL-1 KO mice in comparison to wild-type (WT) mice [81]. It was shown that strong inflammation mediated by IL-1β is essential for tumor development, which was impaired in IL-1β KO mice and accentuated in IL-1Ra KO mice, which express non-attenuated IL-1 levels. In contrast, IL-1α was less important in this inflammatory response, as IL-1α KO mice displayed tumorigenicity patterns similar to WT mice. Our results described, for the first time, that 3-MCA-induced carcinogenesis is inflammation-dependent, while previously it had been suggested that, in this system, tumor development is controlled by immune surveillance mechanisms that eliminate the arising malignant cells [reviewed in [82]]. We have hypothesized that both inflammation and immunosurveillance operate concomitantly and the balance between them determines the outcome of the malignant process. The role of immunosurveillance in 3-MCA- carcinogenesis was observed in IL-1α KO mice. We found that transplantable 3-MCA induced fibrosarcoma cell lines obtained from IL-1α KO mice failed to generate tumors in immune intact mice, whereas tumor development was evident in immunocompetent sublethally irradiated mice [83]. Thus, we demonstrated an impaired immunoedditing in mice deficient in IL-1α. We assume that the membrane-associated form of IL-1α mediates its immunostimulatory effects. IL-1α expression on the membrane possibly acts as a focused adjuvant that ligates with IL-1R1 that is abundant on immune cells, at low levels of expression below those that are toxic to the host. Previously, impaired immunoediting has been demonstrated in various immunodeficient mice that lack immunosurveillance cells, especially innate cells or critical cytokines for the development of anti-tumor cell immunity [reviewed in [84]]. In the absence of these components, immunogenic tumor cells are not destroyed during the process of tumorigenesis and they prevail in the arising tumors.
Other studies have also described in detail the role of the IL-1 molecules in promoting carcinogenesis, as previously reviewed by us [85–88] and have emphasized mainly its alarmin role. Indeed, IL-1 released from local epithelial cells further acts to induce secretion of growth factors that induce cell proliferation that promotes tumor development.
Effect of the IL-1 Molecules on Tumor Invasiveness
Invasiveness of tumors is largely affected by patterns of IL-1 expression by the malignant cells, the host or both. In experimental tumor models in WT mice and in cancer patients, increased local levels of IL-1 at tumor sites usually correlate with tumor invasiveness and a bad prognosis [reviewed in [33, 78–80]]. Very little is known about interactions between IL-1α and IL-1β expressed at tumor sites either by malignant or microenvironment cells. Most studies usually assess only one of the IL-1 agonistic molecules and do not discriminate between its expression in the malignant cells versus the microenvironment. Our studies have included fibrosarcoma cells overexpressing IL-1α or IL-1β, as well as cell lines that are deficient in these cytokines that were transplanted into WT or specific IL-1 KO mice. Overall, IL-1β expressed by the malignant or the host cells promotes tumor invasiveness and immunosuppression, mediated by myeloid-derived suppressor cells (MDSCs) [89, 90]. On the other hand, overexpression of malignant cell ProIL-1α, which is located in the cytosol or membrane-associated, and not secreted, is usually immunostimulatory and induces an anti-tumor immune response, mainly by recruitment of CD8+ T cells, leading to tumor regression [90–92]. Other studies have also emphasized the effectiveness of membrane-associated cytokines expressed on engineered tumor cells (i.e., IFNγ, GM-CSF, M-CSF, TNFα and IL-12) [93–95]. When both IL-1 agonistic molecules are expressed and sufficient amounts of IL-1β are secreted, they induce a MDSC-mediated suppressive response that either prevents development of anti-tumor immunity or obscures it, thus resulting in progressive tumor growth. In the absence of IL-1β, local IL-1α, possibly due to its compartmentalization and low levels of expression, promotes anti-tumor cell immunity. If both IL-1 molecules exist, usually the pro-tumorigenic effects of IL-1β are dominant [81, 83, 96–98]. Our studies have also demonstrated the necessity of IL-1β for tumor-mediated angiogenesis, which is essential for tumor invasiveness [98, 99].
IL-1 as a Biomarker of IBD and CRC Development
The expression of IL-1 by both myeloid and epithelial cells of the mucosa during IBD was reported and its levels correlated with the severity of inflammation in experimental models of disease [100] and in patients [101–108]. In contrast, expression of IL-1Ra is significantly decreased in patients with IBD [101]. In accordance, expression of inhibitory sIL-1RII was associated with amelioration of CD; sIL-1RI was suggested as a biomarker for disease activity in CD patients [109]. In addition, neutralization of IL-1 in experimental colitis, by IL-1Ra or caspase-1 inhibitors, alleviated colitis [110–114]. Thus, due to IL-1 inflammation-promoting features, its involvement in the pathogenesis of IBD can be via its effects on infiltration and activation of myeloid cells. IL-1 was also shown to be necessary for maintenance of mucosal homeostasis by handling microbiota and by protecting the integrity of the epithelial barrier through effects on tight junctions [115, 116]. It was found that excessive secretion of pro-inflammatory cytokines, for example, TNFα and IL-1 in the tumor microenvironment, accelerates production of DNA-damage molecules, such as ROS and nitric oxide that induce mutations in the colon epithelium and thus promote cancer development [reviewed in [12, 117]]. Dysregulated secretion of these pro-inflammatory cytokines, like IL-1, can potentiate Wnt/β-catenin signaling in IECs, which is a major regulator of IEC proliferation in normal and transformed colon cells [117]. Katsurano et.al. found a number of measurable epigenetic alterations detected very early in the colon tissue before the appearance of visible tumors [118]. In a mouse colitis model induced by dextran sulfate sodium (DSS), they identified three CpG islands that are specifically methylated during inflammation in colonic epithelial cells. Methylation levels started to increase as early as 8 weeks after DSS treatment and continued to increase until colon tumors appeared at 15 weeks. In contrast to the temporal profile of DNA methylation levels, infiltration of inflammatory cells spiked immediately after DSS treatment and then gradually decreased. Comparative analysis of inflammation-related genes showed that IFNγ, IL-1β and iNOS2 are expressed concordant with methylation induction, whereas IL-2, IL-6, IL-10 and TNFα did not. These results show that an epigenetic field defect is formed at early stages of colitis-associated carcinogenesis and it affects genes related to innate inflammation, including IL-1β.
