
Abstract
Cancer remains one of the most challenging diseases despite significant advances of early diagnosis and therapeutic treatments. Cancerous tumors are composed of various cell types including cancer stem cells capable of self-renewal, proliferation, differentiation, and invasion of distal tumor sites. Most notably, these cells can enter a dormant cellular state that is resistant to conventional therapies. Thereby, cancer stem cells have the intrinsic potential for tumor initiation, tumor growth, metastasis, and tumor relapse after therapy. Both genetic and epigenetic alterations are attributed to the formation of multiple tumor types. This review is focused on how epigenetic dynamics involving DNA methylation and DNA oxidations are implicated in breast cancer and glioblastoma multiforme. The emergence and progression of these cancer types rely on cancer stem cells with the capacity to enter quiescence also known as a dormant cellular state, which dictates the distinct tumorigenic aggressiveness between breast cancer and glioblastomas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Epigenetic dynamics are essential for normal tissue homeostasis, and their disruption may lead to changes in gene expression networks associated with various types of cancer. This review focuses on epigenetic alterations associated with breast cancer (BC) and glioblastoma multiforme (GBM). BC is the most frequent cancer type among women and remains the leading cause of cancer-related death worldwide. This is in line with an increased trend for BC over the last decade, especially in younger women. In the USA, BC represents over one-fifth of all cancers, with over 268,000 new cases in 2019, according to the national cancer institute [1]. GBM is the sixteenth most common cancer with over 23,000 new cases in 2019, accounting of approximately ~ 50% of primary brain tumors and poor survival rate of 6 to 12 months [2, 3]. Here, we discuss the current literature on BC and GBM with emphasis on the epigenetic changes related to DNA methylation and DNA oxidations that could act as roadblocks for cancer treatments [1,2,3,4,5,6]. We will discuss how these epigenetic dynamics facilitating tumorigenesis could be interrogated to develop new treatment strategies.
A major issue for cancer relapse is the ability of the tumor cells to survive during dormancy state. Tumor dormancy can exist for decades in a pre-diagnostic cancer phase before the cancer becomes clinically discernible [7]. Additionally, tumor dormancy exists during remission and these cells could be the source of tumor recurrence. Dormant cancer cells are equipped to survive therapeutic treatment, thereby increasing the risk of tumor relapse. The dormant cancer cells have intrinsic stem cell–like properties such as pervasive self-renewal, multipotent capacity, and the ability to exit the cell cycle to enter prolonged periods of quiescence, which they used as a therapeutic resistant strategy [8]. Cancer stem cells (CSCs) possess all the aforementioned properties and are currently defined as initiating tumor cells with the capacity to promote neoplastic growth, facilitate metastatic spreading, and escape apoptotic programs [8]. This level of cellular plasticity cannot always be explained by irreversible genetic alterations. Reversible epigenetic modifications may partly explain malignancy by allowing the activation of specific transcriptional networks underlying the various cellular states of CSCs. A plethora of epigenetic dynamics, including DNA methylation and histone modifications, has been attributed to various stages of carcinogenesis [9]. For instance, aberrant expression of epigenetic regulatory factors can reprogram the epigenetic landscape allowing for the activation and repression of oncogenes and tumor suppressors, respectively. Interestingly, multiple epigenetic reprogramming events implicated in tumorigenesis and cancer progression are reminiscent of the ones occurring during embryonic development and cellular reprogramming towards stem cell–like states [10, 11]. For instance, the reprogramming factors Oct4, Sox2, Klf4, and Nanog involved in early stages of embryonic development as well as the formation of induced pluripotent stem cells (iPSCs) are upregulated in various types of cancers and thereby viewed as oncogenes [11,12,13,14,15]. These reprogramming genes are considered pioneer transcription factors capable of recruiting DNA and histone modifying enzymes to remodel the epigenome [16]. The identification of gene expression patterns driven by specific epigenetic programs that are unique to CSCs, and different from normal stem cells, remains largely unclear and thereby an active area of research. Thus, a deeper understanding of the epigenetic regulatory programs that can selectively facilitate the transition to tumor dormancy and the underlying molecular events that culminate in cancer relapse will undoubtedly benefit the development of new cancer therapies. In this review, we focus on two distinct CSC-driven tumors exhibiting cellular dormancy, BC, and glioblastoma. The existence of BC in a dormant cellular state has been studied, but such knowledge is relatively limited for GBM, partly because of the aggressiveness of this cancer type and its poor prognosis. Thereby, dormant cellular states have been challenging to investigate in GBM as compared to BC where dormant cancer cells can survive for decades [17]. However, as new drugs are being developed for GBM, patients will be likely placed into remission. Thus, it is of great importance to discern epigenetic-mediated mechanisms underlying dormancy to develop new therapeutic strategies aiming to prevent tumor recurrence.
Cancer dormancy is a strategic cellular state acquired through epigenetic reprogramming that causes the activation of key survival gene expression networks required to escape cell death and trigger metastatic activating programs. Although CSCs are essential for tumor initiation, progression, and metastasis, they represent < 5% of the total number of cells within various solid tumor types and thereby a challenge to understand their molecular properties. However, rapidly advancing single-cell genome-wide technologies may soon lead to the identification of specific epigenetic-driven transcriptional networks underlying the ability of CSC to acquire dormancy. In this review, we highlight currently known epigenetic programs that are dysregulated in breast cancer and glioblastomas. We highlight the epigenetic reprogramming events involving DNA methylation and DNA oxidations that are relevant to these specific cancer types. Thus, a deeper understanding of any alterations in these epigenetic pathways underlying tumor dormancy and tissue microenvironment interactions is needed for developing new therapeutic strategies.
2 Current limitations in cancer research
Cancer remains a clinical dilemma despite early diagnosis and aggressive treatments including pharmacological inhibitors to disrupt intracellular signaling, ligand antagonism, use of nanoparticles to target stem-like population, chemotherapy, radiation, immune therapy, and epigenetic reprogramming. Despite these improvements, there are increases in cancer relapse. The mechanisms by which current treatments still result in cancer relapse have been focused on genetic mutational analysis relevant to the microenvironment of the primary and secondary tumor sites. Emphasis on epigenetic regulatory processes associated with cancer relapse is currently at an early phase.
BC cells (BCCs) can exist in a dormancy state for decades before growing into metastatic lesions, which are the main cause of BC death. Similar to other cancer types, the existence of CSCs has been reported for BC. However, the mechanisms underlying the formation of CSCs and their ability to enter a dormancy state leading to therapeutic resistance, along with their metastatic potential, are yet to be determined. Epigenetic modifications such as histone and DNA methylation were shown to be deregulated in human BC and therefore of clinical interest to be used as biomarkers for prognosis and therapeutic treatment. GBM is one of the most lethal solid tumors with currently no effective targeted therapy. Chemotherapy can reduce tumor size by targeting rapidly dividing tumor cells, but brain tumor stem cells (BTSCs) can exit the cell cycle and remain in a quiescent state, which gives them resistance to chemotherapy and irradiation treatment leading to tumor relapse. Embryonic stem cell genes such as Sox2 are highly expressed in BTSCs. However, Sox2 is also expressed in normal neural progenitor stem cells (NPCs) and thereby not an ideal therapeutic target. Thus, a better understanding of the epigenetic and transcriptional programs underlying BTSCs propagation that are different from normal NPCs will promote the development of new GBM cancer therapies. Thus, understanding how cancer cells restructure their epigenome to become dormant will serve to develop specific therapies to prevent their escape from immune surveillance and resistance to chemotherapy, thereby diminishing tumor relapse and metastasis.
3 DNA methylation-demethylation dynamics in cell fate
Apart from safeguarding the integrity of our genomic DNA sequence, a plethora of chromatin dynamics constituting the epigenome is capable of modulating gene expression in a highly complex yet organized fashion. One of the main features of the epigenome is its high level of responsiveness towards environmental changes and cellular signaling mechanisms. Notably, epigenetic dynamics are not unidirectional but rather harbor a high degree of plasticity to allow cell fate changes in response the challenges within diverse cellular microenvironments. This concept was experimentally confirmed in one of the most prominent discoveries in modern biology, which is the ability to change cell fate identity through epigenetic remodeling triggered by forced expression of pioneer transcription factors [14]. These pioneer transcription factors, Oct4, Sox2, Klf4, and c-Myc, drive the reprogramming of somatic cells into induced pluripotent stem cells (iPSCs), which, similarly to embryonic stem cells (ESCs), are capable of self-renew and differentiate into all the cell types that constitute an entire organism. Mechanistically, c-MYC associates with histone acetyltransferase complexes allowing OCT4 and SOX2 to recognize their targeted genes, while KLF4 represses the expression of the tumor suppressor p53, which is a roadblock for somatic cellular reprogramming into iPSCs and it is often mutated in multiple cancer types [18,19,20]. Overall, these pluripotency factors can reprogram the epigenome of differentiated somatic cells into a stem cell state that hold an infinite capacity for self-renewal and the ability to differentiate into all cell types [21].
Among the several epigenetic changes that occur during somatic cellular reprogramming, DNA demethylation is essential for restructuring the epigenome to promote the expression of pluripotency genes during formation of iPSCs [22]. Consistently, the ten-eleven translocation enzymes TET1 and TET2, which are dioxygenase involved in an active DNA demethylation process, are critical for promoting cellular reprogramming into iPSCs [23, 24]. More specifically, TET1-mediated reprogramming promotes the expression of the pluripotency gene Oct4, while TET2 targets enhancer regions involved in facilitating the formation of iPSCs [23]. Although epigenetic reprogramming such as DNA methylation/demethylation dynamics are necessary for the conversion of somatic cells into iPSCs, the epigenetic remodeling events required for cellular dedifferentiation processes leading to the formation of other stem cell–like states such as cancer stem cells (CSCs), also known as cancer-initiating cells, remain largely unclear. Furthermore, how epigenetic remodeling impact CSCs to enter a dormant cellular state, which makes them resistant to conventional cancer therapies, remains as a major question. Nevertheless, there are striking similarities between the formation of iPSCs and the process of dedifferentiation into CSCs. For instance, the core pluripotent genes Oct4, Sox2, and Klf4 are upregulated during cellular transformations into oncogenic malignancies and thereby used as biomarkers for cellular dedifferentiation into cancer stem–like cells [11] Apart from dedifferentiation into stem cell–driven oncogenic states, the DNA demethylation–dependent upregulation of these pluripotency genes is likely to promote self-renewal capacity favoring CSC-dependent tumor growth and metastatic invasiveness. In this review, we highlight DNA methylation and demethylation mechanisms underlying cell fate transitions towards CSC dormancy in the context of brain and breast cancers.
4 DNA methyltransferases
DNA methylation occurs at CpG dinucleotides and causes gene repression, which is a critical epigenetic regulatory process involved in the maintenance of cell identity. In mammals, cytosines are methylated at the fifth carbon position leading to the formation of 5-methylcytosine (5mC), which is catalyzed by DNA methyltransferase (DNMT) enzymes [25, 26]. The DNMT family of enzymes is comprised of DNMT1, DNMT3A, and DNMT3B but has distinct capabilities of modulating the epigenome. While DNMT1 is required for the maintenance of DNA methylation, DNMT3A and DNMT3B are needed for de novo DNA methylation at specific genomic loci. A potent mechanism for repressing gene expression is the recruitment of DNMTs at CpG-enriched gene regulatory sequences such as promoters. Overall, DNMT-dependent maintenance and de novo DNA methylation are critical epigenetic regulatory modes to ensure gene expression programs required for sustaining cellular identity. Mechanistically, DNMTs are recruited to specific genomic loci by specialized epigenetic regulators and/or pioneer transcription factors. For instance, the Ubiquitin like PHD and RING Finger domains 1 (UHRF1) factor recruits DNMT1 to preserve the status of DNA methylation during DNA replication, which is impaired upon cellular transformations into malignant states [27, 28].
