
Abstract
Pediatric solid tumors have long been known to shed tumor cells, DNA, RNA, and proteins into the blood. Recent technological advances have allowed for improved capture and analysis of these typically scant circulating materials. Efforts are ongoing to develop “liquid biopsy” assays as minimally invasive tools to address diagnostic, prognostic, and disease monitoring needs in childhood cancer care. Applying these highly sensitive technologies to serial liquid biopsies is expected to advance understanding of tumor biology, heterogeneity, and evolution over the course of therapy, thus opening new avenues for personalized therapy. In this review, we outline the latest technologies available for liquid biopsies and describe the methods, pitfalls, and benefits of the assays that are being developed for children with extracranial solid tumors. We discuss what has been learned in several of the most common pediatric solid tumors including neuroblastoma, sarcoma, Wilms tumor, and hepatoblastoma and highlight promising future directions for the field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Cancer is a group of diseases that arises from genetic inheritance, environmental factors, or accumulated errors in DNA replication that result in uncontrolled cell proliferation [1]. Of these diseases, pediatric solid tumors often have particularly devastating outcomes. Fortunately, increasing knowledge about childhood cancer biology and tumorigenesis has enabled advances in chemotherapy, supportive care, and personalized medicine which have improved outcomes. However, it has become apparent that intensive treatments can negatively impact survivors’ quality of life [2,3,4,5,6,7,8,9] and that therapy for children with solid tumors should be optimally tailored to each child to maximize the likelihood of cure while minimizing short- and long-term toxicity. At population levels, this is best accomplished by stratifying risk according to patient demographics, clinical characteristics, and tumor biology, with escalating treatment for those at the highest risk of relapse [10,11,12,13]. At the individual level, understanding tumor and host biology promises truly personalized care by facilitating targeting of specific drivers of disease [14, 15].
Primary or metastatic tumor biopsies at diagnosis, resection, or relapse are undeniably the gold standard for identifying tumor biology, diagnosing disease, and therapeutic decision-making. However, invasive biopsies are difficult to obtain in children, limiting their ability to monitor disease or determine when alternate treatments may be appropriate. Additionally, all children receive advanced imaging at diagnosis and for disease monitoring that may expose them to ionizing radiation and/or anesthesia [16]. Despite this, many children will relapse despite having no evidence of disease on clinical imaging.
Peripheral blood sampling is an increasingly attractive avenue for developing minimally invasive biomarkers that may complement tissue biopsies and imaging. It has long been known that tumors shed cells, nucleotides, proteins, and vesicles into the blood [17]. Because the material in a liquid biopsy is likely to represent the most aggressive part of a rapidly dividing malignancy, many of the genomic driving aberrations described in tumor biopsies should be identifiable in the peripheral blood [18]. Therefore, assays based on interrogating these circulating analytes, (i.e., “liquid biopsies”) are expected to emerge as tools to diagnose, monitor, and aid in therapeutic decision-making in real time using simple serial blood tests [19]. In adult malignancies, these approaches have led to new screening and diagnostic tests based on known biology [20], and many of the same approaches are currently being adapted for use in children with solid tumors. However, childhood cancers differ considerably from adult malignancies as they infrequently harbor recurrent genomic aberrations [21], driving the need for pediatric-focused initiatives.
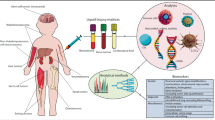
In this review, we will first describe some of the technologies being pursued for circulating analytes such as circulating tumor cells (CTCs) and macromolecular structures (nucleosomes and exosomes), cell-free DNA (cfDNA), and cell-free RNA (cfRNA) (Fig. 1, left). The diversity of approaches for use in liquid biopsies mirrors what has been accomplished using tumor tissue including assessments of genetic mutations, fusions, expression, and epigenetics. Next, we will discuss how liquid biopsies can be used to identify these genomic features and what is currently being evaluated across the spectrum of pediatric solid tumors. We conclude with some of the future directions in the field and potential applications to implement liquid biopsies for diagnostic classification, outcome prediction, disease monitoring, and therapeutic decision making in childhood cancer.

Overview of approaches to capturing and investigating circulating analytes from liquid biopsies. In the presence of malignancy, a liquid biopsy (i.e., blood) has been demonstrated to contain single tumor cells, free nucleic acids, exosomes containing nucleic acids and proteins, and free circulating nucleosomes. These components, sampled by simple phlebotomy, can be analyzed to detect single nucleotide mutations, copy number aberrations, fusions, translocations, and epigenetic changes reflective of tumor genetic heterogeneity. Abbreviations: cfDNA, cell-free DNA; ctDNA, circulating tumor DNA; qPCR, quantitative PCR; RT-qPCR, quantitative reverse transcription PCR; WES, whole exome sequencing; WGS, whole genome sequencing
2 Circulating analytes for liquid biopsy
2.1 CTCs and macromolecular structures
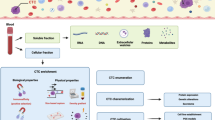
CTCs were first reported in 1869 [22], and are defined as cells circulating freely in the bloodstream with genetic resemblance to their tumor origin. CTCs have now been identified across a range of malignancies, including in patients with metastatic prostate, breast, ovarian, colorectal, and lung cancer [23]. Therefore, CTCs have become appealing analytes for liquid biopsies in childhood cancers as well. CTCs constitute a very small fraction of cells in the bloodstream (5 to 1281 CTCs per mL of blood) and are challenging to identify and capture [24]. Thus, early research efforts were aimed at circulating tumor cell detection, enrichment, and enumeration techniques, while more recent approaches have probed the contents of CTCs, mainly DNA and RNA.
Exosomes are small extracellular vesicles that are lipid bilayer-coated bodies containing DNA, RNA, and proteins, with membrane surface molecules that include MHC proteins [25,26,27] (Fig. 1, left). Secreted by many living cell types including cancer cells, exosomes facilitate cell-cell communication by allowing for intercellular exchange of signaling RNAs such as long non-coding RNAs (lncRNAs), micro RNAs (miRNAs), mitochondrial DNA, single- and double-stranded genomic DNA, and proteins [25, 28, 29]. Greater than 109 exosomes per mL have been observed in blood [30, 31], though they are also present in serum, urine, saliva, cerebral spinal fluid, and amniotic fluid, and can cross the blood-brain barrier [25,26,27]. Early studies focused on detecting and quantifying exosomal presence in the blood; however, it was demonstrated that cancer patients could not be reliably distinguished from healthy individuals by total exosome quantity or exosomal size alone [32]. Therefore, most current studies focus on the accurate capture of exosomes and assay their nucleic acid and protein content.
Circulating nucleosomes and histones are nuclear components observed in blood (Fig. 1, left). Each nucleosome is comprised of DNA wrapped 1.65 times around an eight-histone core, and the N-terminal histone tail protrudes from the nucleosome core and is a site for post-translational modifications [33]. Histone modifications are epigenetic means by which gene expression is regulated via chromatin accessibility and nucleosome positioning [34]. Cell death results in chromatin fragmentation and shedding of nucleosomes and histones into the bloodstream, which can then be detected and interrogated for liquid biopsy applications [35, 36]. Thus, circulating cfDNA, nucleosomes, and histones are closely connected. Indeed, around the time that tumor DNA was first observed in cancer patient plasma, increased levels of circulating nucleosomes were also reported in several adult cancers compared with those in controls [36, 37]. As many as 200 ng per mL of nucleosomes have been quantified in metastatic colorectal cancer [38]. Emerging methods for studying nucleosomes will be discussed in the technologies section.
2.2 Circulating nucleic acids
The probing of circulating cfDNA and cfRNA has been of significant interest to liquid biopsy development [39] (Fig. 1, left). cfDNA was first reported in 1948 [17] and later appreciated in the 1990s for its potential applications in detecting cancer with circulating tumor DNA (ctDNA) [40, 41] and non-invasive prenatal testing with fetal-derived cfDNA [42]. Released into the bloodstream through apoptosis and/or necrosis of cancer cells themselves (circulating tumor cells, primary tumor, or metastatic lesions [43]) or from dying cells in the surrounding tumor microenvironment, cfDNA has been demonstrated to be either free double-stranded DNA fragments or nucleosome-associated [44]. In cancer patients, 0–5 ng to over 1000 ng cfDNA can be isolated per mL of plasma [39]; and for healthy individuals, 0–100 ng per mL of plasma can be extracted [45]. The proportion of tumor-derived cfDNA often varies among patients depending on tumor burden [46]. The short half-life of cfDNA (16–90 min) allows for real-time assessments, including responsiveness to tumor-directed therapy [47]. Unlike tissue biopsies, cfDNA may better represent the diverse tumor cell population and tumor heterogeneity [48, 49].
Cell-free messenger RNA (mRNA), lncRNA, and miRNA are also shed by dying cells and have been detected in blood in spite of endogenous ribonucleases. Circulating mRNA was first reported in cancer patient serum in 1999 [50]. Later, IncRNAs were detected in plasma. As they are over 200-bp in size, it is hypothesized that RNA secondary structure protects the fraction of non-vesicle bound lncRNAs from degradation [51, 52]. miRNAs, which are small (~ 22 base-pair) non-coding RNAs that function as translational inhibitors were observed extracellularly [53] and in plasma within Argonaute2 complexes or associated with lipoproteins [54, 55]. As the roles of lncRNAs and miRNAs are better elucidated, their potential application as liquid biomarkers will be defined.