Polymorphism of the IL-1β gene, accompanied with elevated levels of IL-1β has been associated with an increased risk of colon cancer development [119]. In contrast, single-nucleotide polymorphisms (SNPs) associated with abundant expression of IL-1Ra leads to a greater survival of patients with advanced CRC [120]. A mutation in NOD2, which correlates with severe forms of CD, resulted in increased production and secretion of IL-1β, indicating its role in the pathogenesis of severe IBD and possibly its progression to CRC [121].
These studies have shown an association between colon IL-1β levels, severity of IBD and increased development and invasiveness of CRC. However, in these studies, no parallel expression of IL-1α was examined. In normal cells, IL-1α is mainly expressed in cell-associated forms. However, in malignant cells, IL-1α might be secreted by epithelial cells, as part of the aberrations of the malignant process, and this has been associated with increased tumor invasiveness [reviewed in [33]]. However, the comparative role of IL-1α and IL-1β in chronic colon inflammation and carcinogenesis, as well as in invasiveness of CRC is not yet known.In recent genome-wide association studies (GWAS) and SNP analyzes, some of the components of the inflammasome or its processed cytokines or TLR/IL-1R signaling, such as NLRP3, CARD8, NOD2 and IL-18 and MyD38 were strongly associated with increased susceptibility for IBD development [6, 8, 10, 11]. This has stimulated intensive studies on the role of the inflammasome and its derived cytokines in colon inflammation and its progression to CRC [122–125].
IL-1 in Colon Inflammation and Carcinogenesis
Following the characterization of extensive association between inflammasome activation and susceptibility to IBD, as described above [6, 8, 10, 11], mechanistic studies were performed on the role of inflammasomes on colon inflammation and carcinogenesis [122–125]. KO mice lacking specific components of the inflammasome and its secreted cytokines were treated with Dextran Sodium Sulfate (DSS) to induce acute or chronic colon inflammation. Colon tumorigenesis was initiated with the carcinogen azoxymethane (AOM) and promoted with chronic DSS treatment, as described [126]. Specifically, in KO mice deficient in components of the inflammasome, such as caspase-1 [127, 128], NLRP3 [127, 129, 130], NLRC4 [128], NLRP6 [131], ASC [132], or in IL-18 and IL-18R KO mice [133], acute colitis and the development of colon adenomas following AOM/DSS treatment are exacerbated, as compared to WT mice. This was intuitively in contrast with the notion that progression of colon inflammation to carcinogenesis depends on the inflammatory potential of BM-derived infiltrating myeloid cells, as well as IECs and their secreted cytokines/pro-inflammatory products. If major pro-inflammatory cytokines, such as IL-1β or IL-18 are deleted, disease severity should be attenuated in the above-mentioned KO mice. However, it became apparent that inflammasome-processed pro-inflammatory cytokines are expressed in IECs, especially IL-18, and are crucial for IEC homeostasis and repair of the mucosa. Healing of the epithelial cell compartment, which is highly protective against bacterial invasion, is also essential to prevent CRC development; aberrant healing of IECs may promote colon carcinogenesis. These functions of IL-18 include proliferation/differentiation of IECs, expression of tight junctions that enable the closure of the epithelial barrier of the colon, secretion of anti-bacterial peptides and production of protective mucous by goblet cells. Indeed, it was recently shown that the NLRP6 inflammasome, which is mainly expressed in IECs, governs mucus secretion in goblet cells; NLRP6 KO mice display a deficiency to clear enteric pathogens due to impaired mucus secretion [134]. In these studies, the IEC healing capacity was attributed to IL-18 rather than IL-1β, as in IL-1R KO mice, colitis and colon carcinogenesis patterns are similar to those observed in WT mice [133]. In some of these KO mice, such as caspase-1, IL-18, IL-18R and MyD88 KO mice, the severity of colon inflammation and carcinogenesis could be alleviated by injection of recombinant IL-18, but not IL-1β [129, 132, 133]. Therefore, IL-18 expression in mucosa of CD patients was examined [135–137]. A causal correlation between serum and tissue levels of IL-18 and colon epithelial repair was shown in colitis induced in caspase-1 KO mice compared to WT mice. In the former, impaired IEC proliferation, low defensin secretion and low levels of serum and colon tissue IL-18 were observed, as compared to high levels in WT mice [130, 132]. Surprisingly, there was no difference in IL-1β levels between caspase-1 KO mice and WT mice. Several mechanisms were suggested for IL-18 involvement in healing of the IEC compartment, acting by itself or through the IL-22 pathway. IL-22 is a member of the IL-10 family, which plays an important role in maintaining the colonic epithelial barrier and it is also a key regulator of epithelial cell homeostasis in other organs, such as the skin, liver and lungs. It was shown that IL-18 produced by IECs in the inflamed colon increases IL-22 signaling by a negative feedback loop, downregulating expression and secretion of the IL-22-binding protein by macrophages [138]. An additional positive loop between IL-18 and IL-22 was also suggested; IL-18 enhances transcription of IL-22 in lymphocytes that subsequently promote IL-18 production by IECs [138, 139]. In addition, it was recently shown that IL-22 secreted from Th22 cells in the microenvironment of colorectal cancer, promotes the stemness of malignant cells in an experimental model and in patients and thus increases invasiveness and predicts a poor prognosis. This occurs through activation of the transcription factor STAT3 and induction of DOT1L methyltransferase, resulting in expression of core stem cell genes, such as NANOG, SOX2 and Pou5F1, and an increased concomitant tumorigenic potential [140, 141].