Genomic regions with high CpG content termed CpG islands (CGIs) overlap with the promoters of about 70% of all human genes [29, 30]. Proper maintenance of DNA methylation by UHRF1-DNMT1 interactions is crucial for preserving genomic imprinting and overall epigenetic memory during cell divisions [31]. This level of epigenetic memory allows cells to sense exogenous stressors and adapt to challenging microenvironment without compromising their cellular identity [32]. Interestingly, triple negative BCCs overexpressing UHRF1 exhibit an accelerated cell growth rate due to shortening of the cell cycle at G1 phase and thereby UHRF1-DNA-binding domain has been proposed as a drug target for cancer therapy [33, 34]. In contrast to DNMT1, DNMT3A and DNMT3B are able to establish de novo DNA methylation. Interestingly, apart from DNMT1, UHRF1 can also interact with DNMT3A and DNMT3B to silence gene promoters in embryonic stem cells (ESCs) [35]. Although DNMT enzymes have distinct roles to maintain and establish overall DNA methylation status, they can also collaborate among themselves to safeguard DNA methylation throughout the genome. For instance, DNMT3A targets unmethylated cytosines to create hemimethylated CpG sites, which become new targets for DNMT1 to preserve DNA methylation [36, 37]. Apart from maintaining the integrity of the epigenome, these dynamic changes are essential for adaptation to extracellular challenges such as microenvironment diversity. Multiple epigenetic changes work in concert to establish the necessary gene expression patterns that are linked to various aspects of cellular physiology including metabolic demands, cellular signaling, and environmental adaptation. For example, DNMT3A can recognize trimethylated histone H3 at lysine 36 (H3K36me3) or methylated histone H3 at lysine 4 (H3K4me) prior to initiating DNA methylation [35, 38]. This interplay between DNMTs and histone methylation is essential for regulating specific gene expression patterns in the context of health and disease. In this regard, the interactions between DNMT1, UHRF1, and the histone methyltransferase enzyme G9a were elucidated from cultured BCCs [39, 40]. Additionally, G9a-dependent methylation of histone H3 at lysine 9 (H3K9me) is recognized by the heterochromatin protein 1 (HP1) and consequently causes the recruitment of DNMT3 for de novo DNA methylation [41]. Thus, the chromatin microenvironment harboring histone modifications can facilitate methylation of CpGs by promoting the recruitment of DNMTs. Collectively, the crosstalk between methylation of both DNA and histones can work in concert to maintain or modify the epigenetic landscape in response to cellular and environmental challenges. Alterations to this epigenetic crosstalk are associated with both BC and GBM, as highlighted in more detailed below.
5 TET enzymes
DNA methylation dynamics rely not only on DNMTs but also on specific chemical modifications occurring at methylated cytosines. DNA methylation can be reversed by passive demethylation during DNA replication cycles, where specific DNA methylation sites are not maintained and thereby diluted by successive cellular divisions. Notably, DNA methylation can be erased by an active DNA demethylation process driven by the ten-eleven translocation (TET) family of dioxygenase enzymes (TET1, TET2, TET3) initially discovered as fusion partners of the MLL gene in acute myeloid leukemia and other forms of hematological cancers [42, 43]. Several lines of evidence support the role of TET enzymes in an active DNA demethylation pathway leading to de-repression or activation of gene expression [44,45,46,47]. TET-dependent DNA demethylation occurs as a series of successive oxidation reactions where 5-methylcytosine (5mC) is converted to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), in which 5fC and 5caC can be recognized by DNA-based excision repair factors leading to unmodified cytosines and thereby DNA demethylation [48, 49]. Apart from functioning as intermediates of DNA demethylation, TET-mediated DNA oxidations per se can function as epigenetic elements to modulate gene expression [48]. In vitro analysis using chemically modified oligonucleotides enriched for either 5mC or 5hmC showed they can be targeted by a discrete number of epigenetic readers [30]. Notably, 5fC and 5caC can be directly recognized by RNA polymerase II (Pol II) to slow down transcription elongation by inducing Pol II pausing [49, 50]. This particular finding supports the idea of TET-mediated DNA oxidations functioning as epigenetic determinants regulating gene expression by directly modulating transcription. Thus, TET-mediated DNA oxidation can essentially rewire the epigenome by serving as intermediates of DNA demethylation or as epigenetic elements interacting with the transcriptional machinery. DNA oxidations such as 5hmC are ubiquitously found in multiple cell types and tissues but are particularly abundant in ESCs and neural tissues [30, 48, 51, 52]. The mechanisms, by which these DNA oxidations affect gene expression in the context of health and disease, remain largely unclear [53]. The relevance of TET enzymes in epigenetic remodeling has been investigated in embryonic development and somatic cellular reprogramming into iPSCs [29]. However, it is unknown how TET-mediated DNA oxidations functioning as epigenetic elements impact the epigenome in the context of cancer initiation, progression, and metastatic invasion.
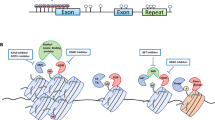
A prominent aspect of TET function involves their recruitment to specific genomic loci. In contrast to TET1 and TET3, the CXXC zinc finger DNA–binding domain is absent in TET2 [54]. A CXXC-containing protein, IDAX, can recruit TET2 to unmethylated CpG sequences within promoters and CGIs [55,56,57]. Interestingly, this IDAX-dependent recruitment causes a caspase-mediated degradation of TET2 bound to CpG sites [57]. Notably, aberrant IDAX expression is found in different types of cancers such as colon adenomas and renal carcinomas [26, 58]. Other CXXC-containing proteins including isoforms of the transcription factors MBD1 and MBD2 can recruit TET1 to specific genomic loci. For instance, MBD1 can recognize methylated CGIs and modify gene expression by enhancing TET1-dependent conversion of 5mC into 5hmC [59]. On the other hand, MBD2 protects 5mC from TET1-mediated DNA oxidations [60, 61]. Thus, the recruitment of TET enzymes is a highly regulated mechanism involving accessory DNA-binding factors to make the epigenome responsive to intracellular and microenvironmental demands. Plausibly, aberrant expression of these accessory factors could negatively alter the recruitment and function of TET enzymes leading to miss regulated gene expression such as activation of oncogenes and/or silencing of tumor suppressor genes. In support of this notion, aberrant expression of MBD1 and MBD2 is found in diverse cancer types [62, 63]. Additionally, other epigenetic factors including the arginine methyltransferase PRMT1 were shown to bind 5hmC produced by TET1 to promote the expression of genes involved in tumorigenic progression of glioblastomas [64]. On the other hand, the oncogenic epidermal growth factor receptor (EGFR) triggers tumor suppressor gene silencing by inhibiting TET1 activity in lung adenocarcinomas and glioblastomas [65]. In this scenario, TET1 induced the expression of tumor suppressor genes via active DNA demethylation. Notably, a new isoform of TET1 lacking CXXC DNA–binding domain was shown to be upregulated in breast cancer and glioblastoma [66]. Although this TET1 isoform can produce 5hmC, it has a poor influence on DNA demethylation at CGI regions. Therefore, unraveling TET-dependent mechanisms of gene regulation could help for the development of new epigenetic-driven cancer therapies or early diagnostic methods for detection of aggressive tumors such as glioblastomas.
6 Interplay among DNA methylation, TET enzymes, and metabolism
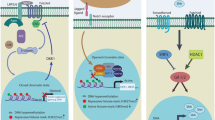
As described above, the overall status of DNA methylation is regulated by the combinatory function of DNMTs and TETs (Fig. 1a). The catalytic activity of these enzymes is dependent on obligatory cofactors including the metabolic intermediates, S-adenosylmethionine (SAM) and α-ketoglutarate (α-KG), respectively [67, 68]. SAM is generated from methionine biosynthesis, and it is the universal donor of methyl groups to both histone and DNA methyltransferases. DNMTs transfer methyl groups donated from SAM onto cytosines at CpG-enriched genomic regions [69]. The intermediate metabolite S-adenosylhomocysteine (SAH), a potent inhibitors of DNMTs, is generated as a byproduct of SAM (reviewed in [70, 71]). SAH can be further hydrolyzed to homocysteine and recycled back to methionine [72, 73]. Thus, changes in methionine metabolism could lead to deregulated DNMT activity and confer cancer cells with proliferative advantages (54).

Crosstalk between metabolites and epigenetics. a S-adenosylmethionine (SAM), which is produced from methionine biosynthesis functions as a methyl group donor for DNMT-dependent DNA methylation. TET enzymes can successively oxidize 5mC into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). Both 5fC and 5caC can be recognized by base-excision repair (BER) factors leading to DNA demethylation. Alpha-ketoglutarate (α-KG), an obligatory cofactor for ten-eleven translocation (TET) enzymes, is generated through the TCA cycle by IDH1 and IDH2 enzymes. Mutated IDH enzymes generate 2-hydroxyglutarate (2-HG), which inhibits TET activity. Generally, DNA methylation is associated with gene repression, while TET-mediated DNA oxidations are involved in gene activation. b TET activity is positively regulated by vitamin C and its obligatory cofactor α-KG. Derivatives of α-KG such as succinate and fumarate inhibit TET activity. In cancer, both succinate and fumarate are accumulated due to mutated versions of succinate dehydrogenase (SDH) and fumarate hydratase (FH) enzymes
The catalytic activity of TET enzymes is strictly dependent on α-KG, which is part of the tricarboxylic acid (TCA) cycle, and it is generated by isocitrate dehydrogenase (IDH) enzymes IDH1 and IDH2 [74, 75]. Mutations in these enzymes cause the production of the oncometabolite 2-hydroxyglutarate (2-HG) functioning as a competitive inhibitor of α-KG found in brain tumors [76] (Fig. 1a). On the other hand, the catalytic activity of TET enzymes can be inhibited by succinate and fumarate, which are intermediate metabolites of the TCA cycle that are downstream of α-KG [74] (Fig. 1b). The catalytic function of TET enzymes is also dependent on vitamin C (ascorbate), which has been proposed as an anti-cancer treatment [77]. Thus, changes in cellular metabolism can influence the activity of DNMTs and TETs by regulating the availability of their cofactors. Generally, alterations in DNA methylation are a hallmark of cancer. For instance, dysregulated DNA methylation at specific genomic loci including promoter regions correlates with multiple types of cancers including breast cancer and glioblastomas [78, 79]. More specifically, genes involved in the regulation of cell cycle and tumor suppression are silenced by DNA hypermethylation in breast cancer due to overexpression DNMT3B [80]. For example, the tumor suppressor PTEN (phosphatase and tensin homolog), which is one of the main regulators of cell cycle progression, is downregulated due to elevated levels of DNMT1 and SAM [81]. On the other hand, high levels of TET expression determined from 162 breast cancer tissues correlated with increased patient survival, possibly due to DNA demethylation–dependent upregulation of tumor suppressor genes [82]. Interestingly, the catalytic activity rather than the expression of TET enzymes is altered in secondary high-grade glioblastomas due to IDH mutations. As mentioned above, IDH mutants generate 2-HG, which results in hypermethylation of tumor suppressor genes [83]. Additional metabolic changes influencing DNA methylation/demethylation dynamics in both breast cancers and glioblastomas are dependent on oxygen levels. For instance, hypoxic conditions in both glioblastomas and breast cancers trigger the upregulation of the hypoxia-inducible factor-1α (HIF-1α), which can interact with TET1 under hypoxic conditions [84]. Tumor hypoxia causes an increase in cellular proliferation, epithelial-mesenchymal transition (EMT), metastasis, and chemo-resistance, which are hallmarks for cellular dedifferentiation towards cancer stem cells. As mentioned above, TET enzymes are strictly dependent on α-KG, which becomes decreasingly available in cancer types expressing mutated fumarate hydratase (FH) and succinate dehydrogenase (SDH) enzymes, thereby leading to an overall decrease in TET activity [74, 85,86,87]. Generally, dysregulated enrichment of DNA methylation at promoter regions is correlated with various types of cancers, including breast cancer and glioblastomas [78, 79]. For instance, microarray analysis shows that breast cancer cell lines characterized by genomic hypermethylation overexpress the de novo DNA methyltransferase enzyme DNMT3B. DNA hypermethylation can also occur in the context of IDH mutations found in brain tumors and breast cancer [88,89,90]. Thereby, metabolic dynamics can affect the epigenome by regulating the activity of TET enzymes during carcinogenesis.