3 Technologies to assess analytes for liquid biopsy
3.1 Detection of macromolecular structures: CTCs, exosomes, and nucleosomes
CTCs and other macromolecular structures are detected and counted in liquid biopsies (Fig. 1, right and Table 1). As of 2019, 265 clinical trials in the USA were being conducted on CTCs [69]. Indeed, the CellSearch™ CTC (Menarini Silicon Biosystems) enumeration assay was reported in 2010 [56] and is now FDA-approved for diagnosing metastatic breast, prostate, and colorectal cancer. The assay enriches EpCAM+/CD45− CTCs using immunomagnetic separation to filter CTCs followed by flow-cytometric analysis for cell counting. Higher CTC numbers (> 5 CTCs per 7.5 mL of blood) were associated with poor progression-free survival (PFS) in metastatic breast and prostate cancers, and conversely lower CTC counts were associated with improved overall survival [70,71,72]. Capture and enrichment systems that have been developed, such as CellSieve™ (Creatv MicroTech) and ClearCell® FX (Genomax Technologies), allow for the investigation of EpCAM-negative CTCs by using physical differences to isolate them from the bloodstream [73, 74]. Once isolated, CTCs can be assessed by assaying commonly secreted proteins. For example, in the EPISPOT assay, viable CTCs were isolated from patients with breast and prostate cancer by culturing cells on membranes with antibodies against CD19, MUC1, PSA, and FGF2 [57]. Limitations for bulk analyses of CTCs include the need for a high yield and purity of samples; therefore, single-cell approaches probing CTC contents, especially CTC-derived nucleic acids, have emerged and will be an area of active research [69].
Isolation of exosomes faces technical challenges because traditional approaches are slow and consume large quantities of material. Exosome isolation methods include serial differential ultracentrifugation, antibody-based affinity purification (e.g., CD63, EpCAM), or size filtration [75]. Newer methods to decrease both processing time and required amount of sample include nPLEX, an assay that uses a surface plasmon resonance-based quantitative and high-throughput assay to achieve label-free exosome isolation [59]. As technologies have improved to capture exosomes, assays to probe exosomal contents have been developed. The first commercially available laboratory-developed tests for exosomes, the ExoDx® Lung assays (Exosome Diagnostics), investigates the RNA content of exosomes and will be discussed further in the transcriptomic assays section.
Cancer-specific histone post-translational modifications and nucleosome positioning have been extensively investigated [76, 77]. ELISA-based quantitation of circulating nucleosomes continues to be an area of research for liquid biopsy [61]. Focused assays test for histone methylation levels on circulating nucleosomes [78, 79], while broader nucleosome footprinting is an emerging technique to study genomic positioning in cfDNA analytes [44]. Recently, nucleosomal occupancy mapping on cfDNA was found to correlate with gene expression, cancer type, and cellular state [80].
3.2 Genomic assays
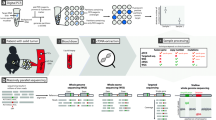
CTC/exosome-derived DNA or cfDNA can be assessed for cancer-associated mutations, copy number variants (CNVs), or single-nucleotide polymorphisms (SNPs) using targeted PCR-based or genome-wide next-generation sequencing (NGS) approaches. One of the few FDA-approved liquid biopsies is the cobas® EGFR Mutation Test v2 (Roche), a real-time quantitative PCR (qPCR) companion diagnostic assay that screens a panel of known EGFR mutations and deletions [62]. While qPCR is used in this and other panel-based approaches, the technology is generally limited by the low nanogram amounts of plasma ctDNA present in some cancer patients. Therefore, digital PCR platforms have been developed to analyze specific mutations and deletions that improve analytical sensitivity, which include methods such as microfluidic droplet digital PCR (ddPCR) and BEAMing (beads, emulsion, amplification, and magnetics) [81,82,83] (Fig. 1, right and Table 1). Briefly, these approaches divide DNA templates into near single-molecule/single-target PCR reactions for quantitative detection without a standard curve. Recently, O’Leary et. al. compared ddPCR with BEAMing in cfDNA from breast cancer patients in the PALOMA-3 trial and found good concordance between the two methods [84]. While these technologies require a pre-existing knowledge of mutations, advantages of ddPCR and BEAMing include low cost, short assay time, and the ability to multiplex mutation assessment.
Whole exome-sequencing (WES) offers a broader method to analyze cfDNA by sequencing the coding region contained in exons which are approximately 1% of the genome [85, 86] (Fig. 1, right and Table 1). In a seminal 2013 cfDNA study evaluating serial plasma samples from breast, lung, and ovarian cancers patients, WES identified genes with known association with acquired chemoresistance (e.g., PIK3CA), and clonal evolution of the cancers in response to therapy could be followed [49]. While WES can aid in biomarker discovery, most applications of WES in liquid biopsy are used to detect known mutations and CNVs. Additionally, because WES generally has lower depth of coverage than targeted sequencing panels, it may have limited ability to detect mutations below 5% allele frequencies [87].
Compared with WES, whole-genome sequencing (WGS) offers insight into the entire genetic landscape (Fig. 1, right and Table 1). WGS covers mutations in non-coding regions which include introns, regulatory elements, and non-coding RNAs that WES does not capture [88]. This allows for better assessment of CNVs, SNPs, and larger structural variations. WGS was first reported on cfDNA from colorectal cancer and breast cancer patients to identify chromosomal rearrangements and copy number changes, including ERBB2 and CDK6 amplification [63]. WGS on cfDNA was soon after applied to demonstrate applicability of minimal residual disease detection in pre- and post-operative cancer patients [89]. Sequencing coverage, along with its associated costs, can be a barrier to WES and WGS approaches. Therefore, low-coverage sequencing (i.e., 0.1x coverage) and associated computational approaches continue to be an area of active research for NGS-based cfDNA liquid biopsies [90,91,92].
3.3 Epigenetic-based assays
Liquid biopsies that focus solely on detecting tumor-specific, low-frequency mutant alleles face challenges if the mutations are lower than the limit of detection in the sampled cfDNA. In contrast, epigenetic modifications are ubiquitous, and the deposition of epigenetic marks is organ- and cell-type specific [93, 94]. Thus, epigenetic analysis of cfDNA can also be used to investigate the tumor microenvironment and metabolic and immune responses to cancers (Fig. 1, right and Table 1). Furthermore, mapping of these epigenetic features can enhance cancer diagnosis, prognosis, and relapse detection [95].
The most common DNA modification is cytosine methylation (5mC), which occurs throughout CpG islands and is associated with transcriptional repression. Despite an overall decrease in 5mC levels reported in cancers [96], aberrant focal 5mC deposition has been observed, supporting models of epigenetic influence on cancer initiation, progression, and invasion [33, 97, 98]. For individual or multiplexed targeting of specific methylated regions, techniques that employ methylation-sensitive restriction endonuclease (MSRE)-PCR or methylation-specific-PCR (MS-PCR) are utilized [99, 100]. Most of the cfDNA 5mC-based assays that are the furthest in the clinical validation pipeline are based on MS-PCR using candidate methylated genes. Indeed, the only epigenetic-based FDA-approved liquid biopsy available is Epi ProColon® (Epigenomics), which consists of MethyLight PCR detection of a single target, methylated SEPT9 [64].
The general approaches for genome-wide DNA methylome mapping include the following: (1) methods that use MSRE coupled to sequencing [101, 102]; (2) enrichment and affinity capture of 5mC-containing DNA fragments including MeDIP-seq [103]), MBD-Seq [104], or a 5mC chemical labeling strategy (e.g. Tet-assisted 5mC sequencing [105]); and (3) bisulfite-conversion-based sequencing methods (BS-Seq) that achieve single-base resolution differentiation of unmethylated and methylated cytosines [106]. To date, most whole-genome methylation studies applied to cfDNA are currently either biomarker discovery or proof-of-concept studies. The application of whole-genome BS-seq [107, 108], MeDIP-seq [109, 110], and other genome-wide methylation mapping technologies to cfDNA for liquid biopsies are likely to continue to emerge.
Other promising epigenetic marks for liquid biopsy detection include oxidized derivatives of 5mC: 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), which are produced by ten-eleven-translocation (TET) dioxygenase catalysis in mammalian cells [111]. Of these cytosine modifications, 5hmC has been most developed as a potential liquid biopsy [112]. Differential deposition of 5hmC has been observed in primary tumor DNA, suggesting that genomic patterns of 5hmC capture tumor heterogeneity. Affinity-based [113, 114] and single-base resolution methods [115,116,117,118,119] have been developed to analyze genome-wide 5hmC; for targeted 5hmC detection, most of these technologies could be coupled to PCR or arrays. A promising method for liquid biopsy in particular is nano-hmC-seal [120]. Nano-hmC-seal modified the hmC-seal approach (chemical labeling, pull-down, and enrichment of 5hmC-modified DNA fragments) [114] to successfully perform it on 1–10 nanogram quantities of DNA. Nano-hmC-Seal has been applied to cfDNA to develop diagnostic biomarkers in hepatocellular carcinoma (HCC) [66, 121], diffuse large B-cell lymphoma [122], and colorectal [65, 66, 123], gastric [65], lung [66, 124], pancreatic [66], breast [66], and esophageal cancers [125]. Given these extensive cfDNA applications, it is likely that clinical validation studies of these whole-genome or targeted panels of 5hmC-modified loci will emerge.
3.4 Transcriptomic assays
Analogous to cfDNA assays, targeted PCR or broader sequencing approaches can be applied to complimentary DNA (cDNA) synthesized by reverse transcription from RNA templates. In general, targeted quantitative reverse-transcriptase PCR (RT-PCR)-based assays are used for detecting specific or multiple transcripts. To expand RT-PCR methodologies to low-input samples, dPCR approaches have been applied to cDNA synthesized from circulating cell-free or exosome/CTC-derived RNA, with efforts focused on improving reverse transcriptase efficiency [67, 126]. The ExoDx® Lung assays (T790M, ALK, or panel EGFR, Exosome Diagnostics), for example, use an RT-dPCR approach on exosomal RNA, combined with analysis of cfDNA, to assess patients with lung cancer for EGFR mutations [60].