The role of IL-1β as a major pleiotropic pro-inflammatory cytokine in colon inflammation and carcinogenesis has not been shown in the above-mentioned studies. In contrast to the observations described above on induction of DSS-induced colitis and colon carcinogenesis in NLRP3 KO mice [127, 129, 130, 135, 136], some studies have shown that NLRP3 KO mice are significantly protected from colitis, as compared to WT mice [130, 138, 142]. The protective effect of NLRP3 deficiency was most pronounced at early time points of the disease, indicating an important role of inflammasome signaling during the initiation of the inflammatory response. Furthermore, it was shown that in vitro, macrophages stimulated with DSS secrete IL-1β that is dependent on NLRP3 activation and upstream events, such as phagocytosis, lysosomal maturation, cathepsins B and L, as well as ROS. In macrophages deficient in NLRP3, ASC or caspase-1, IL-1β secretion was abrogated. The authors suggested that this reflects what occurs in vivo during acute colitis, where macrophage-derived IL-1β mediates the inflammatory response. The protective effect of NLRP3 in mice associated with an increased frequency of CD103+ LP dendritic cells, expressing a tolerogenic phenotype under steady-state conditions, which render these mice more resistant to colitis development [142].
In subsequent studies on responses to microorganisms or tissue damage products, in which activation of the inflammasome is involved, the role of IL-1β was demonstrated. Thus, in a model of infectious colitis induced by Citrobacter rodentium in IL-1R or MyD88 KO mice, an exacerbated form of disease was observed in the KO mice, indicating that IL-1R signaling is essential for colon repair and resolution of colitis [143].
To directly assess the role of the IL-1 molecules in colon inflammation, we induced colitis in IL-1 KO mice, lacking either IL-1α or IL-1β, and examined their patterns of acute DSS-induced colitis [144]. To our surprise, we found distinct, non-redundant functions of both IL-1 major agonistic molecules in acute colon inflammation. In mice deficient in IL-1α, a very mild and rapidly self-healing inflammatory response was observed, as compared to WT mice. Similar results were found when IL-1α was conditionally deleted only in IECs. These findings indicate that in DSS-induced colitis, following the release of IL-1α from necrotizing IECs, it acts as an alarmin and initiates colon inflammation. Thus, for inducing colon inflammation, IL-1α of IEC origin is sufficient. Indeed, BM transplantation experiments have indicated that both IL-1α and IL-1β from later appearing BM infiltrating cells possibly amplify and sustain the inflammatory response. Others have also described the alarmin function of IEC-derived IL-1α in activation of colon myofibroblasts [145, 146]. In contrast, IL-1β KO mice were much more susceptible to DSS-induced colitis, as seen by lower survival rates and manifestation of severe and non-healing tissue damage, with only a marginal inflammatory response, as compared to WT mice. This may indicate that in colitis, IL-1β may play a dominant role in colon repair; in its absence, tissue-damage does not resolve, as previously indicated by Lebeis et al., showing the role of IL-1R in IEC healing [143]. Furthermore, in mice deficient in SIGRR (also named TIR8), which is a negative regulator of TLR4 and IL-1R signaling, exacerbated colitis and development of CRC occurs. Specific restoration of gut epithelium by SIGRR, by its specific knock-in into IECs of SIGGR KO mice, restored the homeostasis of IECs, possibly by limitation of their response to the microbiota or local IL-1 [147]. T cell infiltration into damaged crypts, especially Treg and CD8+ T cells, correlates with repair that is apparent in IL-1α, but not IL-1β KO mice after termination of acute colitis. Treatment with anti-IL-1α antibodies, rather than anti-IL-1β or the IL-1Ra, ameliorates colitis. The contrasting role of the major agonistic IL-1 molecules in colitis may explain the fact that in IL-1R KO mice patterns of colitis are similar to WT mice [133]. Further understanding the differential role of IL-1α and IL-1β in colitis may lead to understanding their involvement in the mechanisms of CRC development/progression and may help to devise novel therapeutical targets.