As mentioned above, IDH mutations altering the levels of α-KG are found in brain tumors. IDH mutations in glioblastoma producing 2-HG are found in 70% of low-grade tumors [91,92,93]. Gliomas of increased malignancy show a decrease in TET1 activity that correlate with lower levels of 5hmC in patient-derived tumor samples. On the other hand, decitabine, a DNA methyltransferase inhibitor, has been shown to reduce cellular proliferation of IDH1 mutant glioma cells [94]. Interestingly, glioma cancer stem cells, which are resistant to most treatments targeting cellular replication, have increased levels of 5fC and 5caC [95]. However, the mechanisms underlying the role of these DNA oxidations in brain tumors remain to be determined. As mentioned above, TET enzymes are dioxygenases whose activity is dependent on oxygen availability. A pan-cancer analysis shows that TET activity in hypoxic tumors is inversely correlated with hypermethylation [83]. Hypoxia is a hallmark of cancer, in which oxygen deprivation can alter epigenetic regulatory programs of cancer cells causing an increase in metastatic competence and by facilitating cellular transitions towards stem cell–like states [96,97,98]. Collectively, TET-mediated DNA oxidations are highly dependent on metabolic states conferring cancer cells the ability to reprogram their epigenome to control gene expression networks that promote tumor growth and metastasis.
7 CSCs
The initiation of malignancy is a complex process that may be due to a combination of the following: viral infection, hereditary gene mutation, somatic mutation, and/or environmental factors that might be sporadic [1, 2]. Tumors are comprised of a heterogeneous population of cells [5]. There is an upsurge of interest in a small subset of cancer cells with self-renewal capabilities that where first identified over 20 years ago in acute leukemia [99]. These cells, designated as cancer stem cells (CSCs), are presumed to be the source of cancer relapse, which sparked clinical interest in their therapeutic targeting. CSCs are a subpopulation of tumor cells that share properties with normal stem cells, such as self-renewal, asymmetric division, hierarchical differentiation, prolonged doubling time, expression of core stem cell genes (i.e., Oct4a, Nanog, Notch, and Sox2), and their ability to exit their cell cycle towards a dormant cellular state [8, 100, 101]. The origin of CSCs could be attributed to mutations within the adult stem cell niche or dedifferentiation of somatic cells via cellular reprogramming into a stem cell–like state [102].
CSCs have increased expression of multidrug resistant genes such as ATP-binding cassette (ABC) transporters, allowing the efflux of toxins and chemicals and causing the evasion of therapeutic treatments [103, 104]. CSCs were initially classified as CD44+/CD24−/low, CD133+, and aldehyde dehydrogenase 1 (ALDH1+) [105,106,107]. However, there is evidence indicating that these markers are not reliable for the identification of CSCs. In our lab, we have stratified BCC subsets based on Octamer-4a (Oct4a) gene expression, allowing for improved identification of CSCs, which constitute about 5% of the total BCC population [8, 108].
Two different mechanisms have been proposed to describe the origin of CSCs. Initially, it was established that mutations in cancer cells resulted in their dedifferentiation into cells with a stem cell–like phenotype (Fig. 2a) [109]. Conversely, it was hypothesized that mutations in core pluripotent genes of normal stem cells contribute to the development of CSCs [110, 111]. Regardless of the origin, CSCs exhibit similar properties to healthy stem cells, which impede their therapeutic targeting due to potential harm to non-malignant stem cells. Furthermore, as mentioned above, CSCs can adopt a dormant phenotype, allowing them to evade conventional treatment and persist in a quiescent state within a particular niche for extended periods of time [112]. In this section, we will address the epigenetic regulation of CSCs from breast cancer and GBMs with respect to their DNA methylome.

Generation of CSCs is mediated by genetic and epigenetic mechanisms. a Mutations in core pluripotency genes promote the acquisition of a malignant phenotype in healthy stem cells. b Secretome from tissue niche facilitates the de-differentiation of progenitor cancer cells into CSCs. c Dysregulation of epigenetic mechanisms, such as DNA methylation and oxidations in both healthy stem cells and progenitor cancer cells, causes formation of CSCs
A major issue for targeting CSCs is their functional similarities to normal stem cells. These include their intrinsic self-renewal capacity, differentiation into functional mature cells, low proliferative potential, and drug resistance, which make them a challenging target for current cancer therapies [3, 5, 99]. Additionally, CSCs, in particular those from BC, choose the bone marrow (BM) as a preferential microenvironmental niche where cancer cells can exist in a dormancy state [113]. This cellular niche preferred by dormant BCCs imposes difficulties for establishing therapeutic targeting strategies since the BM niche is the home for endogenous hematopoietic stem cells [113, 114]. It is therefore important to understand the interaction between CSCs and the microenvironment since this could allow for the development of new therapeutic interventions [2, 3, 5]. Thus, we propose that cancer niches such as the BM will cause changes in the epigenetic “machinery” of cancer cells leading to their reprogramming into CSCs (Fig. 2b). The identification of such facilitators could result in the development of new pharmacological treatments.
8 BM secretome
BM is a complex organ comprising of hematopoietic and non-hematopoietic cells, soluble factors, extracellular matrices, and microvesicles [115]. Hematopoietic stem cells (HSC) and mesenchymal stem cell (MSC) are two major resident stem cells in BM [114, 116]. HSCs are the source of immune and blood cells, whereas MSCs can support HSC function [114, 116, 117]. Additional supporting cells include stromal cells that are generally grouped as fibroblasts, macrophages, endothelial cells, and adipocytes [114]. Cellular support of hematopoiesis includes soluble and insoluble secretome that mediate intercellular communication among hematopoietic and non-hematopoietic cells [108, 118]. BCCs can take advantage of the normal hematopoietic system for their survival as dormant tumor cells and when provided with specific microenvironmental cues their dormancy state can be reversed to facilitate the formation of metastatic cells [119, 120]. Among the microenvironmental cues facilitating a dormant cellular state are mediators of intercellular crosstalk such as cytokines and exosomes [3, 119, 121].
This review focuses on exosomal secretome, which are small microvesicles of endocytic origin [119]. Exosomes can shuttle cargo such as RNA, proteins, lipids, and DNA into recipient cells [121]. Initially, exosomes were thought to be part of a cellular garbage disposal system. These vesicles have since been shown to elicit phenotypic and functional cellular changes, including hematopoietic regulation [119]. Cancers such as BC and GBM cells have also utilized exosomes to facilitate their survival by promoting cellular interactions with the cells within the BM microenvironment [3, 5]. Thus, disseminated tumor cells (DTCs) such as BC show preference for the BM by taking advantage of the microenvironment for dormancy to keep the BCCs in BM and to also use the same BM niche cells to reverse dormancy to release the dormant cells to tertiary metastatic sites (Fig. 3a) [122]. As mentioned above, exosomes can facilitate BCCs to undergo cycling states of quiescence (Fig. 3b).

BM microenvironment in support of BCC dormancy. a Overview of the BM microenvironment and the support of BC behavior in different regions of the cavity. b Mesenchymal stem cells release exosomes containing cargo that modulate the epigenetic landscape of BCCs. A potential mechanism is by enrichment of DNA methylation patterns mediated by DNMT1 and modulation of miRNAs. c At the endosteal niche, BM stroma establish gap junction intercellular communication with CSCs to maintain dormancy. This communication facilitates the exchanges of molecules that could modulate the epigenome of CSCs and thus provide chemoresistance properties
The secretome within the BM niche including small RNAs inside exosomal vesicles can mediate intercellular communication to facilitate dormancy of BCCs [119]. This can occur by the transport of molecules such as miRNA, proteins, and mitochondrias through gap junctions between tumor cells and microenvironment niche cells [123,124,125,126]. Such communication is not limited to BM since similar crosstalk has been reported between astrocytes and glioma cells [127]. Cancer cells can also communicate with each other to transfer drug resistance molecules [128]. Such communication can occur via gap junction due to high expression of Cx43 (GJA1) on BC and GBM (Fig. 3c) [126, 129]. Direct interactions between CSCs and BM stroma are critical for establishment of dormancy at the endosteal niche (Fig. 3c). This communication is characteristic of the late stages of dormancy, and there is minimal understanding of the epigenetic regulation of CSCs during such process.
MiRNA cargo within exosomes has been well-studied with respect to tumor dormancy. These miRNAs are ~ 23 nucleotides in length with significant roles in gene regulation mostly at the post-transcriptional level [130,131,132,133]. They can also induce epigenetic changes involving DNA methylation and histone modifications by targeting epigenetic regulatory factors. These epigenetic factors are associated with chromatin dynamics capable of altering gene expression patterns and play pivotal roles in tumor initiation, progression and invasion [134, 135]. Examples of epigenetic regulators that can be targeted by miRNAs as well as other relevant effects are briefly summarized in Table 1. Since miRNAs are mostly translational suppressor, their involvement could suppress the expression of the epigenes. On the other hand, if the miRNAs suppress the translation of a negative regulator, this would enhance the expression of the epigene. Mutation of such proteins has been linked to decrease in patient prognosis, underscoring the importance of dissecting the function of miRNAs in epigenetic regulation [134, 135].
9 DNA methylation in BCC subsets
BCCs do not solely rely on genetic alterations to persist within a specific niche. Modifications of the epigenome were shown to be beneficial for BCC survival due to increased cellular plasticity, which allows them to adapt to diverse microenvironments. Epigenetic alterations within BCCs are required for cell-fate transitions and might facilitate their reprogramming into CSCs (Fig. 2c). The capacity of CSCs to display epigenetic plasticity could account for cancer aggressiveness and resurgence. Hence, studying the epigenetic mechanisms involved in the regulation and generation of CSCs is a critical step towards successful eradication of these malignant cells.