To more broadly assess RNA in liquid biopsies, transcript microarrays and whole-transcriptome NGS are used. The transcriptomic analog to WGS, RNA-seq, has been transformational in molecular biology [127, 128]. Methods to expand RNA-seq for biomarker discovery from low-quantity or rare cell populations have been developed. Smart-seq, a single-cell level mRNA-seq method, has been tested on mRNA isolated from CTCs to analyze genome wide expression patterns [58]. Considerable effort has been made to establish workflows and appropriate standards for RNA-seq performed on low-input samples, such as miRNAs from patient plasma samples [129] and other cell-free RNA species [68, 130, 131]. In summary, the analytes available in circulating plasma are myriad, but the technologies to probe these analytes and discovered biomarkers need additional clinical validation prior to universal adoption.
4 Applications of liquid biopsy in pediatric solid tumors
4.1 Neuroblastoma
Neuroblastoma is the fourth most common pediatric tumor and the most common extracranial solid malignancy in children. As a neural crest cell-derived cancer, it typically presents in the first few years of life and is characterized by phenotypic and biologic heterogeneity. Patients with low-risk disease can often be monitored with observation alone [132], while patients with aggressive high-risk neuroblastoma can expect a three-year event-free survival of 60% despite high-intensity, multimodal therapy [133]. Diagnosis and monitoring of neuroblastoma are typically accomplished through tumor biopsy followed by serial computed tomography scans, 123I-metaiodobenzylguanidine radionucleotide scanning, urine catecholamines, and bone marrow biopsies and aspirates. Once patients are assigned to high-risk therapy, there are no blood biomarkers to monitor disease or determine response to therapy. Despite the discovery over thirty years ago that neuron specific enolase, gangliosides, and neuropeptide Y were elevated in plasma from patients with neuroblastoma [134,135,136,137,138,139], these circulating biomarkers have not proven reliable enough to augment or supplant imaging and biopsies (Table 2). Therefore, recent efforts have focused on harnessing the latest technology to identify novel liquid biopsy biomarkers of high-risk neuroblastoma.
Neuroblastoma is known to frequently shed CTCs in the blood both at diagnosis and during therapy [140, 183]. Historically, neuroblastoma CTCs were identified either with RT-PCR of CTC-derived mRNA or immunoprecipitation of cell-surface proteins [141, 184]. While it was determined that the presence or absence of CTCs may serve as a circulating biomarker of treatment response and likelihood of progression [142], immunologic purging of CTCs did not improve the survival of children with high-risk neuroblastoma in a randomized phase III trial [185].
Early efforts toward the genomic identification of neuroblastoma in liquid biopsies relied heavily on RT-PCR, which identified mRNA transcripts possibly shed by CTCs. Increased steady-state tyrosine hydroxylase mRNA in newly diagnosed neuroblastoma patients [143, 186, 187] is one example of over twenty different transcripts to show potential as a circulating neuroblastoma biomarker [188]. Indeed, multi-transcript panels [144,145,146,147, 189] and newer NGS approaches to detect miRNA (freely circulating or exosome-derived) [148, 149] demonstrate promise and feasibility, but there have been numerous impediments to clinical implementation including the difficulty of obtaining high-quality RNA in the clinic, prioritizing development of the many gene sets, and a lack of clear understanding of the assays’ minimum detection limits.
More recently, advances in parallel genomic sequencing have made it possible to perform assessment of common genomic aberrations from cfDNA, particularly CNVs [150,151,152], which are well-described as biomarkers of aggressive neuroblastoma [190, 191]. Shallow WGS of cfDNA was evaluated as a relatively economical way to assess CNVs and confirmed that changes identified from liquid biopsies mirrored those from primary tumors [151, 153]. In particular, amplification of the MYCN oncogene, a well-established driver of half of high-risk neuroblastoma tumors [192], can be identified in cfDNA with a variety of technologies [154,155,156,157,158,159]. Gain of 17q and loss of 11q are also readily detectable [160] in serum at diagnosis [160, 161], and amplifications of the ALK gene has been identified using ddPCR [159]. Researchers have also implemented WES and WGS to identify specific genomic alterations in genes such as TERT, ATRX, and ALK that may have therapeutic implications [152], though it remains unclear how effective such approaches will be for delineating patients in complete remission from those with minimal disease states.
It is postulated that neuroblastoma is driven in large part by epigenetic modifications due to the combination of few driving somatic mutations [193] and diverse clinical phenotypes [194, 195]. Indeed, focal and genome-wide profiling of cytosine and histone modifications can recapitulate neuroblastoma risk groups and identify drivers of tumor biology [196,197,198,199,200,201,202,203]. The potential for biomarker development by identifying altered methylation patterns on specific genes in cfDNA has been demonstrated for RASSF1A and DRC2 [162, 163] and efforts are ongoing to explore genome-wide 5mC profiling from cfDNA for biomarker development in neuroblastoma.
In contrast to 5mC, 5hmC is associated with open chromatin, and increased deposition on gene bodies has been shown to correlate with gene expression. Thus, assays of genome-wide 5hmC have the potential to serve a DNA surrogate for gene expression, particularly of regulatory genes [114]. In neuroblastoma, 5hmC profiles generated by nano-hmC-seal from over a hundred diagnostic tumor samples delineated high-risk from non-high-risk disease [196]. This same profiling technology was recently applied to cfDNA from 129 serially collected samples from mostly high-risk patients during treatment and follow-up [204]. 5hmC profiles were highly correlated with metastatic disease burden in both discovery and validation cohorts regardless of underlying tumor biology, but were also able to identify samples from patients with MYCN-amplification. Furthermore, 5hmC cfDNA profiles differentiated some patients who responded to initial chemotherapy from those who did not and detected minimal amounts of disease in patients who were classified as achieving complete remission using established clinical response criteria. This promising methodology will be prospectively validated in larger, independent cohorts.
4.2 Sarcoma
Sarcomas represent 20% of all pediatric solid malignancies [205] and are the most common malignancies of bone diagnosed in the first three decades of life [206]. Tissue biopsy is the gold standard for diagnosis, but because of its invasiveness it is not feasible for serial monitoring of disease prior to surgical resection. No liquid biopsy approaches have been approved for integration into clinical care, and an expanding number of studies are assessing their utility for diagnosis, monitoring, and management. For example, across high-grade sarcomas, Hayashi et. al. showed that CTCs were detectable and quantifiable at the time of diagnosis, decreased with treatment, and correlated with risk of radiographic relapse [164].
When considering circulating cfDNA as an analyte for osteosarcoma, the lack of recurrent translocations limits the application of mutation-detection assays such as ddPCR and WES. In spite of this, McBride et. al. designed patient-specific PCR primers based on DNA mutations found in primary tumor DNA to detect tumor-derived DNA in plasma from one osteosarcoma patient [207]. Additionally, Barris et. al. demonstrated the use of targeted ultra-deep NGS to identify and track ctDNA burden of osteosarcoma patients using common aberrant genes such as TP53 and ATRX [165]. Others have demonstrated that ultra-low-passage WGS of plasma can identify tumor material in a majority of patients with osteosarcoma [166]. Circulating RNA, including miRNA, has also been profiled and shown to be prognostic in patients with osteosarcoma, though validation studies are needed to confirm correlation with outcome [167, 168].
Another pediatric bone malignancy, Ewing sarcoma, has characteristic translocations that can be detected in liquid biopsy samples [153]. In an early study, RT-PCR of fusion gene transcripts EWS-FLI1 or EWS-ERG were used to identify occult tumor cells in plasma, and occult cell presence was associated with micrometastases and adverse outcomes [169]. However, the prognostic significance of this observation has yet to be validated [208]. More recently, in patients with newly diagnosed Ewing sarcoma, EWSR1 translocations and other mutations such as TP53 and STAG2 were detected in cfDNA using ultra-low-passage WGS [166]. This study also explored the prognostic potential of a translocation-based liquid biopsy, showing that the half of patients with detectable ctDNA at diagnosis had inferior three-year event-free survival compared to those without detectable ctDNA at diagnosis, an association likely related to increased rates of metastatic disease in those with detectable ctDNA. Hayashi et. al. developed a highly sensitive and targeted approach to detect ctDNA by using PCR primers for tumor-specific EWS-ETS fusion gene breakpoints and subsequently applied ddPCR to detect the fusion gene in patient plasma [170]. ddPCR was also used to assess EWSR1-FLI1 fusion levels in cfDNA. Increased EWSR1-FLI1 levels were associated with disease burden and relapse, while decreased levels were observed in patients who responded to initial chemotherapy [171]. The EWS-FLI1 translocation has also been identified from cfRNA using RT-PCR [172].
Rhabdomyosarcoma, the most common tissue sarcoma, is also associated with fusion genes in a subset of cases. Eguchi-Ishimae et. al. reported that the PAX3-FOXO1 translocation could be detected by qPCR on cfDNA extracted from serial blood samples of a single patient with alveolar rhabdomyosarcoma. Notably, the translocation was detected in cfDNA during clinical remission, prior to a relapse that was not radiographically evident for several additional months [173]. In addition to fusion gene detection, Miyachi et. al. used RT-PCR to identify circulating miRNAs (miR-1, miR-133a, miR-133b, and miR-206), with miR-206 having the highest sensitivity and specificity to distinguish rhabdomyosarcoma from other solid tumors [174].