In recent years, the physiological significance of the microbiota in inflammasome activation and inflammasome-dependent cytokines, i.e., IL-1β and IL-18, in intestinal homeostasis, as well as in disease (IBD and CRC) has been highlighted. Recent studies demonstrated that during colitis in inflammasome deficient mice, there is overgrowth of commensal bacteria and antibiotic treatment ameliorated the exacerbated disease in these mice [129]. Pioneering studies by Elinav et al. demonstrated that the exacerbated colitis phenotype in NLRP6 KO mice is accompanied by changes in phyla of certain commensal bacteria in the colon, with prevalence of Bacteroides (prevotellacae) and TMV7. Furthermore, susceptibility to colitis could be transferred from NLRP6 KO mice to WT mice by co-housing [131], clearly indicating the crucial role of the microbiota in the pathogenesis of colitis and CRC development. The results also indicate that susceptibility to colitis and colon carcinogenesis does not involve only an endogenous defect in inflammasomes in IECs and impaired secretion of cytokines, but it also involved in interactions between the modified microbiota and with diverse cells in the colon. Furthermore, dysbiosis, as detected by 16S RNA sequencing, was observed by the Flavell group in NLRP3, ASC, Caspase-1 and IL-18 KO mice with an exacerbated form of colitis, as described above. In these strains of mice, co-housing with WT mice altered the colitis phenotype of WT mice to a severe one, due to acquisition of a dysbiotic microbiota [131]. Antibiotic treatment reversed the colitis phenotype of inflammasome-deficient mice, indicating the role of a communicable dysbiotic microbiota that enhances susceptibility to colitis. Similar results were reported on the transfer of susceptibility to colitis from susceptible WT mice to resistant NLRP3 KO mice by co-housing [142]. As a unifying hypothesis, the Flavell group has suggested that interactions between the microbiota (healthy or dysbiotic), functional inflammasomes and generation of inflammasome-derived cytokines (IL-1β and IL-18) determine patterns of colon inflammation and carcinogenesis. In some mouse colonies, strains that lack inflammasomes contain dysbiotic microbiota for reasons not yet known. The activation of diverse inflammasomes in distinct cells of the colon (myeloid cells, IECs, stromal myofibroblasts) at defined time intervals can result in a plethora of cytokines and mechanisms that control colitis or colon carcinogenesis. How inflammasomes and their secreted cytokines in different cell types act in a coordinated manner in steady-state homeostasis of the intestine and in disease is largely unknown and should be clarified. This will help to understand the mechanisms of IBD and CRC, using experimental mouse models and specimens from relevant patients.
It is not yet known whether interactions of defined species of bacteria with innate intestinal cells would yield in similar or unique outcomes. The role of specific microbiota on the differentiation of lymphoid cells in the colon was recently described. Thus, the ratio between Th17 cells (IL-17+) and FoxP3+ Treg cells, which is involved in intestinal homeostasis, is largely affected by the Clostridia-related segmented filamentous bacteria (SFB) [reviewed in [148]]. SFB stimulates polarization of Th17 cells that secrete IL-17 and IL-22, which can further activate IECs to secrete anti-microbial peptides, thus resulting in increased resistance to microorganisms. However, when these cytokines are produced in excess, chronic autoimmune inflammation develops. On the other hand, Treg cells maintain homeostasis in the intestine by suppressing local immunity, through inhibition of pro-inflammatory Th17 cells. These studies represent the first example of a specific commensal species that skews the mucosal effector T cell balance and predisposes the host for altered immunity following insults in the intestine. In another study, the Nunez group demonstrated the unique involvement of IL-1β in the development of steady-state Th17 cells in the intestine [149]. IL-1β is secreted by intestinal macrophages in response to microbiota through the TLR-MyD88 pathway. Subsequently, IL-1β acts directly on IL-1R-expressing T cells and polarizes them into Th17 cells, in a pathway that was previously described [reviewed in [150]]. This does not occur in germ-free (GF) or MyD88 KO mice, as they do not produce IL-1β. Injection of recombinant IL-1β into GF mice induced the production of steady-state Th17 cells. This effect is specific to IL-1β, as other cytokines, such as IL-6, did not activate this pathway.
The Nunez group further studied the mechanisms of IL-1β expression and secretion under steady-state homeostasis and in response to pathogenic bacteria [reviewed in [151]], when phagocytes are anergic to TLR ligands or commensal bacteria. Intestinal resident phagocytes constitutively express ProIL-1β, which is immediately cleaved and secreted only upon infection with pathogenic bacteria (Salmonella or Pseudomonas) through NLRC4 inflammasomes. Under these conditions, IL-6 or TNFα, two typical cytokines of macrophages are not induced. This mechanism is local and specific to the gut, as susceptibility to Salmonella infection was observed in NLRC4 or IL-1R KO mice only upon orogastric, but not intraperitoneal infection. Thus, IL-1β production through NLCR4 inflammasomes discriminates between innate responses elicited by commensal versus pathogenic bacteria. The role of IL-1β in neutralization of the pathogenic bacteria Clostridium difficile, which induces pseudomembranous colitis, was further analyzed [152]. During infection, microbiota translocate across the damaged colon barrier followed by massive recruitment of neutrophils. IL-1β is secreted through NLRP3 activation and IL-1β further induces CXCL1 production, which represents the actual chemoattractant for neutrophils. The role of tissue damage in the intestine in induction of IL-1R-mediated inflammation by commensal bacteria was further demonstrated [153–155]. Thus, in the normal host, Proteus mirabilis is considered as a commensal bacteria (parabiont) and its toxins do not affect the host by inducing inflammation or immunity. However, in mice treated with DSS, which induces intestinal injury, the toxin hemolysin hpmA of P. micrabilis interacts with circulating LyC6highCCR2+ inflammatory monocytes, rather than with anergic colon resident macrophages, which results in activation of the NLRP3 inflammasome and robust secretion of IL-1β, in amounts equivalent to those induced by pathogenic Salmonella. This leads to a potent inflammatory response in the colon. Many species of isolated, commensal bacteria induced expression of mRNA of ProIL-1β in inflammatory macrophages, but only P.mirabilis and pathogenic Salmonella activated the secretion of mature IL-1β. As P. mirabilis is a parabiont bacterium, which becomes pathogenic under certain circumstances, it is not yet clear whether intestinal damage induced by DSS transformed it to a pathogenic bacterium. The unique patterns of IL-1β expression in response to various bacterial species or the different molecules originated from tissue damage may point to this possibility.