As mentioned previously, one of the major caveats in the field is the proper and specific identification of CSCs. For instance, CSCs were phenotypically identified as CD44+/CD24−; however, not all cells harboring this phenotype display functional properties of CSCs. Subsequently, breast CSCs were characterized as CD44high/CD24low/ALDHhigh and only a small population had long-term repopulating capacities. Several reports have shown that CSCs exhibit distinct epigenetic features that can be used as identification markers. For example, breast CSCs possess elevated levels of DNMT1, which is critical for their self-renewal (Fig. 2c) [136]. Moreover, differentially expressed methylation patterns observed in young BC patients (< 35 years) versus old patients are associated with the aggressiveness of the disease [137]. In addition, specific DNA methylation patterns confer breast CSCs resistance to treatment by modulating their TGF-β signaling pathway, which is involved in cellular growth [138]. Notably, modulation of methylated patterns at specific miRNAs promoter allows the enrichment of breast CSCs. More specifically, DNA methylation–dependent repression of miR-203 induces self-renewal capacities in BCCs and abrogates epithelial-to-mesenchymal cellular transitions (EMT) [139]. Importantly, inhibition of 5mC levels at miR-203 promoter regions by 5-azacytidine resulted in decreased tumor formation and enhanced migratory properties of BCCs [139]. Another miRNA that displays enriched methylation is the miR-200c/141cluster, allowing transcriptional activation of the oncogene Twist, an EMT promoting protein that confers invasive properties in BCCs [140]. In human ductal breast carcinoma, aberrant methylation at CpG islands within ERα and E-cadherin gene promoters are observed right before the tumor obtains invasive capacities increasing cancer progression [141]. Importantly, heritable DNA methylation profiles from peripheral blood circulating DNA are associated with susceptibility to BC and serve as a prognostic for clinical assessment [142]. Interestingly, mapping of DNA methylation patterns in BCCs revealed that enhancer regions of the genes ERα, FOXA1, and GATA3 are enriched with 5mC, allowing for the identification of a specific subset of BCs [143]. Another study indicated that breast CSCs exhibit hypomethylated CpGs in genes involved in Jak-STAT signaling and thereby critical for self-renewal [144]. Additionally, circulating tumor cells derived from patients with metastatic BC shown methylation of tumor suppressor genes such as cystatin M and breast cancer metastasis suppressor 1 (BRMS1) [145].
One of the reasons for CSCs to persist in a microenvironment niche after treatment is due to their immune evasive properties. Mechanistically, this can occur by upregulation of immune checkpoints such as programmed death-ligand 1 (PD-L1). Evaluation of PD-L1 promoter in BCCs revealed enhanced hypomethylation and displayed significant enrichment of 5hmC mediated by TET3 [146]. Another feature that favors CSC survival is their ability to exist in hypoxic environments. Hypoxia-inducible factors are transcription factors that respond to low oxygen levels and have been shown to be dysregulated in multiple cancer types [147]. Hypomethylation at non-CpG sites along with acetylation of histone H3 at lysine 9 (H3K9ac) trigger the upregulation of the hypoxia inducible factors HIF-1α in BCCs, which is critical for their survival and maintenance [148]. Overall, DNA methylation dynamics are part of an epigenetic program that sustains the survival and proliferation of BCCs.
10 TET enzymes in BCC subsets
Abrogation of TET enzymes during early stages of BC is associated with poor patient prognosis [82]. Despite the standalone contribution of TET enzymes to BC progression, interplay between TET enzymes and histone modifiers is crucial for regulation of metastasis, cell cycle progression, and dormancy (Fig. 2c). For example, the loss of mixed-lineage leukemia 3 (MLL3), a mediator of H3K4 methylation at enhancers, disrupts the association between TET2 and ERα, resulting in a reduction of TET2 and inhibition of BCC proliferation [149]. In addition, the histone lysine demethylase 2A (KDM2A) has been shown to be a transcriptional repressor of TET2 by interacting with RelA, which inhibits tumor suppressor genes in BC such as epithelial cell adhesion molecule (EpCAM) and E-cadherin [150]. Moreover, EZH2, a member of the polycomb repressive complex 2 (PRC2), which represses transcription via methylation of histone H3 at lysine 27 (H3K27me3), is associated with poor patient prognosis. Notably, EZH2 can decrease TET1 expression, thereby preventing demethylation of the tumor suppressor gene TP53, which results in dysregulated BCC proliferation [151].
As mentioned above, hypoxia regulates tumor dormancy and cellular stemness. Specifically, hypoxia causes TET1 and TET3 enrichment, which is associated with CSC features [152]. Increased 5hmC levels upon hypoxia treatment induce TNF-α-p38-MAPK signaling, which is critical for maintaining a CSC phenotype and for survival of BCCs [152]. Furthermore, hypoxia drives hypermethylation of tumor suppressor genes and reduces function of TET enzymes [153]. TET1 has been shown to promote BCC progression by regulating genes involved in the PI3K/mTOR pathway, resulting in increased cellular migration and proliferation [154]. In addition to its oncogenic role, TET1 can act as a tumor suppressor by regulating the homeobox A gene, HOX9. To do so, TET1 autoregulates its promoter causing enhanced demethylation and enrichment of H3K4me3; as a result, TET1 interacts with HOX9 to restrict BCC growth [155]. Furthermore, TET1 modulates expression of tissue inhibitors of metalloproteinase 2 and 3 to suppress breast and prostate tumors [155]. Equivalently, loss of TET2 restricts BCC growth in an estrogen-dependent manner [48].
A mechanism by which tumor cells persist within a niche is via immune cells recruitment. However, a recent study demonstrated that p65-mediated downregulation of TET1 decreases expression of immune markers in BCCs, affecting recruitment of immune cells to the tumor microenvironment [156]. As mentioned above, vitamin C is a cofactor for TET activity. Interestingly, vitamin C promotes the enrichment of 5hmC inducing apoptosis in BCCs via TRAIL pathway [157]. These studies proposed TET-dependent mechanisms that halt tumor growth. Collectively, the interplay between DNA methylation and demethylation functions as a critical epigenetic program regulating CSCs.
11 DNA methylation in GBM
GBM, a grade IV astrocytoma, is one of the most aggressive brain tumors with patient survival being limited to only ~ 15 months after diagnosis. Currently, there is no effective therapy against GBM; thus, a deeper understanding of its development and progression is required to eradicate these oncogenic cells to increase patient lifespan. The fast progression of GBM could be explained by epigenetic dynamics favoring cellular plasticity, which allows adaptation to specific microenvironments. Hence, targeting of epigenetic mechanisms could serve as a plausible avenue for GBM treatment.
Tumor heterogeneity among GBM cells can arise as a result of alterations in DNA methylation patterns (Fig. 4) [158]. To further complicate the scheme of this cancer type, specific DNA methylation marks are observed in GBM CSCs. For instance, DNA methylation causing transcriptional repression of the tumor suppressor genes SPINT2, NEFM, and PENK2 are present in GBM CSCs [159]. Notably, overexpression of SPINT2 altered GBM growth resulting in decrease cellular stemness and proliferation, thereby restricting tumor progression [159]. Acquisition of a CSC phenotype by GBM progenitor cells occurs by increased expression of SOX2 and FOXG1 genes. Importantly, enhanced DNA methylation at the promoter region of downstream targets of SOX2:FOXG1 such as Foxo3 promotes self-renewal of GBM CSCs and facilitates cellular dormancy [160].

Regulation of DNA methylation in GBM increases susceptibility to TMZ. Upregulation of DNMT1 in GBM cells promotes increase of miRNA20a which confers resistance to treatment. Inhibition of DNMT1 downregulates expression of miRNA resulting in susceptibility to TMZ and cell death [164]
CD133, a membrane glycoprotein, has been used as a surface marker for the isolation of CSCs. However, in GBM progenitor cells, DNA methylation represses expression of CD133, resulting in increased recruitment of methyl-DNA binding proteins [161]. Similarly, DNA hypomethylation of alkaline phosphatase is observed in GBM patients and is associated with poor outcome [162]. Importantly, modulation of the methylome in GBM results in chemosensitivity to temozolomide (TMZ) (Fig. 4). Mechanistically, downregulation of DNMT1 reduced methylation of miR-20a and enhances sensitivity to TMZ (Fig. 4) [163]. In addition, DNMT1 cooperates with EZH2 to promote silencing of miR-200b/a/429 resulting in progression of GBM and gastric cancer [164]. Furthermore, GBM cells harbor increased methylation in apoptosis-related genes allowing their survival [165]. Interestingly, inhibition of DNMT3A by miR-129-5p results in cell cycle arrest, which affected GBM cell proliferation [166]. MiR152-3p has been reported to downregulate the expression of DNMT1, and as a result, expression of neurofibromatosis type 2 (NF2) is increased, allowing tumor suppression [167]. Collectively, these studies demonstrate the relevance of DNA methylation in the regulation of GBM.
12 DNA oxidation in GBM
Accumulation of 5hmC marks across the epigenome is a hallmark encountered in GBM. For instance, elevated levels of TET1 cause an increase of 5hmC in GBM cells, resulting in enhanced proliferation and tumorigenesis. As an epigenetic entity, 5hmC recruits CHTOP, a protein that targets arginine methyltransferases to promote transcriptional activation [64, 168]. Depletion of CHTOP caused a decrease in tumor formation and a reduction in global 5hmC levels. EGFR-mediated repression of TET1 results in downregulation of tumor suppressor genes, allowing survival of GBM [65]. Deposition of 5hmC at enhancer elements is associated with better prognosis [169]. Conversely, genome-wide reduction of 5hmC levels caused by downregulation of TET3 increases proliferation of GBM cells [170]. Concordantly, high expression of TET3 correlates with increased survival in GBM patients [170]. Furthermore, TLX, a nuclear receptor involved in initiation of brain tumors, facilitates CSCs self-renewal and proliferation rate of GBM [171]. Downregulation of TLX potentiates TET3, resulting in a reduction of tumor growth and inhibition of CSC self-renewal properties [172]. Interestingly, TET3 is variably expressed across CSCs derived from multiple cell lines, whereas TET1 and TET2 display downregulation and upregulation, respectively [173]. GBM CSCs show differential expression of HOX genes, which are implicated in survival and apoptotic and proliferative pathways in comparison to normal neural stem cells (NSCs) [173]. In addition, TET2 modulates TNFα signaling to sustain CSCs by providing chemoresistance through inhibition of pro-apoptotic pathways [174]. Indeed, TET2 can act as tumor suppressor by enhancing expression of miRNAs that restricts GBM differentiation [175]. Overall, DNA oxidations are key epigenetic components of GBM and much needs to be unraveled about their mechanisms involved in gene regulation.
In conclusion, modulation of the epigenome via DNA methylation/demethylation dynamics is an integral mechanism involved in cancer dormancy. Although dormant CSCs are present in both BC and GBM, the aggressiveness and resistance to therapy are quite different between these cancer types. However, the mechanisms underlying epigenetic modifications such as DNA methylation and DNA oxidations as drivers of cellular reprogramming towards dormant CSCs in BC versus GBM remain to be elucidated. For instance, the specific recruitment and genes targeted by individual members of the TET family of enzymes remain as major questions. Distinct roles for TET enzymes were described in embryonic stem cells (ESCs), in which TET1 is preferentially bound near transcription start sites, whereas TET2 is predominantly associated with gene bodies [176]. Nevertheless, it remains to be determined if this applies to CSCs. Additionally, differential expression levels among individual TETs could play relevant roles in the formation of dormant CSCs in BC versus GBM. Furthermore, the roles of each DNA oxidation, 5hmC, 5fC, and 5caC, generated by individual TET enzymes in the context of cancer dormancy, remain largely unexplored. Finally, it is unknown how the interplay between DNMTs and TETs in the context of specific histone modifications could impact cellular dormancy in BC and GBM. Therefore, a clear understanding of DNA methylation/oxidations as epigenetic determinants of cellular dormancy could sprout new methods for early diagnosis or therapeutic treatments to combat GBM, which is one of the most aggressive cancers exhibiting a high degree of therapeutic resistance.
References
Afifi, A. M., Saad, A. M., Al-Husseini, M. J., Elmehrath, A. O., Northfelt, D. W., & Sonbol, M. B. (2019). Causes of death after breast cancer diagnosis: a US population-based analysis. Cancer. https://doi.org/10.1002/cncr.32648.