4.3 Wilms tumor
Wilms tumor, the most common renal malignancy in childhood, is primarily managed with two distinct approaches. In much of Europe, and in some patients for whom upfront resection is contraindicated, neoadjuvant chemotherapy is often initiated without a tissue biopsy, resulting in mismatched treatment in up to 5% of children [209]. In contrast, North American trials have demonstrated prognostic pre-treatment biologic features to stratify higher risk patients for intensified therapy, and upfront resection or tumor biopsy is preferred [12]. The development of liquid biopsies for Wilms tumor diagnosis may help to standardize therapy by providing valuable risk-stratification information. In a biomarker discovery study, WES was performed on cfDNA obtained from pre-nephrectomy patients at diagnosis and identified tumor-specific mutations across all kidney tumor histologies [175]. A recent study used low passage WGS of ctDNA from eight patients with Wilms tumors and identified four patients with detectable ctDNA, two of whom had 1q gain, a prognostic marker in Wilms tumor [153]. Others have focused on identifying serum miRNAs using RT-PCR. Circulating miR-143-3p and miR-129-5p differentiated Wilms tumors from neuroblastoma [176], whereas miR-130b-3p, miR-100-5p, and miR-143-3p differentiated patients with Wilms tumor from healthy controls with an accuracy of 84.5% [177]. These data are promising, yet additional diagnostic liquid biopsy needs remain, including the ability to reliably differentiate Wilms tumor from benign lesions, nephrogenic rests or nephroblastomatosis, and other pediatric renal pathologies (e.g., clear cell sarcoma of the kidney or rhabdoid tumor). Further, an ideal liquid biopsy will be able to detect prognostic molecular biomarkers of favorable histology Wilms tumors, such as 1p and 16q loss of heterozygosity [12].
Several technologies using cfDNA to monitor disease have been tested with variable success for patients with Wilms tumor. While a signature of 176 circulating miRNAs was diagnostic of Wilms tumor at diagnosis and could distinguish healthy controls, it did not accurately reflect treatment effects [178]. In patients with anaplastic histology Wilms tumor, Treger et al. identified TP53 mutations using ddPCR in matched tumor, blood, and urine samples at various time points during treatment, although the correlation with prognosis has yet to be explored [179]. Finally, as somatic epigenetic drivers are common in Wilms tumors, Charlton et. al. detected tumor-derived methylated loci in cfDNA from patients with Wilms tumor [180]. These methylated sites were successfully identified in cfDNA at the time of diagnosis, and in some cases, during neoadjuvant chemotherapy, after resection, and during adjuvant chemotherapy [180]. Taken together, the genetic, transcriptomic, and epigenetic probing of circulating nucleic acids show promise for liquid biopsies from patients with Wilms tumor.
4.4 Hepatoblastoma
Hepatoblastoma, the most common liver tumor of childhood, is often diagnosed in association with underlying cancer predisposition syndromes such as Beckwith-Wiedemann syndrome. Early detection of malignancies is especially important for these children [210], and screening typically involves periodic imaging and measuring of serum alpha fetal protein (AFP) [210]. Thus, advanced liquid biopsies could be combined with imaging/AFP to improve screening sensitivity. To begin to address this, Murray et. al. performed RT-PCR of miRNA isolated from whole blood samples collected from patients with hepatoblastoma compared to healthy controls and children with other tumors and identified that miR-122-5p, miR-483-3p, and miR-205-5p could be used to differentiate patients with hepatoblastoma from those with neuroblastoma [176]. Liu et. al. performed RT-PCR to measure the expression levels of miR-21 in the plasma and the exosomes of hepatoblastoma patients and demonstrated that exosomal miR-21 was able to diagnose hepatoblastoma more accurately than serum AFP levels [181]. In this study, miR-21 was an independent predictor of event-free survival for hepatoblastoma patients [181]. In contrast, Jiao et. al. found that circulating miR-34a, miR-34b, and miR-34c was not diagnostically superior to AFP, but the presence of these miRNAs were the significant predictors of outcome, even when controlled for PRETEXT stage IV, presence of metastases, and presence of vascular invasion [182]. The potential application of liquid biopsies in hepatoblastoma requires additional effort to identify optimal analytes and assays for further development.
5 Future directions
The applications reviewed above include those for cancer diagnostics, risk stratification, and predictive modeling to guide pediatric personalized medicine. There remains a significant unmet need in children with solid tumors to titrate the intensity and duration of therapy based on clinical and biologic features of disease, including risk of recurrence. Liquid biopsies offer potential to track tumor burden complementary or independent of radiologic or surgical findings. For example, when treatment for rhabdomyosarcoma is completed, a portion of patients may have persistent circulating tumor analytes, and the detection of this through liquid biopsy may justify maintenance-like chemotherapy for these patients [211]. Serially collected liquid biopsies could also provide insight about the association between disease burden and chemosensitivity, i.e., a liquid biopsy indication of rising tumor burden while on conventional therapy could trigger clinical decisions to augment or shift treatment. Additional applications of liquid biopsies will likely focus on monitoring patients at high risk of developing cancer (including children with genetic predisposition or cancer history) without exposure to ionizing radiation or anesthesia.
Assaying circulating tumor material offers unique opportunity to expand our understanding of cancer biology and pathogenesis through the study of tumor heterogeneity and evolution. While a tissue biopsy provides cellular and molecular insight about the sampled region, a liquid biopsy captures the diverse landscape of a tumor [212], and therefore deep sequencing, for example, may identify disease-causal and persistent subclones that are chemo-resistant. Serial liquid biopsies may facilitate the application of spatiotemporal genomics and gene expression profiling, or the study of how tumors evolve over time, which will allow for the identification of the most highly relevant disease-associated genes and pathways, potentially enabling personalized medicine approaches. The addition of whole-genome epigenetic profiling may also complement genomic approaches, as will evolving technologies to profile metabolites.
Childhood cancer researchers are poised to advance the study of liquid biopsies and circulating biomarkers. While clinical validation of new assays through randomized controlled trials will be necessary for clinical adoption, the process will be supported by the nearly universal cooperation of providers and patients for clinical trial enrollment, and the increased emphasis on developing robust, clinically annotated biorepositories. Well established, centralized infrastructure to handle sample processing, storage, and distribution can aid in the comprehensive study of these rare diseases, and this should remain a priority amongst researchers and funding organizations.
The technologies supporting liquid biopsies in pediatric solid tumors continue to expand and the clinical applications are being refined by many independent research groups. Robust biorepositories will help ensure that as novel technologies to probe circulating analytes emerge, the use of patient sample and patient information will be maximized for comprehensive validation and clinical adoption.
References
Tomasetti, C., Li, L., & Vogelstein, B. (2017). Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science, 355(6331), 1330–1334. https://doi.org/10.1126/science.aaf9011.
Applebaum, M. A., Vaksman, Z., Lee, S. M., Hungate, E. A., Henderson, T. O., London, W. B., et al. (2017). Neuroblastoma survivors are at increased risk for second malignancies: a report from the International Neuroblastoma Risk Group Project. European Journal of Cancer, 72, 177–185. https://doi.org/10.1016/j.ejca.2016.11.022.
Ginsberg, J. P., Goodman, P., Leisenring, W., Ness, K. K., Meyers, P. A., Wolden, S. L., et al. (2010). Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. Journal of the National Cancer Institute, 102(16), 1272–1283. https://doi.org/10.1093/jnci/djq278.
Oeffinger, K. C., & Bhatia, S. (2009). Second primary cancers in survivors of childhood cancer. Lancet, 374(9700), 1484–1485. https://doi.org/10.1016/S0140-6736(09)61885-7.
Oeffinger, K. C., Mertens, A. C., Sklar, C. A., Kawashima, T., Hudson, M. M., Meadows, A. T., et al. (2006). Chronic health conditions in adult survivors of childhood cancer. The New England Journal of Medicine, 355(15), 1572–1582. https://doi.org/10.1056/NEJMsa060185.
Kirchhoff, A. C., Nipp, R., Warner, E. L., Kuhlthau, K., Leisenring, W. M., Donelan, K., et al. (2018). "Job Lock" Among Long-term Survivors of Childhood Cancer: a report from the childhood cancer survivor study. JAMA Oncology, 4(5), 707–711. https://doi.org/10.1001/jamaoncol.2017.3372.
Nagarajan, R., Kamruzzaman, A., Ness, K. K., Marchese, V. G., Sklar, C., Mertens, A., et al. (2011). Twenty years of follow-up of survivors of childhood osteosarcoma: a report from the Childhood Cancer Survivor Study. Cancer, 117(3), 625–634. https://doi.org/10.1002/cncr.25446.
Termuhlen, A. M., Tersak, J. M., Liu, Q., Yasui, Y., Stovall, M., Weathers, R., et al. (2011). Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatric Blood & Cancer, 57(7), 1210–1216. https://doi.org/10.1002/pbc.23090.
Zheng, D. J., Krull, K. R., Chen, Y., Diller, L., Yasui, Y., Leisenring, W., et al. (2018). Long-term psychological and educational outcomes for survivors of neuroblastoma: a report from the Childhood Cancer Survivor Study. Cancer, 124(15), 3220–3230. https://doi.org/10.1002/cncr.31379.
Cohn, S. L., Pearson, A. D., London, W. B., Monclair, T., Ambros, P. F., Brodeur, G. M., et al. (2009). The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of Clinical Oncology, 27(2), 289–297. https://doi.org/10.1200/JCO.2008.16.6785.
Twist, C. J., Naranjo, A., Schmidt, M. L., Tenney, S. C., Cohn, S. L., Meany, H. J., et al. (2019). Defining Risk Factors for Chemotherapeutic Intervention in Infants With Stage 4S Neuroblastoma: a Report From Children's Oncology Group Study ANBL0531. Journal of Clinical Oncology, 37(2), 115–124. https://doi.org/10.1200/JCO.18.00419.
Dome, J. S., Perlman, E. J., & Graf, N. (2014). Risk stratification for wilms tumor: current approach and future directions. American Society of Clinical Oncology Educational Book, 215–223. https://doi.org/10.14694/EdBook_AM.2014.34.215.