In summary, results show that the IL-1 molecules play a major role in host defense mechanisms against microorganisms. During infection with pathogens, tissue damage occurs and microorganisms stimulate IL-1β production and inflammation. Some of these conditions also occur during chronic colon inflammation and carcinogenesis. We observed the distinctive role of IL-1α and IL-1β in acute colon inflammation. Studies in progress are aimed at assessing the role of both IL-1α and IL-1β in chronic colon inflammation and carcinogenesis, as well as in invasiveness of CRC. These results will hopefully lead to therapy based on neutralization of specific IL-1 molecules. These include the IL-1Ra, soluble receptors, specific antibodies and inhibitors of specific inflammasomes. Many of these agents are FDA-approved and currently used safely and efficiently in the treatment of autoinflammatory disorders, as well as some autoimmune diseases, such as Rheumatoid arthritis (RA). There are agents being now tested in a variety of diseases with a pronounced inflammatory component, such as myocardial infarction (MI) and Diabetes type I.
References
Pancione M, Giordano G, Remo A, Febbraro A, Sabatino L, Manfrin E, Ceccarelli M, Colantuoni V (2014) Immune escape mechanisms in colorectal cancer pathogenesis and liver metastasis. J Immunol Res 2014:686879
Westbrook AM, Szakmary A, Schiestl RH (2010) Mechanisms of intestinal inflammation and development of associated cancers: lessons learned from mouse models. Mutat Res 705:40–59
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867
Vendramini-Costa DB, Carvalho JE (2012) Molecular link mechanisms between inflammation and cancer. Curr Pharm Des 18:3831–3852
Engel MA, Neurath MF (2010) New pathophysiological insights and modern treatment of IBD. J Gastroenterol 45:571–583
Hamilton MJ, Snapper SB, Blumberg RS (2012) Update on biologic pathways in inflammatory bowel disease and their therapeutic relevance. J Gastroenterol 47:1–8
Strober W, Fuss I, Mannon P (2007) The fundamental basis of inflammatory bowel disease. J Clin Invest 117:514–521
Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, Rossmann P, Hrncir T, Kverka M, Zakostelska Z, Klimesova K, Pribylova J, Bartova J, Sanchez D, Fundova P, Borovska D, Srutkova D, Zidek Z, Schwarzer M, Drastich P, Funda DP (2011) The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol 8:110–120
Ullman TA, Itzkowitz SH (2011) Intestinal inflammation and cancer. Gastroenterology 140:1807–1816
Yang VW, Lewis J, Wang TC, Rustgi AK (2010) Colon cancer: an update and future directions. Gastroenterology 138:2027–2028
Grivennikov SI (2013) Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol 35:229–244
Rogler G (2014) Chronic ulcerative colitis and colorectal cancer. Cancer Lett 345:235–241
Manicassamy S, Pulendran B (2011) Dendritic cell control of tolerogenic responses. Immunol Rev 241:206–227
Abraham C, Medzhitov R (2011) Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 140:1729–1737
Franchi L, Munoz-Planillo R, Nunez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332
MacDonald TT, Monteleone I, Fantini MC, Monteleone G (2011) Regulation of homeostasis and inflammation in the intestine. Gastroenterology 140:1768–1775
Lebeer S, Vanderleyden J, De Keersmaecker SC (2010) Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat Rev Microbiol 8:171–184
Lotz M, Gutle D, Walther S, Menard S, Bogdan C, Hornef MW (2006) Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med 203:973–984
Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD (2005) Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 115:66–75
Uematsu S, Akira S (2008) Toll-Like receptors (TLRs) and their ligands. Handb Exp Pharmacol 1–20
Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M (2009) Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 10:1178–1184
Bamias G, Arseneau KO, Cominelli F (2014) Cytokines and mucosal immunity. Curr Opin Gastroenterol 30:547–552
Cario E (2010) Heads up! How the intestinal epithelium safeguards mucosal barrier immunity through the inflammasome and beyond. Curr Opin Gastroenterol 26:583–590
Erreni M, Mantovani A, Allavena P (2011) Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron 4:141–154
Harrison OJ, Maloy KJ (2011) Innate immune activation in intestinal homeostasis. J Innate Immunity 3:585–593
Maloy KJ, Powrie F (2011) Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474:298–306
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118:229–241
Saleh M, Trinchieri G (2010) Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol 11:9–20
Strober W, Fuss IJ (2011) Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140:1756–1767
Terzic J, Grivennikov S, Karin E, Karin M (2010) Inflammation and colon cancer. Gastroenterology 138(2101–2114), e2105
Xavier RJ, Podolsky DK (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434
Apte RN, Voronov E (2008) Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol Rev 222:222–241
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550
Dinarello CA (2011) A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol 41:1203–1217
Gabay C, Lamacchia C, Palmer G (2010) IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 6:232–241
Garlanda C, Anders HJ, Mantovani A (2009) TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol 30:439–446
O’Neill LA (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18
Sims JE, Smith DE (2010) The IL-1 family: regulators of immunity. Nat Rev Immunol 10:89–102
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Palm NW, Medzhitov R (2009) Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227:221–233
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Carta S, Castellani P, Delfino L, Tassi S, Vene R, Rubartelli A (2009) DAMPs and inflammatory processes: the role of redox in the different outcomes. J Leukoc Biol 86:549–555
Rock KL, Kono H (2008) The inflammatory response to cell death. Annu Rev Pathol 3:99–126
Srikrishna G, Freeze HH (2009) Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11:615–628
Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W (2014) Inflammasomes in cancer: a double-edged sword. Protein Cell 5:12–20
Vanaja SK, Rathinam VA, Fitzgerald KA (2015) Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 25:308–315
Eisenbarth SC, Flavell RA (2009) Innate instruction of adaptive immunity revisited: the inflammasome. EMBO Mol Med 1:92–98
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241–247
Latz E (2010) The inflammasomes: mechanisms of activation and function. Curr Opin Immunol 22:28–33
Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832
Vermeire S, Van Assche G, Rutgeerts P (2010) Inflammatory bowel disease and colitis: new concepts from the bench and the clinic. Curr Opin Gastroenterol 27:32–37
Gagliani N, Huber S, Flavell RA (2012) The Intestine: where amazing things happen. Cell Res 22:277–279
de Zoete MR, Flavell RA (2013) Interactions Between nod-like receptors and intestinal bacteria. Front Immunol 4:462
Strowig T, Henao-Mejia J, Elinav E, Flavell R (2012) Inflammasomes in health and disease. Nature 481:278–286
Zitvogel L, Kepp O, Galluzzi L, Kroemer G (2012) Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol 13:343–351
Miao EA, Rajan JV, Aderem A (2011) Caspase-1-induced pyroptotic cell death. Immunol Rev 243:206–214
Wang KK (2000) Calpain and caspase: can you tell the difference? Trends Neurosci 23:20–26
Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, Kundig TM (2011) Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci U S A 108:18055–18060
Gross O (2012) Measuring the inflammasome. Methods Mol Biol 844:199–222
Keller M, Ruegg A, Werner S, Beer HD (2008) Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818–831
Dinarello CA (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117:3720–3732
Hacham M, Argov S, White RM, Segal S, Apte RN (2002) Different patterns of interleukin-1alpha and interleukin-1beta expression in organs of normal young and old mice. Eur Cytokine Netw 13:55–65
Hacham M, Cristal N, White RM, Segal S, Apte RN (1996) Complementary organ expression of IL-1 vs. IL-6 and CSF-1 activities in normal and LPS-injected mice. Cytokine 8:21–31
Lotze MT, Deisseroth A, Rubartelli A (2007) Damage associated molecular pattern molecules. Clin Immunol 124:1–4
Raucci A, Palumbo R, Bianchi ME (2007) HMGB1: a signal of necrosis. Autoimmunity 40:285–289
Chen GY, Nunez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10:826–837
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN (2010) Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A 107:2574–2579
Lukens JR, Gross JM, Kanneganti TD (2012) IL-1 family cytokines trigger sterile inflammatory disease. Front Immunol 3:315
Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN (2011) IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 187:4835–4843
Rider P, Carmi Y, Voronov E, Apte RN (2013) Interleukin-1alpha. Semin Immunol 25:430–438
Rock KL, Lai JJ, Kono H (2011) Innate and adaptive immune responses to cell death. Immunol Rev 243:191–205
Rider P, Kaplanov I, Romzova M, Bernardis L, Braiman A, Voronov E, Apte RN (2012) The transcription of the alarmin cytokine interleukin-1 alpha is controlled by hypoxia inducible factors 1 and 2 alpha in hypoxic cells. Front Immunol 3:290
Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC (2013) Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 38:285–295
Di Paolo NC, Shayakhmetov DM (2013) Interleukin-1 receptor 2 keeps the lid on interleukin-1alpha. Immunity 38:203–205
Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, Azam T, Kim S, Dinarello CA (2013) The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front Immunol 4:391
Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E (2006) The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev 25:387–408
Apte RN, Krelin Y, Song X, Dotan S, Recih E, Elkabets M, Carmi Y, Dvorkin T, White RM, Gayvoronsky L, Segal S, Voronov E (2006) Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur J Cancer 42:751–759
Voronov E, Dotan S, Krelin Y, Song X, Elkabets M, Carmi Y, Rider P, Idan C, Romzova M, Kaplanov I, Apte RN (2013) Unique versus redundant functions of IL-1alpha and IL-1beta in the tumor microenvironment. Front Immunol 4:177
Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA, Apte RN (2007) Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res 67:1062–1071
Smyth MJ, Dunn GP, Schreiber RD (2006) Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol 90:1–50
Elkabets M, Krelin Y, Dotan S, Cerwenka A, Porgador A, Lichtenstein RG, White MR, Zoller M, Iwakura Y, Dinarello CA, Voronov E, Apte RN (2009) Host-derived interleukin-1alpha is important in determining the immunogenicity of 3-methylcholantrene tumor cells. J Immunol 182:4874–4881
Chow MT, Moller A, Smyth MJ (2012) Inflammation and immune surveillance in cancer. Semin Cancer Biol 22:23–32
Arwert EN, Lal R, Quist S, Rosewell I, van Rooijen N, Watt FM (2010) Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. Proc Natl Acad Sci U S A 107:19903–19908
Cataisson C, Salcedo R, Hakim S, Moffitt BA, Wright L, Yi M, Stephens R, Dai RM, Lyakh L, Schenten D, Yuspa HS, Trinchieri G (2012) IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J Exp Med 209:1689–1702
Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, Liu J, Lemischka IR, Hung MC, Chiao PJ (2012) KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 21:105–120
Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC (2008) Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14:408–419
Elkabets M, Ribeiro VSG, Dinarello CA, Ostrand-Rosenberg S, Di Santo J, Apte RN, Vosshenrich CAJ (2010) IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol 40
Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN (2005) CD11b+/Gr-1+ Immature Myeloid Cells Mediate Suppression of T Cells in Mice Bearing Tumors of IL-1{beta}-Secreting Cells. J Immunol 175:8200–8208