Tamimi, A. F., & Juweid, M. (2017). Epidemiology and outcome of glioblastoma. In S. De Vleeschouwer (Ed.), Glioblastoma Brisbane (AU). Chapter 8, 143–154
Lathia, J. D., Mack, S. C., Mulkearns-Hubert, E. E., Valentim, C. L., & Rich, J. N. (2015). Cancer stem cells in glioblastoma. Genes & Development, 29(12), 1203–1217. https://doi.org/10.1101/gad.261982.115.
Omuro, A., & DeAngelis, L. M. (2013). Glioblastoma and other malignant gliomas: a clinical review. JAMA, 310(17), 1842–1850. https://doi.org/10.1001/jama.2013.280319.
Kahlert, U. D., Mooney, S. M., Natsumeda, M., Steiger, H. J., & Maciaczyk, J. (2017). Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. International Journal of Cancer, 140(1), 10–22. https://doi.org/10.1002/ijc.30259.
Han, H. R., Park, S. A., Ahn, S., Jeun, S. S., & Ryu, C. H. (2019). Evaluation of combination treatment effect with TRAIL-secreting mesenchymal stem cells and compound C against glioblastoma. Anticancer Research, 39(12), 6635–6643. https://doi.org/10.21873/anticanres.13878.
Pantel, K., Alix-Panabieres, C., & Riethdorf, S. (2009). Cancer micrometastases. Nature Reviews. Clinical Oncology, 6(6), 339–351. https://doi.org/10.1038/nrclinonc.2009.44.
Patel, S. A., Ramkissoon, S. H., Bryan, M., Pliner, L. F., Dontu, G., Patel, P. S., et al. (2012). Delineation of breast cancer cell hierarchy identifies the subset responsible for dormancy. Scientific Reports, 2, 906–906. https://doi.org/10.1038/srep00906.
Chrun, E. S., Modolo, F., & Daniel, F. I. (2017). Histone modifications: a review about the presence of this epigenetic phenomenon in carcinogenesis. Pathology, Research and Practice, 213(11), 1329–1339. https://doi.org/10.1016/j.prp.2017.06.013.
Ben-Porath, I., Thomson, M. W., Carey, V. J., Ge, R., Bell, G. W., Regev, A., et al. (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics, 40(5), 499–507. https://doi.org/10.1038/ng.127.
Suva, M. L., Riggi, N., & Bernstein, B. E. (2013). Epigenetic reprogramming in cancer. Science, 339(6127), 1567–1570. https://doi.org/10.1126/science.1230184.
Loh, K. M., & Lim, B. (2011). A precarious balance: pluripotency factors as lineage specifiers. Cell Stem Cell, 8(4), 363–369. https://doi.org/10.1016/j.stem.2011.03.013.
Thomson, M., Liu, S. J., Zou, L. N., Smith, Z., Meissner, A., & Ramanathan, S. (2011). Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell, 145(6), 875–889. https://doi.org/10.1016/j.cell.2011.05.017.
Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126(4), 663–676. https://doi.org/10.1016/j.cell.2006.07.024.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131(5), 861–872. https://doi.org/10.1016/j.cell.2007.11.019.
Iwafuchi-Doi, M., & Zaret, K. S. (2014). Pioneer transcription factors in cell reprogramming. Genes & Development, 28(24), 2679–2692. https://doi.org/10.1101/gad.253443.114.
Zhang, W., Bado, I., Wang, H., Lo, H. C., & Zhang, X. H. (2019). Bone metastasis: find your niche and fit in. Trends Cancer, 5(2), 95–110. https://doi.org/10.1016/j.trecan.2018.12.004.
Rowland, B. D., Bernards, R., & Peeper, D. S. (2005). The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nature Cell Biology, 7(11), 1074–1082. https://doi.org/10.1038/ncb1314.
Utikal, J., Polo, J. M., Stadtfeld, M., Maherali, N., Kulalert, W., Walsh, R. M., et al. (2009). Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature, 460(7259), 1145–1148. https://doi.org/10.1038/nature08285.
Hanna, J., Saha, K., Pando, B., van Zon, J., Lengner, C. J., Creyghton, M. P., et al. (2009). Direct cell reprogramming is a stochastic process amenable to acceleration. Nature, 462(7273), 595–601. https://doi.org/10.1038/nature08592.
Hochedlinger, K., & Jaenisch, R. (2015). Induced pluripotency and epigenetic reprogramming. Cold Spring Harbor Perspectives in Biology, 7(12). https://doi.org/10.1101/cshperspect.a019448.
De Carvalho, D. D., You, J. S., & Jones, P. A. (2010). DNA methylation and cellular reprogramming. Trends in Cell Biology, 20(10), 609–617. https://doi.org/10.1016/j.tcb.2010.08.003.
Gao, Y., Chen, J., Li, K., Wu, T., Huang, B., Liu, W., et al. (2013). Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell, 12(4), 453–469. https://doi.org/10.1016/j.stem.2013.02.005.
Sardina, J. L., Collombet, S., Tian, T. V., Gomez, A., Di Stefano, B., Berenguer, C., et al. (2018). Transcription factors drive Tet2-mediated enhancer demethylation to reprogram cell fate. Cell Stem Cell, 23(5), 727–741.e729. https://doi.org/10.1016/j.stem.2018.08.016.
Du, Q., Wang, Z., & Schramm, V. L. (2016). Human DNMT1 transition state structure. Proceedings of the National Academy of Sciences of the United States of America, 113(11), 2916–2921. https://doi.org/10.1073/pnas.1522491113.
Okashita, N., Kumaki, Y., Ebi, K., Nishi, M., Okamoto, Y., Nakayama, M., et al. (2014). PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development, 141(2), 269–280. https://doi.org/10.1242/dev.099622.
Bostick, M., Kim, J. K., Esteve, P. O., Clark, A., Pradhan, S., & Jacobsen, S. E. (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science, 317(5845), 1760–1764. https://doi.org/10.1126/science.1147939.
Stathopoulou, A., Chhetri, J. B., Ambrose, J. C., Esteve, P. O., Ji, L., Erdjument-Bromage, H., et al. (2017). A novel requirement for DROSHA in maintenance of mammalian CG methylation. Nucleic Acids Research, 45(16), 9398–9412. https://doi.org/10.1093/nar/gkx695.
Grosser, C., Wagner, N., Grothaus, K., & Horsthemke, B. (2015). Altering TET dioxygenase levels within physiological range affects DNA methylation dynamics of HEK293 cells. Epigenetics, 10(9), 819–833. https://doi.org/10.1080/15592294.2015.1073879.
Spruijt, C. G., Gnerlich, F., Smits, A. H., Pfaffeneder, T., Jansen, P. W., Bauer, C., et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell, 152(5), 1146–1159. https://doi.org/10.1016/j.cell.2013.02.004.
Liu, L., Mao, S. Q., Ray, C., Zhang, Y., Bell, F. T., Ng, S. F., et al. (2015). Differential regulation of genomic imprinting by TET proteins in embryonic stem cells. Stem Cell Research, 15(2), 435–443. https://doi.org/10.1016/j.scr.2015.08.010.
Skiles, W. M., Kester, A., Pryor, J. H., Westhusin, M. E., Golding, M. C., & Long, C. R. (2018). Oxygen-induced alterations in the expression of chromatin modifying enzymes and the transcriptional regulation of imprinted genes. Gene Expression Patterns, 28, 1–11. https://doi.org/10.1016/j.gep.2018.01.001.
Li, X. L., Xu, J. H., Nie, J. H., & Fan, S. J. (2012). Exogenous expression of UHRF1 promotes proliferation and metastasis of breast cancer cells. Oncology Reports, 28(1), 375–383. https://doi.org/10.3892/or.2012.1792.
Unoki, M., Brunet, J., & Mousli, M. (2009). Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochemical Pharmacology, 78(10), 1279–1288. https://doi.org/10.1016/j.bcp.2009.05.035.
Maenohara, S., Unoki, M., Toh, H., Ohishi, H., Sharif, J., Koseki, H., et al. (2017). Role of UHRF1 in de novo DNA methylation in oocytes and maintenance methylation in preimplantation embryos. PLoS Genetics, 13(10), e1007042. https://doi.org/10.1371/journal.pgen.1007042.
Gordon, C. A., Hartono, S. R., & Chedin, F. (2013). Inactive DNMT3B splice variants modulate de novo DNA methylation. PLoS One, 8(7), e69486. https://doi.org/10.1371/journal.pone.0069486.
Noh, K. M., Wang, H., Kim, H. R., Wenderski, W., Fang, F., Li, C. H., et al. (2015). Engineering of a histone-recognition domain in Dnmt3a alters the epigenetic landscape and phenotypic features of mouse ESCs. Molecular Cell, 59(1), 89–103. https://doi.org/10.1016/j.molcel.2015.05.017.
Jeltsch, A., & Jurkowska, R. Z. (2016). Allosteric control of mammalian DNA methyltransferases - a new regulatory paradigm. Nucleic Acids Research, 44(18), 8556–8575. https://doi.org/10.1093/nar/gkw723.
Duvall-Noelle, N., Karwandyar, A., Richmond, A., & Raman, D. (2016). LASP-1: a nuclear hub for the UHRF1-DNMT1-G9a-Snail1 complex. Oncogene, 35(9), 1122–1133. https://doi.org/10.1038/onc.2015.166.
Xue, B., Zhao, J., Feng, P., Xing, J., Wu, H., & Li, Y. (2019). Epigenetic mechanism and target therapy of UHRF1 protein complex in malignancies. Oncotargets and Therapy, 12, 549–559. https://doi.org/10.2147/ott.S192234.
Snowden, A. W., Gregory, P. D., Case, C. C., & Pabo, C. O. (2002). Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Current Biology, 12(24), 2159–2166. https://doi.org/10.1016/s0960-9822(02)01391-x.
Ko, M., An, J., Pastor, W. A., Koralov, S. B., Rajewsky, K., & Rao, A. (2015). TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunological Reviews, 263(1), 6–21. https://doi.org/10.1111/imr.12239.
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324(5929), 930–935. https://doi.org/10.1126/science.1170116.
Hu, X., Zhang, L., Mao, S. Q., Li, Z., Chen, J., Zhang, R. R., et al. (2014). Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell, 14(4), 512–522. https://doi.org/10.1016/j.stem.2014.01.001.
Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., et al. (2016). Editing DNA methylation in the mammalian genome. Cell, 167(1), 233–247.e217. https://doi.org/10.1016/j.cell.2016.08.056.
Wu, M. J., Kim, M. R., Chen, Y. S., Yang, J. Y., & Chang, C. J. (2017). Retinoic acid directs breast cancer cell state changes through regulation of TET2-PKCzeta pathway. Oncogene, 36(22), 3193–3206. https://doi.org/10.1038/onc.2016.467.
Xu, Y., Liu, S. Y., Li, J., Zhang, L., Chen, D., Zhang, J. P., et al. (2018). Real-time sensing of TET2-mediated DNA demethylation in vitro by metal-organic framework-based oxygen sensor for mechanism analysis and stem-cell behavior prediction. Analytical Chemistry, 90(15), 9330–9337. https://doi.org/10.1021/acs.analchem.8b01941.
Rasmussen, K. D., & Helin, K. (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes & Development, 30(7), 733–750. https://doi.org/10.1101/gad.276568.115.
Wang, L., Zhou, Y., Xu, L., Xiao, R., Lu, X., Chen, L., et al. (2015). Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex. Nature, 523(7562), 621–625. https://doi.org/10.1038/nature14482.
Kellinger, M. W., Song, C. X., Chong, J., Lu, X. Y., He, C., & Wang, D. (2012). 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nature Structural & Molecular Biology, 19(8), 831–833. https://doi.org/10.1038/nsmb.2346.