Meyers, R. L., Maibach, R., Hiyama, E., Haberle, B., Krailo, M., Rangaswami, A., et al. (2017). Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. The Lancet Oncology, 18(1), 122–131. https://doi.org/10.1016/S1470-2045(16)30598-8.
Harris, M. H., DuBois, S. G., Glade Bender, J. L., Kim, A., Crompton, B. D., Parker, E., et al. (2016). Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: the Individualized Cancer Therapy (iCat) Study. JAMA Oncology. https://doi.org/10.1001/jamaoncol.2015.5689.
Mody, R. J., Wu, Y. M., Lonigro, R. J., Cao, X., Roychowdhury, S., Vats, P., et al. (2015). Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA, 314(9), 913–925. https://doi.org/10.1001/jama.2015.10080.
Weiser, D. A., Kaste, S. C., Siegel, M. J., & Adamson, P. C. (2013). Imaging in childhood cancer: a Society for Pediatric Radiology and Children's Oncology Group Joint Task Force report. Pediatric Blood & Cancer, 60(8), 1253–1260. https://doi.org/10.1002/pbc.24533.
Mandel, P., & Metais, P. (1948). Les acides nucleiques du plasma sanguin chez l'homme. Comptes Rendus des Seances de la Societe de Biologie et de Ses Filiales, 142(3-4), 241–243.
Wan, J. C. M., Massie, C., Garcia-Corbacho, J., Mouliere, F., Brenton, J. D., Caldas, C., et al. (2017). Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nature Reviews. Cancer, 17(4), 223–238. https://doi.org/10.1038/nrc.2017.7.
Abbou, S. D., Shulman, D. S., DuBois, S. G., & Crompton, B. D. (2019). Assessment of circulating tumor DNA in pediatric solid tumors: the promise of liquid biopsies. Pediatric Blood & Cancer, 66(5), e27595. https://doi.org/10.1002/pbc.27595.
Rossi, G., & Ignatiadis, M. (2019). Promises and Pitfalls of Using Liquid Biopsy for Precision Medicine. Cancer Research, 79(11), 2798–2804. https://doi.org/10.1158/0008-5472.CAN-18-3402.
Grobner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature, 555(7696), 321–327. https://doi.org/10.1038/nature25480.
Ashworth, T. R. (1869). A case of cancer in which cells similar to those in the tumours were seen in the blood after death. The Medical Journal of Australia, 14, 146–147.
Allard, W. J., Matera, J., Miller, M. C., Repollet, M., Connelly, M. C., Rao, C., et al. (2004). Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clinical Cancer Research, 10(20), 6897–6904. https://doi.org/10.1158/1078-0432.CCR-04-0378.
Nagrath, S., Sequist, L. V., Maheswaran, S., Bell, D. W., Irimia, D., Ulkus, L., et al. (2007). Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature, 450(7173), 1235–1239. https://doi.org/10.1038/nature06385.
Peterson, M. F., Otoc, N., Sethi, J. K., Gupta, A., & Antes, T. J. (2015). Integrated systems for exosome investigation. Methods, 87, 31–45. https://doi.org/10.1016/j.ymeth.2015.04.015.
Contreras-Naranjo, J. C., Wu, H. J., & Ugaz, V. M. (2017). Microfluidics for exosome isolation and analysis: enabling liquid biopsy for personalized medicine. Lab on a Chip, 17(21), 3558–3577. https://doi.org/10.1039/c7lc00592j.
Jiang, N., Pan, J., Fang, S., Zhou, C., Han, Y., Chen, J., et al. (2019). Liquid biopsy: circulating exosomal long noncoding RNAs in cancer. Clinica Chimica Acta, 495, 331–337. https://doi.org/10.1016/j.cca.2019.04.082.
Whiteside, T. L. (2016). Tumor-Derived Exosomes and Their Role in Cancer Progression. Advances in Clinical Chemistry, 74, 103–141. https://doi.org/10.1016/bs.acc.2015.12.005.
Kalluri, R. (2016). The biology and function of exosomes in cancer. The Journal of Clinical Investigation, 126(4), 1208–1215. https://doi.org/10.1172/JCI81135.
He, M., & Zeng, Y. (2016). Microfluidic Exosome Analysis toward Liquid Biopsy for Cancer. Journal of Laboratory Automation, 21(4), 599–608. https://doi.org/10.1177/2211068216651035.
Kowal, J., Tkach, M., & Thery, C. (2014). Biogenesis and secretion of exosomes. Current Opinion in Cell Biology, 29, 116–125. https://doi.org/10.1016/j.ceb.2014.05.004.
Melo, S. A., Luecke, L. B., Kahlert, C., Fernandez, A. F., Gammon, S. T., Kaye, J., et al. (2015). Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature, 523(7559), 177–182. https://doi.org/10.1038/nature14581.
Esteller, M. (2007). Cancer epigenomics: DNA methylomes and histone-modification maps. Nature Reviews. Genetics, 8(4), 286–298. https://doi.org/10.1038/nrg2005.
Wang, Z., Zang, C., Rosenfeld, J. A., Schones, D. E., Barski, A., Cuddapah, S., et al. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nature Genetics, 40(7), 897–903. https://doi.org/10.1038/ng.154.
Xu, J., Zhang, X., Pelayo, R., Monestier, M., Ammollo, C. T., Semeraro, F., et al. (2009). Extracellular histones are major mediators of death in sepsis. Nature Medicine, 15(11), 1318–1321. https://doi.org/10.1038/nm.2053.
Holdenrieder, S., Stieber, P., Bodenmuller, H., Busch, M., Fertig, G., Furst, H., et al. (2001). Nucleosomes in serum of patients with benign and malignant diseases. International Journal of Cancer, 95(2), 114–120. https://doi.org/10.1002/1097-0215(20010320)95:2<114::aid-ijc1020>3.0.co;2-q.
Kuroi, K., Tanaka, C., & Toi, M. (1999). Plasma Nucleosome Levels in Node-Negative Breast Cancer Patients. Breast Cancer, 6(4), 361–364.
Fahmueller, Y. N., Nagel, D., Hoffmann, R. T., Tatsch, K., Jakobs, T., Stieber, P., et al. (2012). Predictive and prognostic value of circulating nucleosomes and serum biomarkers in patients with metastasized colorectal cancer undergoing Selective Internal Radiation Therapy. BMC Cancer, 12, 5. https://doi.org/10.1186/1471-2407-12-5.
Schwarzenbach, H., Hoon, D. S., & Pantel, K. (2011). Cell-free nucleic acids as biomarkers in cancer patients. Nature Reviews. Cancer, 11(6), 426–437. https://doi.org/10.1038/nrc3066.
Chen, X. Q., Stroun, M., Magnenat, J. L., Nicod, L. P., Kurt, A. M., Lyautey, J., et al. (1996). Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nature Medicine, 2(9), 1033–1035. https://doi.org/10.1038/nm0996-1033.
Nawroz, H., Koch, W., Anker, P., Stroun, M., & Sidransky, D. (1996). Microsatellite alterations in serum DNA of head and neck cancer patients. Nature Medicine, 2(9), 1035–1037. https://doi.org/10.1038/nm0996-1035.
Lo, Y. M., Corbetta, N., Chamberlain, P. F., Rai, V., Sargent, I. L., Redman, C. W., et al. (1997). Presence of fetal DNA in maternal plasma and serum. Lancet, 350(9076), 485–487. https://doi.org/10.1016/S0140-6736(97)02174-0.
Jahr, S., Hentze, H., Englisch, S., Hardt, D., Fackelmayer, F. O., Hesch, R. D., et al. (2001). DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Research, 61(4), 1659–1665.
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M., & Shendure, J. (2016). Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell, 164(1-2), 57–68. https://doi.org/10.1016/j.cell.2015.11.050.
Thierry, A. R., El Messaoudi, S., Gahan, P. B., Anker, P., & Stroun, M. (2016). Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Reviews, 35(3), 347–376. https://doi.org/10.1007/s10555-016-9629-x.
Diehl, F., Li, M., Dressman, D., He, Y., Shen, D., Szabo, S., et al. (2005). Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proceedings of the National Academy of Sciences of the United States of America, 102(45), 16368–16373. https://doi.org/10.1073/pnas.0507904102.
Stewart, C. M., & Tsui, D. W. Y. (2018). Circulating cell-free DNA for non-invasive cancer management. Cancer Genetics, 228-229, 169–179. https://doi.org/10.1016/j.cancergen.2018.02.005.
Diaz Jr., L. A., Williams, R. T., Wu, J., Kinde, I., Hecht, J. R., Berlin, J., et al. (2012). The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature, 486(7404), 537–540. https://doi.org/10.1038/nature11219.
Murtaza, M., Dawson, S. J., Tsui, D. W., Gale, D., Forshew, T., Piskorz, A. M., et al. (2013). Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature, 497(7447), 108–112. https://doi.org/10.1038/nature12065.
Kopreski, M. S., Benko, F. A., Kwak, L. W., & Gocke, C. D. (1999). Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clinical Cancer Research, 5(8), 1961–1965.
Reis, E. M., & Verjovski-Almeida, S. (2012). Perspectives of Long Non-Coding RNAs in Cancer Diagnostics. Frontiers in Genetics, 3, 32. https://doi.org/10.3389/fgene.2012.00032.
Qi, P., Zhou, X. Y., & Du, X. (2016). Circulating long non-coding RNAs in cancer: current status and future perspectives. Molecular Cancer, 15(1), 39. https://doi.org/10.1186/s12943-016-0524-4.
Reid, G., Kirschner, M. B., & van Zandwijk, N. (2011). Circulating microRNAs: association with disease and potential use as biomarkers. Critical Reviews in Oncology/Hematology, 80(2), 193–208. https://doi.org/10.1016/j.critrevonc.2010.11.004.