Dvorkin T, Song X, Argov S, White RM, Zoller M, Segal S, Dinarello CA, Voronov E, Apte RN (2006) Immune phenomena involved in the in vivo regression of fibrosarcoma cells expressing cell-associated IL-1{alpha}. J Leukoc Biol 80:96–106
Song X, Voronov E, Dvorkin T, Fima E, Cagnano E, Benharroch D, Shendler Y, Bjorkdahl O, Segal S, Dinarello CA, Apte RN (2003) Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J Immunol 171:6448–6456
el-Shami KM, Tzehoval E, Vadai E, Feldman M, Eisenbach L (1999) Induction of antitumor immunity with modified autologous cells expressing membrane-bound murine cytokines. J Interf Cytokine Res: Off J Int Soc Interf Cytokine Res 19:1391–1401
Marr RA, Addison CL, Snider D, Muller WJ, Gauldie J, Graham FL (1997) Tumour immunotherapy using an adenoviral vector expressing a membrane-bound mutant of murine TNF alpha. Gene Ther 4:1181–1188
Soo Hoo W, Lundeen KA, Kohrumel JR, Pham NL, Brostoff SW, Bartholomew RM, Carlo DJ (1999) Tumor cell surface expression of granulocyte-macrophage colony-stimulating factor elicits antitumor immunity and protects from tumor challenge in the P815 mouse mastocytoma tumor model. J Immunol 162:7343–7349
Marhaba R, Nazarenko I, Knofler D, Reich E, Voronov E, Vitacolonna M, Hildebrand D, Elter E, Apte RN, Zoller M (2008) Opposing effects of fibrosarcoma cell-derived IL-1alpha and IL-1beta on immune response induction. Int J Cancer 123:134–145
Nazarenko I, Marhaba R, Reich E, Voronov E, Vitacolonna M, Hildebrand D, Elter E, Rajasagi M, Apte RN, Zoller M (2008) Tumorigenicity of IL-1alpha- and IL-1beta-deficient fibrosarcoma cells. Neoplasia 10:549–562
Voronov E, Reich E, Dotan S, Dransh P, Cohen I, Huszar M, Fogel M, Kleinman HK, White RM, Apte RN (2009) Effects of IL-1 molecules on growth patterns of 3-MCA-induced cell lines: An interplay between immunogenicity and invasive potential. J Immunotoxicol
Carmi Y, Rinott G, Dotan S, Elkabets M, Rider P, Voronov E, Apte RN (2011) Microenvironment-derived IL-1 and IL-17 interact in the control of lung metastasis. J Immunol 186:3462–3471
Berglund M, Melgar S, Kobayashi KS, Flavell RA, Hornquist EH, Hultgren OH (2010) IL-1 receptor-associated kinase M downregulates DSS-induced colitis. Inflamm Bowel Dis 16:1778–1786
Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F (1995) Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol 154:2434–2440
Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, Thompson RC, Dinarello CA (1990) Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest 86:972–980
Kaczmarek E, Banasiewicz T, Seraszek-Jaros A, Krokowicz P, Grochowalski M, Majewski P, Zurawski J, Paszkowski J, Drews M (2014) Digital image analysis of inflammation markers in colorectal mucosa by using a spatial visualization method. Pathol Res Pract 210:147–154
Klampfer L (2011) Cytokines, inflammation and colon cancer. Curr Cancer Drug Targets 11:451–464
Lin WW, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117:1175–1183
Lopetuso LR, Chowdhry S, Pizarro TT (2013) Opposing functions of classic and novel IL-1 family members in gut health and disease. Front Immunol 4:181
Mahida YR, Wu K, Jewell DP (1989) Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut 30:835–838
Stronati L, Negroni A, Pierdomenico M, D’Ottavio C, Tirindelli D, Di Nardo G, Oliva S, Viola F, Cucchiara S (2010) Altered expression of innate immunity genes in different intestinal sites of children with ulcerative colitis. Dig Liver Dis : Off J Ital Soc Gastroenterol Ital Assoc Study Liver 42:848–853
Ludwiczek O, Vannier E, Borggraefe I, Kaser A, Siegmund B, Dinarello CA, Tilg H (2004) Imbalance between interleukin-1 agonists and antagonists: relationship to severity of inflammatory bowel disease. Clin Exp Immunol 138:323–329
Bauer C, Loher F, Dauer M, Mayer C, Lehr HA, Schonharting M, Hallwachs R, Endres S, Eigler A (2007) The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-alpha mRNA expression. Dig Dis Sci 52:1642–1652
Cominelli F, Nast CC, Duchini A, Lee M (1992) Recombinant interleukin-1 receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology 103:65–71
Ferretti M, Casini-Raggi V, Pizarro TT, Eisenberg SP, Nast CC, Cominelli F (1994) Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J Clin Invest 94:449–453
Loher F, Bauer C, Landauer N, Schmall K, Siegmund B, Lehr HA, Dauer M, Schoenharting M, Endres S, Eigler A (2004) The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J Pharmacol Exp Ther 308:583–590
Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA (2001) IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 98:13249–13254
Reuter BK, Pizarro TT (2004) Commentary: the role of the IL-18 system and other members of the IL-1R/TLR superfamily in innate mucosal immunity and the pathogenesis of inflammatory bowel disease: friend or foe? Eur J Immunol 34:2347–2355
Al-Sadi RM, Ma TY (2007) IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol 178:4641–4649
Westbrook AM, Wei B, Braun J, Schiestl RH (2009) Intestinal mucosal inflammation leads to systemic genotoxicity in mice. Cancer Res 69:4827–4834
Katsurano M, Niwa T, Yasui Y, Shigematsu Y, Yamashita S, Takeshima H, Lee MS, Kim YJ, Tanaka T, Ushijima T (2012) Early-stage formation of an epigenetic field defect in a mouse colitis model, and non-essential roles of T- and B-cells in DNA methylation induction. Oncogene 31:342–351
Gunter MJ, Canzian F, Landi S, Chanock SJ, Sinha R, Rothman N (2006) Inflammation-related gene polymorphisms and colorectal adenoma. Cancer Epidemiol Biomark Prev : Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol 15:1126–1131
Graziano F, Ruzzo A, Canestrari E, Loupakis F, Santini D, Rulli E, Humar B, Galluccio N, Bisonni R, Floriani I, Maltese P, Falcone A, Tonini G, Catalano V, Fontana A, Giustini L, Masi G, Vincenzi B, Alessandroni P, Magnani M (2009) Variations in the interleukin-1 receptor antagonist gene impact on survival of patients with advanced colorectal cancer. Pharmacogenomics J 9:78–84
Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M (2005) Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 307:734–738