Globisch, D., Munzel, M., Muller, M., Michalakis, S., Wagner, M., Koch, S., et al. (2010). Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One, 5(12), e15367. https://doi.org/10.1371/journal.pone.0015367.
Zhang, L. Y., Han, C. S., Li, P. L., & Zhang, X. C. (2016). 5-Hydroxymethylcytosine expression is associated with poor survival in cervical squamous cell carcinoma. Japanese Journal of Clinical Oncology, 46(5), 427–434. https://doi.org/10.1093/jjco/hyw002.
Zhang, P., Huang, B., Xu, X., & Sessa, W. C. (2013). Ten-eleven translocation (Tet) and thymine DNA glycosylase (TDG), components of the demethylation pathway, are direct targets of miRNA-29a. Biochemical and Biophysical Research Communications, 437(3), 368–373. https://doi.org/10.1016/j.bbrc.2013.06.082.
Putiri, E. L., Tiedemann, R. L., Thompson, J. J., Liu, C., Ho, T., Choi, J. H., et al. (2014). Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biology, 15(6), R81. https://doi.org/10.1186/gb-2014-15-6-r81.
Iyer, L. M., Abhiman, S., & Aravind, L. (2011). Natural history of eukaryotic DNA methylation systems. Progress in Molecular Biology and Translational Science, 101, 25–104. https://doi.org/10.1016/b978-0-12-387685-0.00002-0.
Iyer, L. M., Tahiliani, M., Rao, A., & Aravind, L. (2009). Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle, 8(11), 1698–1710. https://doi.org/10.4161/cc.8.11.8580.
Ko, M., An, J., Bandukwala, H. S., Chavez, L., Aijo, T., Pastor, W. A., et al. (2013). Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature, 497(7447), 122–126. https://doi.org/10.1038/nature12052.
Nguyen, A. V., Albers, C. G., & Holcombe, R. F. (2010). Differentiation of tubular and villous adenomas based on Wnt pathway-related gene expression profiles. International Journal of Molecular Medicine, 26(1), 121–125. https://doi.org/10.3892/ijmm_00000443.
Zhang, P., Rausch, C., Hastert, F. D., Boneva, B., Filatova, A., Patil, S. J., et al. (2017). Methyl-CpG binding domain protein 1 regulates localization and activity of Tet1 in a CXXC3 domain-dependent manner. Nucleic Acids Research, 45(12), 7118–7136. https://doi.org/10.1093/nar/gkx281.
Ludwig, A. K., Zhang, P., Hastert, F. D., Meyer, S., Rausch, C., Herce, H. D., et al. (2017). Binding of MBD proteins to DNA blocks Tet1 function thereby modulating transcriptional noise. Nucleic Acids Research, 45(5), 2438–2457. https://doi.org/10.1093/nar/gkw1197.
Rausch, C., Hastert, F. D., & Cardoso, M. C. (2019). DNA modification readers and writers and their interplay. Journal of Molecular Biology. https://doi.org/10.1016/j.jmb.2019.12.018.
Li, L., Chen, B. F., & Chan, W. Y. (2015). An epigenetic regulator: methyl-CpG-binding domain protein 1 (MBD1). International Journal of Molecular Sciences, 16(3), 5125–5140. https://doi.org/10.3390/ijms16035125.
Wood, K. H., & Zhou, Z. (2016). Emerging molecular and biological functions of MBD2, a reader of DNA methylation. Frontiers in Genetics, 7, 93. https://doi.org/10.3389/fgene.2016.00093.
Takai, H., Masuda, K., Sato, T., Sakaguchi, Y., Suzuki, T., Suzuki, T., et al. (2014). 5-Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP-methylosome complex. Cell Reports, 9(1), 48–60. https://doi.org/10.1016/j.celrep.2014.08.071.
Forloni, M., Gupta, R., Nagarajan, A., Sun, L. S., Dong, Y., Pirazzoli, V., et al. (2016). Oncogenic EGFR represses the TET1 DNA demethylase to induce silencing of tumor suppressors in cancer cells. Cell Reports, 16(2), 457–471. https://doi.org/10.1016/j.celrep.2016.05.087.
Good, C. R., Madzo, J., Patel, B., Maegawa, S., Engel, N., Jelinek, J., et al. (2017). A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Research, 45(14), 8269–8281. https://doi.org/10.1093/nar/gkx435.
Inoue-Choi, M., Nelson, H. H., Robien, K., Arning, E., Bottiglieri, T., Koh, W. P., et al. (2013). Plasma S-adenosylmethionine, DNMT polymorphisms, and peripheral blood LINE-1 methylation among healthy Chinese adults in Singapore. BMC Cancer, 13, 389. https://doi.org/10.1186/1471-2407-13-389.
Kohli, R. M., & Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature, 502(7472), 472–479. https://doi.org/10.1038/nature12750.
Cavuoto, P., & Fenech, M. F. (2012). A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treatment Reviews, 38(6), 726–736. https://doi.org/10.1016/j.ctrv.2012.01.004.
Soda, K. (2018). Polyamine metabolism and gene methylation in conjunction with one-carbon metabolism. International Journal of Molecular Sciences, 19(10). https://doi.org/10.3390/ijms19103106.
Mahmoud, A. M., & Ali, M. M. (2019). Methyl donor micronutrients that modify DNA methylation and cancer outcome. Nutrients, 11(3). https://doi.org/10.3390/nu11030608.
Gut, P., & Verdin, E. (2013). The nexus of chromatin regulation and intermediary metabolism. Nature, 502(7472), 489–498. https://doi.org/10.1038/nature12752.
Kaelin Jr., W. G., & McKnight, S. L. (2013). Influence of metabolism on epigenetics and disease. Cell, 153(1), 56–69. https://doi.org/10.1016/j.cell.2013.03.004.
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H., et al. (2012). Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & Development, 26(12), 1326–1338. https://doi.org/10.1101/gad.191056.112.
Xu, Y., Wu, F., Tan, L., Kong, L., Xiong, L., Deng, J., et al. (2011). Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Molecular Cell, 42(4), 451–464. https://doi.org/10.1016/j.molcel.2011.04.005.
Flavahan, W. A., Drier, Y., Liau, B. B., Gillespie, S. M., Venteicher, A. S., Stemmer-Rachamimov, A. O., et al. (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature, 529(7584), 110–114. https://doi.org/10.1038/nature16490.
Cimmino, L., Neel, B. G., & Aifantis, I. (2018). Vitamin C in stem cell reprogramming and cancer. Trends in Cell Biology, 28(9), 698–708. https://doi.org/10.1016/j.tcb.2018.04.001.
Aoki, K., & Natsume, A. (2019). Overview of DNA methylation in adult diffuse gliomas. Brain Tumor Pathology, 36(2), 84–91. https://doi.org/10.1007/s10014-019-00339-w.
Bouras, E., Karakioulaki, M., Bougioukas, K. I., Aivaliotis, M., Tzimagiorgis, G., & Chourdakis, M. (2019). Gene promoter methylation and cancer: an umbrella review. Gene, 710, 333–340. https://doi.org/10.1016/j.gene.2019.06.023.
Roll, J. D., Rivenbark, A. G., Sandhu, R., Parker, J. S., Jones, W. D., Carey, L. A., et al. (2013). Dysregulation of the epigenome in triple-negative breast cancers: basal-like and claudin-low breast cancers express aberrant DNA hypermethylation. Experimental and Molecular Pathology, 95(3), 276–287. https://doi.org/10.1016/j.yexmp.2013.09.001.
Phuong, N. T., Kim, S. K., Lim, S. C., Kim, H. S., Kim, T. H., Lee, K. Y., et al. (2011). Role of PTEN promoter methylation in tamoxifen-resistant breast cancer cells. Breast Cancer Research and Treatment, 130(1), 73–83. https://doi.org/10.1007/s10549-010-1304-2.
Yang, L., Yu, S. J., Hong, Q., Yang, Y., & Shao, Z. M. (2015). Reduced expression of TET1, TET2, TET3 and TDG mRNAs are associated with poor prognosis of patients with early breast cancer. PLoS One, 10(7), e0133896. https://doi.org/10.1371/journal.pone.0133896.
Thienpont, B., Galle, E., & Lambrechts, D. (2016). TET enzymes as oxygen-dependent tumor suppressors: exciting new avenues for cancer management. Epigenomics, 8(11), 1445–1448. https://doi.org/10.2217/epi-2016-0126.
Kao, S. H., Wu, K. J., & Lee, W. H. (2016). Hypoxia, epithelial-mesenchymal transition, and TET-mediated epigenetic changes. Journal of Clinical Medicine, 5(2). https://doi.org/10.3390/jcm5020024.
Adam, J., Yang, M., Soga, T., & Pollard, P. J. (2014). Rare insights into cancer biology. Oncogene, 33(20), 2547–2556. https://doi.org/10.1038/onc.2013.222.
Laukka, T., Mariani, C. J., Ihantola, T., Cao, J. Z., Hokkanen, J., Kaelin Jr., W. G., et al. (2016). Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. The Journal of Biological Chemistry, 291(8), 4256–4265. https://doi.org/10.1074/jbc.M115.688762.
Liu, Y., Jiang, W., Liu, J., Zhao, S., Xiong, J., Mao, Y., et al. (2012). IDH1 mutations inhibit multiple alpha-ketoglutarate-dependent dioxygenase activities in astroglioma. Journal of Neuro-Oncology, 109(2), 253–260. https://doi.org/10.1007/s11060-012-0914-4.
Agnihotri, S., Aldape, K. D., & Zadeh, G. (2014). Isocitrate dehydrogenase status and molecular subclasses of glioma and glioblastoma. Neurosurgical Focus, 37(6), E13. https://doi.org/10.3171/2014.9.Focus14505.
Budczies, J., & Denkert, C. (2016). Tissue-based metabolomics to analyze the breast cancer metabolome. Recent Results in Cancer Research, 207, 157–175. https://doi.org/10.1007/978-3-319-42118-6_7.
Hervouet, E., Peixoto, P., Delage-Mourroux, R., Boyer-Guittaut, M., & Cartron, P. F. (2018). Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clinical Epigenetics, 10, 17. https://doi.org/10.1186/s13148-018-0450-y.
Inoue, S., Li, W. Y., Tseng, A., Beerman, I., Elia, A. J., Bendall, S. C., et al. (2016). Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell, 30(2), 337–348. https://doi.org/10.1016/j.ccell.2016.05.018.
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. The New England Journal of Medicine, 360(8), 765–773. https://doi.org/10.1056/NEJMoa0808710.
Muller, T., Gessi, M., Waha, A., Isselstein, L. J., Luxen, D., Freihoff, D., et al. (2012). Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. The American Journal of Pathology, 181(2), 675–683. https://doi.org/10.1016/j.ajpath.2012.04.017.
Turcan, S., Fabius, A. W., Borodovsky, A., Pedraza, A., Brennan, C., Huse, J., et al. (2013). Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT inhibitor decitabine. Oncotarget, 4(10), 1729–1736. https://doi.org/10.18632/oncotarget.1412.
Zhou, Z., Li, H. Q., & Liu, F. (2018). DNA methyltransferase inhibitors and their therapeutic potential. Current Topics in Medicinal Chemistry, 18(28), 2448–2457. https://doi.org/10.2174/1568026619666181120150122.
Chen, A., Sceneay, J., Godde, N., Kinwel, T., Ham, S., Thompson, E. W., et al. (2018). Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene, 37(31), 4214–4225. https://doi.org/10.1038/s41388-018-0259-3.
Flavahan, W. A., Gaskell, E., & Bernstein, B. E. (2017). Epigenetic plasticity and the hallmarks of cancer. Science, 357(6348). https://doi.org/10.1126/science.aal2380.