Vickers, K. C., Palmisano, B. T., Shoucri, B. M., Shamburek, R. D., & Remaley, A. T. (2011). MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nature Cell Biology, 13(4), 423–433. https://doi.org/10.1038/ncb2210.
Arroyo, J. D., Chevillet, J. R., Kroh, E. M., Ruf, I. K., Pritchard, C. C., Gibson, D. F., et al. (2011). Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proceedings of the National Academy of Sciences of the United States of America, 108(12), 5003–5008. https://doi.org/10.1073/pnas.1019055108.
Miller, M. C., Doyle, G. V., & Terstappen, L. W. (2010). Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer. Journal of Oncology, 2010, 617421. https://doi.org/10.1155/2010/617421.
Alix-Panabieres, C. (2012). EPISPOT assay: detection of viable DTCs/CTCs in solid tumor patients. Recent Results in Cancer Research, 195, 69–76. https://doi.org/10.1007/978-3-642-28160-0_6.
Ramskold, D., Luo, S., Wang, Y. C., Li, R., Deng, Q., Faridani, O. R., et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology, 30(8), 777–782. https://doi.org/10.1038/nbt.2282.
Im, H., Shao, H., Park, Y. I., Peterson, V. M., Castro, C. M., Weissleder, R., et al. (2014). Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nature Biotechnology, 32(5), 490–495. https://doi.org/10.1038/nbt.2886.
Castellanos-Rizaldos, E., Grimm, D. G., Tadigotla, V., Hurley, J., Healy, J., Neal, P. L., et al. (2018). Exosome-Based Detection of EGFR T790M in Plasma from Non-Small Cell Lung Cancer Patients. Clinical Cancer Research, 24(12), 2944–2950. https://doi.org/10.1158/1078-0432.CCR-17-3369.
McAnena, P., Brown, J. A., & Kerin, M. J. (2017). Circulating Nucleosomes and Nucleosome Modifications as Biomarkers in Cancer. Cancers (Basel), 9(1). https://doi.org/10.3390/cancers9010005.
Gale, D., Lawson, A. R. J., Howarth, K., Madi, M., Durham, B., Smalley, S., et al. (2018). Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS One, 13(3), e0194630. https://doi.org/10.1371/journal.pone.0194630.
Leary, R. J., Sausen, M., Kinde, I., Papadopoulos, N., Carpten, J. D., Craig, D., et al. (2012). Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Science Translational Medicine, 4(162), 162ra154. https://doi.org/10.1126/scitranslmed.3004742.
Wasserkort, R., Kalmar, A., Valcz, G., Spisak, S., Krispin, M., Toth, K., et al. (2013). Aberrant septin 9 DNA methylation in colorectal cancer is restricted to a single CpG island. BMC Cancer, 13, 398. https://doi.org/10.1186/1471-2407-13-398.
Li, W., Zhang, X., Lu, X., You, L., Song, Y., Luo, Z., et al. (2017). 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Research, 27(10), 1243–1257. https://doi.org/10.1038/cr.2017.121.
Song, C. X., Yin, S., Ma, L., Wheeler, A., Chen, Y., Zhang, Y., et al. (2017). 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Research, 27(10), 1231–1242. https://doi.org/10.1038/cr.2017.106.
Sanders, R., Mason, D. J., Foy, C. A., & Huggett, J. F. (2013). Evaluation of digital PCR for absolute RNA quantification. PLoS One, 8(9), e75296. https://doi.org/10.1371/journal.pone.0075296.
Giraldez, M. D., Spengler, R. M., Etheridge, A., Godoy, P. M., Barczak, A. J., Srinivasan, S., et al. (2018). Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nature Biotechnology, 36(8), 746–757. https://doi.org/10.1038/nbt.4183.
Lim, S. B., Di Lee, W., Vasudevan, J., Lim, W. T., & Lim, C. T. (2019). Liquid biopsy: one cell at a time. NPJ Precision Oncology, 3, 23. https://doi.org/10.1038/s41698-019-0095-0.
Pantel, K., Brakenhoff, R. H., & Brandt, B. (2008). Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nature Reviews. Cancer, 8(5), 329–340. https://doi.org/10.1038/nrc2375.
Bidard, F. C., Peeters, D. J., Fehm, T., Nole, F., Gisbert-Criado, R., Mavroudis, D., et al. (2014). Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. The Lancet Oncology, 15(4), 406–414. https://doi.org/10.1016/S1470-2045(14)70069-5.
Bidard, F. C., Michiels, S., Riethdorf, S., Mueller, V., Esserman, L. J., Lucci, A., et al. (2018). Circulating tumor cells in breast cancer patients treated by neoadjuvant chemotherapy: a meta-analysis. Journal of the National Cancer Institute, 110(6), 560–567. https://doi.org/10.1093/jnci/djy018.
Adams, D. L., Stefansson, S., Haudenschild, C., Martin, S. S., Charpentier, M., Chumsri, S., et al. (2015). Cytometric characterization of circulating tumor cells captured by microfiltration and their correlation to the CellSearch((R)) CTC test. Cytometry. Part A, 87(2), 137–144. https://doi.org/10.1002/cyto.a.22613.
Lee, Y., Guan, G., & Bhagat, A. A. (2018). ClearCell(R) FX, a label-free microfluidics technology for enrichment of viable circulating tumor cells. Cytometry. Part A, 93(12), 1251–1254. https://doi.org/10.1002/cyto.a.23507.
Li, X., Corbett, A. L., Taatizadeh, E., Tasnim, N., Little, J. P., Garnis, C., et al. (2019). Challenges and opportunities in exosome research-Perspectives from biology, engineering, and cancer therapy. APL Bioengineering, 3(1), 011503. https://doi.org/10.1063/1.5087122.
Fraga, M. F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., et al. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics, 37(4), 391–400. https://doi.org/10.1038/ng1531.
Seligson, D. B., Horvath, S., Shi, T., Yu, H., Tze, S., Grunstein, M., et al. (2005). Global histone modification patterns predict risk of prostate cancer recurrence. Nature, 435(7046), 1262–1266. https://doi.org/10.1038/nature03672.
Gezer, U., Yoruker, E. E., Keskin, M., Kulle, C. B., Dharuman, Y., & Holdenrieder, S. (2015). Histone Methylation Marks on Circulating Nucleosomes as Novel Blood-Based Biomarker in Colorectal Cancer. International Journal of Molecular Sciences, 16(12), 29654–29662. https://doi.org/10.3390/ijms161226180.
Bauden, M., Pamart, D., Ansari, D., Herzog, M., Eccleston, M., Micallef, J., et al. (2015). Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clinical Epigenetics, 7, 106. https://doi.org/10.1186/s13148-015-0139-4.
Ulz, P., Thallinger, G. G., Auer, M., Graf, R., Kashofer, K., Jahn, S. W., et al. (2016). Inferring expressed genes by whole-genome sequencing of plasma DNA. Nature Genetics, 48(10), 1273–1278. https://doi.org/10.1038/ng.3648.
Vogelstein, B., & Kinzler, K. W. (1999). Digital PCR. Proceedings of the National Academy of Sciences of the United States of America, 96(16), 9236–9241. https://doi.org/10.1073/pnas.96.16.9236.
Zhu, Z., Jenkins, G., Zhang, W., Zhang, M., Guan, Z., & Yang, C. J. (2012). Single-molecule emulsion PCR in microfluidic droplets. Analytical and Bioanalytical Chemistry, 403(8), 2127–2143. https://doi.org/10.1007/s00216-012-5914-x.
Li, M., Diehl, F., Dressman, D., Vogelstein, B., & Kinzler, K. W. (2006). BEAMing up for detection and quantification of rare sequence variants. Nature Methods, 3(2), 95–97. https://doi.org/10.1038/nmeth850.
O'Leary, B., Hrebien, S., Beaney, M., Fribbens, C., Garcia-Murillas, I., Jiang, J., et al. (2019). Comparison of BEAMing and Droplet Digital PCR for Circulating Tumor DNA Analysis. Clinical Chemistry. https://doi.org/10.1373/clinchem.2019.305805.
Choi, M., Scholl, U. I., Ji, W., Liu, T., Tikhonova, I. R., Zumbo, P., et al. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America, 106(45), 19096–19101. https://doi.org/10.1073/pnas.0910672106.
Forshew, T., Murtaza, M., Parkinson, C., Gale, D., Tsui, D. W., Kaper, F., et al. (2012). Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Science Translational Medicine, 4(136), 136ra168. https://doi.org/10.1126/scitranslmed.3003726.
Koeppel, F., Blanchard, S., Jovelet, C., Genin, B., Marcaillou, C., Martin, E., et al. (2017). Whole exome sequencing for determination of tumor mutation load in liquid biopsy from advanced cancer patients. PLoS One, 12(11), e0188174. https://doi.org/10.1371/journal.pone.0188174.
Nakagawa, H., Wardell, C. P., Furuta, M., Taniguchi, H., & Fujimoto, A. (2015). Cancer whole-genome sequencing: present and future. Oncogene, 34(49), 5943–5950. https://doi.org/10.1038/onc.2015.90.
Chan, K. C., Jiang, P., Zheng, Y. W., Liao, G. J., Sun, H., Wong, J., et al. (2013). Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clinical Chemistry, 59(1), 211–224. https://doi.org/10.1373/clinchem.2012.196014.
Pasaniuc, B., Rohland, N., McLaren, P. J., Garimella, K., Zaitlen, N., Li, H., et al. (2012). Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nature Genetics, 44(6), 631–635. https://doi.org/10.1038/ng.2283.
Adalsteinsson, V. A., Ha, G., Freeman, S. S., Choudhury, A. D., Stover, D. G., Parsons, H. A., et al. (2017). Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nature Communications, 8(1), 1324. https://doi.org/10.1038/s41467-017-00965-y.