Elinav E, Henao-Mejia J, Flavell RA (2013) Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol 6:4–13
Gagliani N, Palm NW, de Zoete MR, Flavell RA (2014) Inflammasomes and intestinal homeostasis: regulating and connecting infection, inflammation and the microbiota. Int Immunol 26:495–499
Lissner D, Siegmund B (2011) The multifaceted role of the inflammasome in inflammatory bowel diseases. TheScientificWorldJOURNAL 11:1536–1547
Nunes T, de Souza HS (2013) Inflammasome in intestinal inflammation and cancer. Mediat Inflamm 2013:654963
Murthy SN, Cooper HS, Shim H, Shah RS, Ibrahim SA, Sedergran DJ (1993) Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci 38:1722–1734
Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207:1045–1056
Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA (2010) Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A 107:21635–21640
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32:379–391
Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, McCafferty DM, Rioux KP, Ghosh S, Xavier RJ, Colgan SP, Tschopp J, Muruve D, MacDonald JA, Beck PL (2011) NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis 17:1359–1372
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745–757
Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros P, Beauchemin N, Vallance BA, Saleh M (2010) Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32:367–378
Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O’HUigin C, Marincola FM, Trinchieri G (2010) MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med 207:1625–1636
Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, Elinav E, Finlay BB, Flavell RA (2014) NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156:1045–1059
Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M (2010) Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59:1192–1199
Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr, Foley E, Moskaluk CA, Bickston SJ, Cominelli F (1999) IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol 162:6829–6835
Siegmund B (2010) Interleukin-18 in intestinal inflammation: friend and foe? Immunity 32:300–302
Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W Jr, Murphy AJ, Valenzuela DM, Yancopoulos GD, Booth CJ, Cho JH, Ouyang W, Abraham C, Flavell RA (2012) IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491:259–263
Munoz M, Eidenschenk C, Ota N, Wong K, Lohmann U, Kuhl AA, Wang X, Manzanillo P, Li Y, Rutz S, Zheng Y, Diehl L, Kayagaki N, van Lookeren-Campagne M, Liesenfeld O, Heimesaat M, Ouyang W (2015) Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity 42:321–331
Koltsova EK, Grivennikov SI (2014) IL-22 gets to the stem of colorectal cancer. Immunity 40:639–641
Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S, Zgodzinski W, Majewski M, Wallner G, Fang J, Huang E, Zou W (2014) IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 40:772–784
Bauer C, Duewell P, Lehr HA, Endres S, Schnurr M (2012) Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig Dis 30(Suppl 1):82–90
Lebeis SL, Powell KR, Merlin D, Sherman MA, Kalman D (2009) Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun 77:604–614
Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, Rider P, Kaplanov I, Aychek T, Dinarello CA, Apte RN, Voronov E (2014) Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut 63:598–609
Suwara MI, Green NJ, Borthwick LA, Mann J, Mayer-Barber KD, Barron L, Corris PA, Farrow SN, Wynn TA, Fisher AJ, Mann DA (2014) IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol 7:684–693
Scarpa M, Kessler S, Sadler T, West G, Homer C, McDonald C, de la Motte C, Fiocchi C, Stylianou E (2015) The epithelial danger signal IL-1alpha is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am J Pathol 185:1624–1637
Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, Altuntas CZ, Wald D, Ma C, Zhou H, Tuohy VK, Fairchild RL, de la Motte C, Cua D, Vallance BA, Li X (2007) The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 26:461–475
Ivanov II, Littman DR (2010) Segmented filamentous bacteria take the stage. Mucosal Immunol 3:209–212
Shaw MH, Kamada N, Kim YG, Nunez G (2012) Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med 209:251–258
Zhu J, Yamane H, Paul WE (2010) Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28:445–489
Kamada N, Chen GY, Inohara N, Nunez G (2013) Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 14:685–690
Hasegawa M, Kamada N, Jiao Y, Liu MZ, Nunez G, Inohara N (2012) Protective role of commensals against Clostridium difficile infection via an IL-1beta-mediated positive-feedback loop. J Immunol 189:3085–3091
Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L, Trinchieri G (2015) The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur J Immunol 45:17–31
Dzutsev A, Trinchieri G (2015) Proteus mirabilis: the enemy within. Immunity 42:602–604
Seo SU, Kamada N, Munoz-Planillo R, Kim YG, Kim D, Koizumi Y, Hasegawa M, Himpsl SD, Browne HP, Lawley TD, Mobley HL, Inohara N, Nunez G (2015) Distinct commensals induce interleukin-1beta via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 42:744–755
Acknowledgments
Ron N. Apte and Elena Voronov have been supported by the Israel Ministry of Science (MOS) jointly with the Deutsches Krebsforschungscentrum (DKFZ), Heidelberg, Germany, the Israel Science Foundation funded by the Israel Academy of Sciences and Humanities, the Israel Cancer Association and the Israel Ministry of Health Chief Scientist’s Office, FP7: “Cancer and Inflammation” (INFLA-CARE), The Binational (Israel-USA) Science Foundation (BSF) and the German-Israeli Foundation (GIF). Prof. Ron N. Apte is an incumbent of the Irving Isaac Sklar Chair in Endocrinology and Cancer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Voronov, E., Apte, R.N. IL-1 in Colon Inflammation, Colon Carcinogenesis and Invasiveness of Colon Cancer. Cancer Microenvironment 8, 187–200 (2015). https://doi.org/10.1007/s12307-015-0177-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-015-0177-7