Prasad, P., Mittal, S. A., Chongtham, J., Mohanty, S., & Srivastava, T. (2017). Hypoxia-mediated epigenetic regulation of stemness in brain tumor cells. Stem Cells, 35(6), 1468–1478. https://doi.org/10.1002/stem.2621.
Lapidot, T., Sirard, C., Vormoor, J., Murdoch, B., Hoang, T., Caceres-Cortes, J., et al. (1994). A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature, 367(6464), 645–648. https://doi.org/10.1038/367645a0.
Reya, T., Morrison, S. J., Clarke, M. F., & Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature, 414(6859), 105–111. https://doi.org/10.1038/35102167.
Giordano, A., Gao, H., Cohen, E. N., Anfossi, S., Khoury, J., Hess, K., et al. (2013). Clinical relevance of cancer stem cells in bone marrow of early breast cancer patients. Annals of oncology : official journal of the European Society for Medical Oncology, 24(10), 2515–2521. https://doi.org/10.1093/annonc/mdt223.
Bu, Y., & Cao, D. (2012). The origin of cancer stem cells. Frontiers in Bioscience (Scholar Edition), 4, 819–830. https://doi.org/10.2741/s302.
Prieto-Vila, M., Takahashi, R.-U., Usuba, W., Kohama, I., & Ochiya, T. (2017). Drug resistance driven by cancer stem cells and their niche. International Journal of Molecular Sciences, 18(12). https://doi.org/10.3390/ijms18122574.
Lobo, N. A., Shimono, Y., Qian, D., & Clarke, M. F. (2007). The biology of cancer stem cells. Annual Review of Cell and Developmental Biology, 23(1), 675–699. https://doi.org/10.1146/annurev.cellbio.22.010305.104154.
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., & Clarke, M.F. F. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA, 100(7), 3983–3988
Hwang-Verslues, W. W., Kuo, W.-H., Chang, P.-H., Pan, C.-C., Wang, H.-H., Tsai, S.-T., et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem. Cell Markers. PLoS ONE, 4(12), 8377–8377. https://doi.org/10.1371/journal.pone.0008377.
Figueiredo, E., Zaghloul, K., Zhang, Y., Tan, S. T., Bradshaw, A., Wickremsekera, A., et al. (2016). Cancer stem cell hierarchy in glioblastoma multiforme. 3, 2, doi:https://doi.org/10.3389/fsurg.2016.00021.
Bliss, S. A., Sinha, G., Sandiford, O. A., Williams, L. M., Engelberth, D. J., Guiro, K., et al. (2016). Mesenchymal stem cell–derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Research, 76(19), 5832–5844. https://doi.org/10.1158/0008-5472.CAN-16-1092.
Mani, S. A., Guo, W., Liao, M.-J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 133(4), 704–715. https://doi.org/10.1016/j.cell.2008.03.027.
Sell, S. (2004). Stem cell origin of cancer and differentiation therapy. Critical Reviews in Oncology/Hematology, 51(1), 1–28. https://doi.org/10.1016/J.CRITREVONC.2004.04.007.
Wicha, M. S., Liu, S., & Dontu, G. (2006). Cancer stem cells: an old idea-A paradigm shift. Cancer Research, 66(4), 1883–1890. https://doi.org/10.1158/0008-5472.CAN-05-3153.
Marsden, C. G., Wright, M. J., Carrier, L., Moroz, K., & Rowan, B. G. (2012). Disseminated breast cancer cells acquire a highly malignant and aggressive metastatic phenotype during metastatic latency in the bone. PLoS One, 7(11), e47587–e47587. https://doi.org/10.1371/journal.pone.0047587.
Shiozawa, Y., & Taichman, R. S. (2012). Cancer stem cells and the bone marrow microenvironment. Bonekey Reports, 2012(1). https://doi.org/10.1038/bonekey.2012.48.
Birbrair, A., & Frenette, P. S. (2016). Niche heterogeneity in the bone marrow. Annals of the New York Academy of Sciences, 1370(1), 82–96. https://doi.org/10.1111/nyas.13016.
Morrison, S. J., & Scadden, D. T. (2014). The bone marrow niche for haematopoietic stem cells. Nature, 505(7483), 327–334. https://doi.org/10.1038/nature12984.
Yu, V. W., & Scadden, D. T. (2016). Hematopoietic stem cell and its bone marrow niche. Current Topics in Developmental Biology, 118, 21–44. https://doi.org/10.1016/bs.ctdb.2016.01.009.
Guerra, D. A. P., Paiva, A. E., Sena, I. F. G., Azevedo, P. O., Batista Jr., M. L., Mintz, A., et al. (2018). Adipocytes role in the bone marrow niche. Cytometry. Part A, 93(2), 167–171. https://doi.org/10.1002/cyto.a.23301.
Rameshwar, P. (2010). Breast cancer cell dormancy in bone marrow: potential therapeutic targets within the marrow microenvironment. Expert Review of Anticancer Therapy, 10(2), 129–132. https://doi.org/10.1586/era.10.3.
Eltoukhy, H. S., Sinha, G., Moore, C. A., Gergues, M., & Rameshwar, P. (2018). Secretome within the bone marrow microenvironment: a basis for mesenchymal stem cell treatment and role in cancer dormancy. Biochimie, 155, 92–103. https://doi.org/10.1016/j.biochi.2018.05.018.
Ridge, S. M., Sullivan, F. J., & Glynn, S. A. (2017). Mesenchymal stem cells: key players in cancer progression. Molecular Cancer, 16(1), 31. https://doi.org/10.1186/s12943-017-0597-8.
Ruivo, C. F., Adem, B., Silva, M., & Melo, S. A. (2017). The biology of cancer exosomes: insights and new perspectives. Cancer Research, 77(23), 6480–6488. https://doi.org/10.1158/0008-5472.CAN-17-0994.
Walker, N. D., Elias, M., Guiro, K., Bhatia, R., Greco, S. J., Bryan, M., et al. (2019). Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death & Disease, 10(2), 59. https://doi.org/10.1038/s41419-019-1304-z.
Wu, J. I., & Wang, L. H. (2019). Emerging roles of gap junction proteins connexins in cancer metastasis, chemoresistance and clinical application. Journal of Biomedical Science, 26(1), 8. https://doi.org/10.1186/s12929-019-0497-x.
Zefferino, R., Piccoli, C., Gioia, S. D., Capitanio, N., & Conese, M. (2019). Gap junction intercellular communication in the carcinogenesis hallmarks: is this a phenomenon or epiphenomenon? Cells, 8(8). https://doi.org/10.3390/cells8080896.
Sinyuk, M., Mulkearns-Hubert, E. E., Reizes, O., & Lathia, J. (2018). Cancer connectors: connexins, gap junctions, and communication. Frontiers in Oncology, 8, 646. https://doi.org/10.3389/fonc.2018.00646.
Busby, M., Hallett, M. T., & Plante, I. (2018). The complex subtype-dependent role of connexin 43 (GJA1) in breast cancer. International Journal of Molecular Sciences, 19(3). https://doi.org/10.3390/ijms19030693.
Oliveira, R., Christov, C., Guillamo, J. S., de Bouard, S., Palfi, S., Venance, L., et al. (2005). Contribution of gap junctional communication between tumor cells and astroglia to the invasion of the brain parenchyma by human glioblastomas. BMC Cell Biology, 6(1), 7. https://doi.org/10.1186/1471-2121-6-7.
Munoz, J. L., Bliss, S. A., Greco, S. J., Ramkissoon, S. H., Ligon, K. L., & Rameshwar, P. (2013). Delivery of functional anti-miR-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Molecular Therapy--Nucleic Acids, 2, e126. https://doi.org/10.1038/mtna.2013.60.
Park, J. M., Munoz, J. L., Won, B. W., Bliss, S. A., Greco, S. J., Patel, S. A., et al. (2013). Exogenous CXCL12 activates protein kinase C to phosphorylate connexin 43 for gap junctional intercellular communication among confluent breast cancer cells. Cancer Letters, 331(1), 84–91. https://doi.org/10.1016/j.canlet.2012.12.007.
Poddar, S., Kesharwani, D., & Datta, M. (2017). Interplay between the miRNome and the epigenetic machinery: implications in health and disease. Journal of Cellular Physiology, 232(11), 2938–2945. https://doi.org/10.1002/jcp.25819.
Piletic, K., & Kunej, T. (2016). MicroRNA epigenetic signatures in human disease. Archives of Toxicology, 90(10), 2405–2419. https://doi.org/10.1007/s00204-016-1815-7.
Tu, J., Ng, S. H., Luk, A. C., Liao, J., Jiang, X., Feng, B., et al. (2015). MicroRNA-29b/Tet1 regulatory axis epigenetically modulates mesendoderm differentiation in mouse embryonic stem cells. Nucleic Acids Research, 43(16), 7805–7822. https://doi.org/10.1093/nar/gkv653.
Iwama, H., Kato, K., Imachi, H., Murao, K., & Masaki, T. (2018). Human microRNAs preferentially target genes with intermediate levels of expression and its formation by mammalian evolution. PLoS One, 13(5), e0198142. https://doi.org/10.1371/journal.pone.0198142.
Liu, X., Chen, X., Yu, X., Tao, Y., Bode, A. M., Dong, Z., et al. (2013). Regulation of microRNAs by epigenetics and their interplay involved in cancer. Journal of Experimental & Clinical Cancer Research, 32, 96. https://doi.org/10.1186/1756-9966-32-96.
Daniunaite, K., Dubikaityte, M., Gibas, P., Bakavicius, A., Rimantas Lazutka, J., Ulys, A., et al. (2017). Clinical significance of miRNA host gene promoter methylation in prostate cancer. Human Molecular Genetics, 26(13), 2451–2461. https://doi.org/10.1093/hmg/ddx138.
Pathania, R. a. R. S. a. E. S. a. P. R. a. Y. P. a. C. S. a. V.-K. R. a. A. P. a. G.-P. J. P. a. S. (2015). DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nature Communications, 6(1), 6910. https://doi.org/10.1038/ncomms7910.
Oltra, S. S., Peña-Chilet, M., Flower, K., Martinez, M. T., Alonso, E., Burgues, O., et al. (2019). Acceleration in the DNA methylation age in breast cancer tumours from very young women. Scic Rep, 9(1). https://doi.org/10.1038/s41598-019-51457-6.
El Helou, R., Wicinski, J., Guille, A., Adélaïde, J., Finetti, P., Bertucci, F., et al. (2014). Brief reports: a distinct DNA methylation signature defines breast cancer stem cells and predicts cancer outcome. Stem Cells, 32(11), 3031–3036. https://doi.org/10.1002/stem.1792.
Taube, J. H., Malouf, G. G., Lu, E., Sphyris, N., Vijay, V., Ramachandran, P. P., et al. (2013). Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Scientific Reports, 3, 2687. https://doi.org/10.1038/srep02687.
Neves, R., Scheel, C., Weinhold, S., Honisch, E., Iwaniuk, K. M., Trompeter, H. I., et al. (2010). Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Research Notes, 3, 219. https://doi.org/10.1186/1756-0500-3-219.
Nass, S. J., Herman, J. G., Gabrielson, E., Iversen, P. W., Parl, F. F., Davidson, N. E., et al. (2000). Aberrant methylation of the estrogen receptor and E-cadherin 5′ CpG islands increases with malignant progression in human breast cancer. Cancer Research, 60(16), 4346–4348.
Joo, J. E., Dowty, J. G., Milne, R. L., Wong, E. M., Dugue, P. A., English, D., et al. (2018). Heritable DNA methylation marks associated with susceptibility to breast cancer. Nature Communications, 9(1), 867. https://doi.org/10.1038/s41467-018-03058-6.