Hovelson, D. H., Liu, C. J., Wang, Y., Kang, Q., Henderson, J., Gursky, A., et al. (2017). Rapid, ultra low coverage copy number profiling of cell-free DNA as a precision oncology screening strategy. Oncotarget, 8(52), 89848–89866. https://doi.org/10.18632/oncotarget.21163.
Jaenisch, R., & Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics, 33(Suppl), 245–254. https://doi.org/10.1038/ng1089.
Bernstein, B. E., Meissner, A., & Lander, E. S. (2007). The mammalian epigenome. Cell, 128(4), 669–681. https://doi.org/10.1016/j.cell.2007.01.033.
Kustanovich, A., Schwartz, R., Peretz, T., & Grinshpun, A. (2019). Life and death of circulating cell-free DNA. Cancer Biology & Therapy, 20(8), 1057–1067. https://doi.org/10.1080/15384047.2019.1598759.
Luo, C., Hajkova, P., & Ecker, J. R. (2018). Dynamic DNA methylation: in the right place at the right time. Science, 361(6409), 1336–1340. https://doi.org/10.1126/science.aat6806.
Ehrlich, M. (2006). Cancer-linked DNA hypomethylation and its relationship to hypermethylation. Current Topics in Microbiology and Immunology, 310, 251–274. https://doi.org/10.1007/3-540-31181-5_12.
Ehrlich, M. (2009). DNA hypomethylation in cancer cells. Epigenomics, 1(2), 239–259. https://doi.org/10.2217/epi.09.33.
Melnikov, A. A., Gartenhaus, R. B., Levenson, A. S., Motchoulskaia, N. A., & Levenson Chernokhvostov, V. V. (2005). MSRE-PCR for analysis of gene-specific DNA methylation. Nucleic Acids Research, 33(10), e93. https://doi.org/10.1093/nar/gni092.
Sasaki, M., Anast, J., Bassett, W., Kawakami, T., Sakuragi, N., & Dahiya, R. (2003). Bisulfite conversion-specific and methylation-specific PCR: a sensitive technique for accurate evaluation of CpG methylation. Biochemical and Biophysical Research Communications, 309(2), 305–309. https://doi.org/10.1016/j.bbrc.2003.08.005.
Brunner, A. L., Johnson, D. S., Kim, S. W., Valouev, A., Reddy, T. E., Neff, N. F., et al. (2009). Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Research, 19(6), 1044–1056. https://doi.org/10.1101/gr.088773.108.
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D'Souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature, 466(7303), 253–257. https://doi.org/10.1038/nature09165.
Taiwo, O., Wilson, G. A., Morris, T., Seisenberger, S., Reik, W., Pearce, D., et al. (2012). Methylome analysis using MeDIP-seq with low DNA concentrations. Nature Protocols, 7(4), 617–636. https://doi.org/10.1038/nprot.2012.012.
Aberg, K. A., McClay, J. L., Nerella, S., Xie, L. Y., Clark, S. L., Hudson, A. D., et al. (2012). MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case--control samples. Epigenomics, 4(6), 605–621. https://doi.org/10.2217/epi.12.59.
Zhang, L., Szulwach, K. E., Hon, G. C., Song, C. X., Park, B., Yu, M., et al. (2013). Tet-mediated covalent labelling of 5-methylcytosine for its genome-wide detection and sequencing. Nature Communications, 4, 1517. https://doi.org/10.1038/ncomms2527.
Wang, Q., Gu, L., Adey, A., Radlwimmer, B., Wang, W., Hovestadt, V., et al. (2013). Tagmentation-based whole-genome bisulfite sequencing. Nature Protocols, 8(10), 2022–2032. https://doi.org/10.1038/nprot.2013.118.
Chan, K. C., Jiang, P., Chan, C. W., Sun, K., Wong, J., Hui, E. P., et al. (2013). Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proceedings of the National Academy of Sciences of the United States of America, 110(47), 18761–18768. https://doi.org/10.1073/pnas.1313995110.
Lee, E. J., Luo, J., Wilson, J. M., & Shi, H. (2013). Analyzing the cancer methylome through targeted bisulfite sequencing. Cancer Letters, 340(2), 171–178. https://doi.org/10.1016/j.canlet.2012.10.040.
Shen, S. Y., Singhania, R., Fehringer, G., Chakravarthy, A., Roehrl, M. H. A., Chadwick, D., et al. (2018). Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature, 563(7732), 579–583. https://doi.org/10.1038/s41586-018-0703-0.
Shen, S. Y., Burgener, J. M., Bratman, S. V., & De Carvalho, D. D. (2019). Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nature Protocols, 14(10), 2749–2780. https://doi.org/10.1038/s41596-019-0202-2.
Song, C. X., & He, C. (2013). Potential functional roles of DNA demethylation intermediates. Trends in Biochemical Sciences, 38(10), 480–484. https://doi.org/10.1016/j.tibs.2013.07.003.
Zeng, C., Stroup, E. K., Zhang, Z., Chiu, B. C., & Zhang, W. (2019). Towards precision medicine: advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Communications (Lond), 39(1), 12. https://doi.org/10.1186/s40880-019-0356-x.
Pastor, W. A., Pape, U. J., Huang, Y., Henderson, H. R., Lister, R., Ko, M., et al. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature, 473(7347), 394–397. https://doi.org/10.1038/nature10102.
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nature Biotechnology, 29(1), 68–72. https://doi.org/10.1038/nbt.1732.
Yu, M., Hon, G. C., Szulwach, K. E., Song, C. X., Zhang, L., Kim, A., et al. (2012). Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell, 149(6), 1368–1380. https://doi.org/10.1016/j.cell.2012.04.027.
Petterson, A., Chung, T. H., Tan, D., Sun, X., & Jia, X. Y. (2014). RRHP: a tag-based approach for 5-hydroxymethylcytosine mapping at single-site resolution. Genome Biology, 15(9), 456. https://doi.org/10.1186/s13059-014-0456-5.
Zeng, H., He, B., Xia, B., Bai, D., Lu, X., Cai, J., et al. (2018). Bisulfite-Free, Nanoscale Analysis of 5-Hydroxymethylcytosine at Single Base Resolution. Journal of the American Chemical Society, 140(41), 13190–13194. https://doi.org/10.1021/jacs.8b08297.
Hu, L., Liu, Y., Han, S., Yang, L., Cui, X., Gao, Y., et al. (2019). Jump-seq: genome-Wide Capture and Amplification of 5-Hydroxymethylcytosine Sites. Journal of the American Chemical Society, 141(22), 8694–8697. https://doi.org/10.1021/jacs.9b02512.
Wang, Y., Zhang, X., Wu, F., Chen, Z., & Zhou, X. (2019). Bisulfite-free, single base-resolution analysis of 5-hydroxymethylcytosine in genomic DNA by chemical-mediated mismatch. Chemical Science, 10(2), 447–452. https://doi.org/10.1039/c8sc04272a.
Han, D., Lu, X., Shih, A. H., Nie, J., You, Q., Xu, M. M., et al. (2016). A Highly Sensitive and Robust Method for Genome-wide 5hmC Profiling of Rare Cell Populations. Molecular Cell, 63(4), 711–719. https://doi.org/10.1016/j.molcel.2016.06.028.
Cai, J., Chen, L., Zhang, Z., Zhang, X., Lu, X., Liu, W., et al. (2019). Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut. https://doi.org/10.1136/gutjnl-2019-318882.
Chiu, B. C., Zhang, Z., You, Q., Zeng, C., Stepniak, E., Bracci, P. M., et al. (2019). Prognostic implications of 5-hydroxymethylcytosines from circulating cell-free DNA in diffuse large B-cell lymphoma. Blood Advances, 3(19), 2790–2799. https://doi.org/10.1182/bloodadvances.2019000175.
Gao, P., Lin, S., Cai, M., Zhu, Y., Song, Y., Sui, Y., et al. (2019). 5-Hydroxymethylcytosine profiling from genomic and cell-free DNA for colorectal cancers patients. Journal of Cellular and Molecular Medicine, 23(5), 3530–3537. https://doi.org/10.1111/jcmm.14252.
Zhang, J., Han, X., Gao, C., Xing, Y., Qi, Z., Liu, R., et al. (2018). 5-Hydroxymethylome in Circulating Cell-free DNA as A Potential Biomarker for Non-small-cell Lung Cancer. Genomics, Proteomics & Bioinformatics, 16(3), 187–199. https://doi.org/10.1016/j.gpb.2018.06.002.
Tian, X., Sun, B., Chen, C., Gao, C., Zhang, J., Lu, X., et al. (2018). Circulating tumor DNA 5-hydroxymethylcytosine as a novel diagnostic biomarker for esophageal cancer. Cell Research, 28(5), 597–600. https://doi.org/10.1038/s41422-018-0014-x.
Del Re, M., Marconcini, R., Pasquini, G., Rofi, E., Vivaldi, C., Bloise, F., et al. (2018). PD-L1 mRNA expression in plasma-derived exosomes is associated with response to anti-PD-1 antibodies in melanoma and NSCLC. British Journal of Cancer, 118(6), 820–824. https://doi.org/10.1038/bjc.2018.9.
David, L., Huber, W., Granovskaia, M., Toedling, J., Palm, C. J., Bofkin, L., et al. (2006). A high-resolution map of transcription in the yeast genome. Proceedings of the National Academy of Sciences of the United States of America, 103(14), 5320–5325. https://doi.org/10.1073/pnas.0601091103.
Wang, Z., Gerstein, M., & Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews. Genetics, 10(1), 57–63. https://doi.org/10.1038/nrg2484.