Fleischer, T., Tekpli, X., Mathelier, A., Wang, S., Nebdal, D., Dhakal, H. P., et al. (2017). DNA methylation at enhancers identifies distinct breast cancer lineages. Nature Communications, 8, 1379. https://doi.org/10.1038/s41467-017-00510-x.
Hernandez-Vargas, H., Ouzounova, M., Le Calvez-Kelm, F., Lambert, M. P., McKay-Chopin, S., Tavtigian, S. V., et al. (2011). Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics, 6(4), 428–439. https://doi.org/10.4161/epi.6.4.14515.
Chimonidou, M., Strati, A., Tzitzira, A., Sotiropoulou, G., Malamos, N., Georgoulias, V., et al. (2011). DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clinical Chemistry, 57(8), 1169–1177. https://doi.org/10.1373/clinchem.2011.165902.
Darvin, P., Sasidharan Nair, V., & Elkord, E. (2019). PD-L1 expression in human breast cancer stem cells is epigenetically regulated through posttranslational histone modifications. Journal of Oncology, 2019, 3958908. https://doi.org/10.1155/2019/3958908.
Rankin, E. B., & Giaccia, A. J. (2008). The role of hypoxia-inducible factors in tumorigenesis. Cell Death and Differentiation, 15(4), 678–685. https://doi.org/10.1038/cdd.2008.21.
Guo, C., Chen, L. H., Huang, Y., Chang, C. C., Wang, P., Pirozzi, C. J., et al. (2013). KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget, 4(11), 2144–2153. https://doi.org/10.18632/oncotarget.1555.
Wang, L., Ozark, P. A., Smith, E. R., Zhao, Z., Marshall, S. A., Rendleman, E. J., et al. (2018). TET2 coactivates gene expression through demethylation of enhancers. Science Advances, 4(11), eaau6986. https://doi.org/10.1126/sciadv.aau6986.
Chen, J. Y., Luo, C. W., Lai, Y. S., Wu, C. C., & Hung, W. C. (2017). Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer. Oncogenesis, 6(8), e369. https://doi.org/10.1038/oncsis.2017.71.
Yu, Y., Qi, J., Xiong, J., Jiang, L., Cui, D., He, J., et al. (2019). Epigenetic co-deregulation of EZH2/TET1 is a senescence-countering, actionable vulnerability in triple-negative breast cancer. Theranostics, 9(3), 761–777. https://doi.org/10.7150/thno.29520.
Wu, M. Z., Chen, S. F., Nieh, S., Benner, C., Ger, L. P., Jan, C. I., et al. (2015). Hypoxia drives breast tumor malignancy through a TET-TNFalpha-p38-MAPK signaling axis. Cancer Research, 75(18), 3912–3924. https://doi.org/10.1158/0008-5472.CAN-14-3208.
Thienpont, B., Steinbacher, J., Zhao, H., D’Anna, F., Kuchnio, A., Ploumakis, A., et al. (2016). Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature, 537(7618), 63–68. https://doi.org/10.1038/nature19081.
Ryan Good, C., Panjarian, S., Kelly, A. D., Madzo, J., Patel, B., Jelinek, J., et al. (2018). Genome and epigenome TET1-mediated hypomethylation activates oncogenic signaling in triple-negative. Breast Cancer. https://doi.org/10.1158/0008-5472.CAN-17-2082.
Hsu, C. H., Peng, K. L., Kang, M. L., Chen, Y. R., Yang, Y. C., Tsai, C. H., et al. (2012). TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Reports, 2(3), 568–579. https://doi.org/10.1016/j.celrep.2012.08.030.
Collignon, E., Canale, A., Al Wardi, C., Bizet, M., Calonne, E., Dedeurwaerder, S., et al. (2018). Immunity drives TET1 regulation in cancer through NF-kappaB. Science Advances, 4(6), eaap7309. https://doi.org/10.1126/sciadv.aap7309.
Sant, D. W., Mustafi, S., Gustafson, C. B., Chen, J., Slingerland, J. M., & Wang, G. (2018). Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Scientific Reports, 8(1), 5306. https://doi.org/10.1038/s41598-018-23714-7.
Klughammer, J., Kiesel, B., Roetzer, T., Fortelny, N., Nemc, A., Nenning, K. H., et al. (2018). The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nature Medicine, 24(10), 1611–1624. https://doi.org/10.1038/s41591-018-0156-x.
Lee, E. J., Rath, P., Liu, J., Ryu, D., Pei, L., Noonepalle, S. K., et al. (2015). Identification of global DNA methylation signatures in glioblastoma-derived cancer stem cells. Journal of Genetics and Genomics, 42(7), 355–371. https://doi.org/10.1016/j.jgg.2015.06.003.
Bulstrode, H., Johnstone, E., Marques-Torrejon, M. A., Ferguson, K. M., Bressan, R. B., Blin, C., et al. (2017). Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes & Development, 31(8), 757–773. https://doi.org/10.1101/gad.293027.116.
Gopisetty, G., Xu, J., Sampath, D., Colman, H., & Puduvalli, V. K. (2013). Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by Sp1/myc and promoter methylation. Oncogene, 32(26), 3119–3129. https://doi.org/10.1038/onc.2012.331.
Iwadate, Y., Suganami, A., Tamura, Y., Matsutani, T., Hirono, S., Shinozaki, N., et al. (2017). The pluripotent stem-cell marker alkaline phosphatase is highly expressed in refractory glioblastoma with DNA hypomethylation. Neurosurgery, 80(2), 248–256. https://doi.org/10.1093/neuros/nyw026.
Zhou, D., Wan, Y., Xie, D., Wang, Y., Wei, J., Yan, Q., et al. (2015). DNMT1 mediates chemosensitivity by reducing methylation of miRNA-20a promoter in glioma cells. Experimental & Molecular Medicine, 47, e182. https://doi.org/10.1038/emm.2015.57.
Ning, X., Shi, Z., Liu, X., Zhang, A., Han, L., Jiang, K., et al. (2015). DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Letters, 359(2), 198–205. https://doi.org/10.1016/j.canlet.2015.01.005.
Hervouet, E., Vallette, F. M., & Cartron, P. F. (2010). Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death & Disease, 1, e8. https://doi.org/10.1038/cddis.2009.7.
Gu, X., Gong, H., Shen, L., & Gu, Q. (2018). MicroRNA-129-5p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting DNMT3A. American Journal of Translational Research, 10(9), 2834–2847.
Sun, J., Tian, X., Zhang, J., Huang, Y., Lin, X., Chen, L., et al. (2017). Regulation of human glioma cell apoptosis and invasion by miR-152-3p through targeting DNMT1 and regulating NF2 : MiR-152-3p regulate glioma cell apoptosis and invasion. Journal of Experimental & Clinical Cancer Research, 36(1), 100. https://doi.org/10.1186/s13046-017-0567-4.
Izumikawa, K., Ishikawa, H., Simpson, R. J., & Takahashi, N. (2018). Modulating the expression of Chtop, a versatile regulator of gene-specific transcription and mRNA export. RNA Biology, 15(7), 849–855. https://doi.org/10.1080/15476286.2018.1465795.
Johnson, K. C., Houseman, E. A., King, J. E., von Herrmann, K. M., Fadul, C. E., & Christensen, B. C. (2016). 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nature Communications, 7, 13177. https://doi.org/10.1038/ncomms13177.
Carella, A., Tejedor, J. R., García, M. G., Urdinguio, R. G., Bayón, G. F., Sierra, M., et al. (2020). Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. International Journal of Cancer, 146(2), 373–387. https://doi.org/10.1002/ijc.32520.
Qu, Q., Sun, G., Li, W., Yang, S., Ye, P., Zhao, C., et al. (2010). Orphan nuclear receptor TLX activates Wnt/Β-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nature Cell Biology, 12(1), 31–40. https://doi.org/10.1038/ncb2001.
Cui, Q., Yang, S., Ye, P., Tian, E., Sun, G., Zhou, J., et al. (2016). Downregulation of TLX induces TET3 expression and inhibits glioblastoma stem cell self-renewal and tumorigenesis. Nature Communications, 7, 10637. https://doi.org/10.1038/ncomms10637.
Zhou, D., Alver, B. M., Li, S., Hlady, R. A., Thompson, J. J., Schroeder, M. A., et al. (2018). Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biology, 19(1), 43–43. https://doi.org/10.1186/s13059-018-1420-6.
Puig, I., Tenbaum, S. P., Chicote, I., Arques, O., Martinez-Quintanilla, J., Cuesta-Borras, E., et al. (2018). TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. The Journal of Clinical Investigation, 128(9), 3887–3905. https://doi.org/10.1172/jci96393.
Garcia, M. G., Carella, A., Urdinguio, R. G., Bayon, G. F., Lopez, V., Tejedor, J. R., et al. (2018). Epigenetic dysregulation of TET2 in human glioblastoma. Oncotarget, 9(40), 25922–25934. https://doi.org/10.18632/oncotarget.25406.
Huang, Y., Chavez, L., Chang, X., Wang, X., Pastor, W. A., Kang, J., et al. (2014). Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 111(4), 1361–1366. https://doi.org/10.1073/pnas.1322921111.
Xu, Y., Chao, L., Wang, J., & Sun, Y. (2017). miRNA-148a regulates the expression of the estrogen receptor through DNMT1-mediated DNA methylation in breast cancer cells. Oncology Letters, 14(4), 4736–4740. https://doi.org/10.3892/ol.2017.6803.
Cho, J. H., Dimri, M., & Dimri, G. P. (2015). MicroRNA-31 is a transcriptional target of histone deacetylase inhibitors and a regulator of cellular senescence. The Journal of Biological Chemistry, 290(16), 10555–10567. https://doi.org/10.1074/jbc.M114.624361.
Song, J., Jin, E. H., Kim, D., Kim, K. Y., Chun, C. H., & Jin, E. J. (2015). MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clinical, 3, 79–89. https://doi.org/10.1016/j.bbacli.2014.11.009.
Jiang, S. (2019). MicroRNA Let-7adf in Tet regulation. Aging, 11(14), 4772–4773. https://doi.org/10.18632/aging.102101.
Fu, X., Jin, L., Wang, X., Luo, A., Hu, J., Zheng, X., et al. (2013). MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proceedings of the National Academy of Sciences of the United States of America, 110(44), 17892–17897. https://doi.org/10.1073/pnas.1317397110.
Cheng, J., Guo, S., Chen, S., Mastriano, S. J., Liu, C., D’Alessio, A. C., et al. (2013). An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Reports, 5(2), 471–481. https://doi.org/10.1016/j.celrep.2013.08.050.
Yang, H., Li, Q., Zhao, W., Yuan, D., Zhao, H., & Zhou, Y. (2014). miR-329 suppresses the growth and motility of neuroblastoma by targeting KDM1A. FEBS Letters, 588(1), 192–197. https://doi.org/10.1016/j.febslet.2013.11.036.
Nilsson, E. M., Laursen, K. B., Whitchurch, J., McWilliam, A., Odum, N., Persson, J. L., et al. (2015). MiR137 is an androgen regulated repressor of an extended network of transcriptional coregulators. Oncotarget, 6(34), 35710–35725. https://doi.org/10.18632/oncotarget.5958.
Bao, J., Zou, J. H., Li, C. Y., & Zheng, G. Q. (2016). miR-194 inhibits gastric cancer cell proliferation and tumorigenesis by targeting KDM5B. European Review for Medical and Pharmacological Sciences, 20(21), 4487–4493.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ferrer, A.I., Trinidad, J.R., Sandiford, O. et al. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev 39, 721–738 (2020). https://doi.org/10.1007/s10555-020-09882-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-020-09882-x