Buschmann, D., Haberberger, A., Kirchner, B., Spornraft, M., Riedmaier, I., Schelling, G., et al. (2016). Toward reliable biomarker signatures in the age of liquid biopsies - how to standardize the small RNA-Seq workflow. Nucleic Acids Research, 44(13), 5995–6018. https://doi.org/10.1093/nar/gkw545.
Giraldez, M. D., Spengler, R. M., Etheridge, A., Goicochea, A. J., Tuck, M., Choi, S. W., et al. (2019). Phospho-RNA-seq: a modified small RNA-seq method that reveals circulating mRNA and lncRNA fragments as potential biomarkers in human plasma. The EMBO Journal, 38(11). https://doi.org/10.15252/embj.2019101695.
Yuan, T., Huang, X., Woodcock, M., Du, M., Dittmar, R., Wang, Y., et al. (2016). Plasma extracellular RNA profiles in healthy and cancer patients. Scientific Reports, 6, 19413. https://doi.org/10.1038/srep19413.
Pinto, N. R., Applebaum, M. A., Volchenboum, S. L., Matthay, K. K., London, W. B., Ambros, P. F., et al. (2015). Advances in Risk Classification and Treatment Strategies for Neuroblastoma. Journal of Clinical Oncology, 33(27), 3008–3017. https://doi.org/10.1200/JCO.2014.59.4648.
Park, J. R., Kreissman, S. G., London, W. B., Naranjo, A., Cohn, S. L., Hogarty, M. D., et al. (2019). Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: a Randomized Clinical Trial. JAMA, 322(8), 746–755. https://doi.org/10.1001/jama.2019.11642.
Tsuchida, Y., Honna, T., Iwanaka, T., Saeki, M., Taguchi, N., Kaneko, T., et al. (1987). Serial determination of serum neuron-specific enolase in patients with neuroblastoma and other pediatric tumors. Journal of Pediatric Surgery, 22(5), 419–424. https://doi.org/10.1016/s0022-3468(87)80261-0.
Zeltzer, P. M., Marangos, P. J., Evans, A. E., & Schneider, S. L. (1986). Serum neuron-specific enolase in children with neuroblastoma. Relationship to stage and disease course. Cancer, 57(6), 1230–1234. https://doi.org/10.1002/1097-0142(19860315)57:6<1230::aid-cncr2820570628>3.0.co;2-#.
Kogner, P., Bjork, O., & Theodorsson, E. (1993). Neuropeptide Y in neuroblastoma: increased concentration in metastasis, release during surgery, and characterization of plasma and tumor extracts. Medical and Pediatric Oncology, 21(5), 317–322. https://doi.org/10.1002/mpo.2950210502.
Kogner, P., Theodorsson, E., & Bjork, O. (1991). Plasma neuropeptide Y (NPY): a novel marker of neuroblastoma. Progress in Clinical and Biological Research, 366, 367–373.
Kogner, P., Bjork, O., & Theodorsson, E. (1990). Neuropeptide Y as a marker in pediatric neuroblastoma. Pediatric Pathology, 10(1-2), 207–216. https://doi.org/10.3109/15513819009067108.
Valentino, L., Moss, T., Olson, E., Wang, H. J., Elashoff, R., & Ladisch, S. (1990). Shed tumor gangliosides and progression of human neuroblastoma. Blood, 75(7), 1564–1567.
Moss, T. J., & Sanders, D. G. (1990). Detection of neuroblastoma cells in blood. Journal of Clinical Oncology, 8(4), 736–740. https://doi.org/10.1200/JCO.1990.8.4.736.
Mattano Jr., L. A., Moss, T. J., & Emerson, S. G. (1992). Sensitive detection of rare circulating neuroblastoma cells by the reverse transcriptase-polymerase chain reaction. Cancer Research, 52(17), 4701–4705.
Seeger, R. C., Reynolds, C. P., Gallego, R., Stram, D. O., Gerbing, R. B., & Matthay, K. K. (2000). Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. Journal of Clinical Oncology, 18(24), 4067–4076. https://doi.org/10.1200/JCO.2000.18.24.4067.
Burchill, S. A., Lewis, I. J., Abrams, K. R., Riley, R., Imeson, J., Pearson, A. D., et al. (2001). Circulating neuroblastoma cells detected by reverse transcriptase polymerase chain reaction for tyrosine hydroxylase mRNA are an independent poor prognostic indicator in stage 4 neuroblastoma in children over 1 year. Journal of Clinical Oncology, 19(6), 1795–1801. https://doi.org/10.1200/JCO.2001.19.6.1795.
Viprey, V. F., Gregory, W. M., Corrias, M. V., Tchirkov, A., Swerts, K., Vicha, A., et al. (2014). Neuroblastoma mRNAs predict outcome in children with stage 4 neuroblastoma: a European HR-NBL1/SIOPEN study. Journal of Clinical Oncology, 32(10), 1074–1083. https://doi.org/10.1200/JCO.2013.53.3604.
Stutterheim, J., Gerritsen, A., Zappeij-Kannegieter, L., Yalcin, B., Dee, R., van Noesel, M. M., et al. (2009). Detecting minimal residual disease in neuroblastoma: the superiority of a panel of real-time quantitative PCR markers. Clinical Chemistry, 55(7), 1316–1326. https://doi.org/10.1373/clinchem.2008.117945.
Marachelian, A., Villablanca, J. G., Liu, C. W., Liu, B., Goodarzian, F., Lai, H. A., et al. (2017). Expression of Five Neuroblastoma Genes in Bone Marrow or Blood of Patients with Relapsed/Refractory Neuroblastoma Provides a New Biomarker for Disease and Prognosis. Clinical Cancer Research, 23(18), 5374–5383. https://doi.org/10.1158/1078-0432.CCR-16-2647.
Yanez, Y., Grau, E., Oltra, S., Canete, A., Martinez, F., Orellana, C., et al. (2011). Minimal disease detection in peripheral blood and bone marrow from patients with non-metastatic neuroblastoma. Journal of Cancer Research and Clinical Oncology, 137(8), 1263–1272. https://doi.org/10.1007/s00432-011-0997-x.
Zeka, F., Decock, A., Van Goethem, A., Vanderheyden, K., Demuynck, F., Lammens, T., et al. (2018). Circulating microRNA biomarkers for metastatic disease in neuroblastoma patients. JCI Insight, 3(23). https://doi.org/10.1172/jci.insight.97021.
Morini, M., Cangelosi, D., Segalerba, D., Marimpietri, D., Raggi, F., Castellano, A., et al. (2019). Exosomal microRNAs from Longitudinal Liquid Biopsies for the Prediction of Response to Induction Chemotherapy in High-Risk Neuroblastoma Patients: a Proof of Concept SIOPEN Study. Cancers (Basel), 11(10). https://doi.org/10.3390/cancers11101476.
Chicard, M., Boyault, S., Colmet Daage, L., Richer, W., Gentien, D., Pierron, G., et al. (2016). Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clinical Cancer Research, 22(22), 5564–5573. https://doi.org/10.1158/1078-0432.CCR-16-0500.
Van Roy, N., Van Der Linden, M., Menten, B., Dheedene, A., Vandeputte, C., Van Dorpe, J., et al. (2017). Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clinical Cancer Research, 23(20), 6305–6314. https://doi.org/10.1158/1078-0432.CCR-17-0675.
Chicard, M., Colmet-Daage, L., Clement, N., Danzon, A., Bohec, M., Bernard, V., et al. (2018). Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clinical Cancer Research, 24(4), 939–949. https://doi.org/10.1158/1078-0432.CCR-17-1586.
Klega, K., Imamovic-Tuco, A., Ha, G., Clapp, A. N., Meyer, S., Ward, A., et al. (2018). Detection of Somatic Structural Variants Enables Quantification and Characterization of Circulating Tumor DNA in Children With Solid Tumors. JCO Precision Oncology, 2018. https://doi.org/10.1200/PO.17.00285.
Kojima, M., Hiyama, E., Fukuba, I., Yamaoka, E., Ueda, Y., Onitake, Y., et al. (2013). Detection of MYCN amplification using blood plasma: noninvasive therapy evaluation and prediction of prognosis in neuroblastoma. Pediatric Surgery International, 29(11), 1139–1145. https://doi.org/10.1007/s00383-013-3374-9.
Combaret, V., Hogarty, M. D., London, W. B., McGrady, P., Iacono, I., Brejon, S., et al. (2009). Influence of neuroblastoma stage on serum-based detection of MYCN amplification. Pediatric Blood & Cancer, 53(3), 329–331. https://doi.org/10.1002/pbc.22009.
Gotoh, T., Hosoi, H., Iehara, T., Kuwahara, Y., Osone, S., Tsuchiya, K., et al. (2005). Prediction of MYCN amplification in neuroblastoma using serum DNA and real-time quantitative polymerase chain reaction. Journal of Clinical Oncology, 23(22), 5205–5210. https://doi.org/10.1200/JCO.2005.02.014.
Combaret, V., Bergeron, C., Noguera, R., Iacono, I., & Puisieux, A. (2005). Circulating MYCN DNA predicts MYCN-amplification in neuroblastoma. Journal of Clinical Oncology, 23(34), 8919–8920; author reply 8920. https://doi.org/10.1200/JCO.2005.04.0170.
Combaret, V., Audoynaud, C., Iacono, I., Favrot, M. C., Schell, M., Bergeron, C., et al. (2002). Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Research, 62(13), 3646–3648.
Lodrini, M., Sprussel, A., Astrahantseff, K., Tiburtius, D., Konschak, R., Lode, H. N., et al. (2017). Using droplet digital PCR to analyze MYCN and ALK copy number in plasma from patients with neuroblastoma. Oncotarget, 8(49), 85234–85251. https://doi.org/10.18632/oncotarget.19076.
Combaret, V., Brejon, S., Iacono, I., Schleiermacher, G., Pierron, G., Ribeiro, A., et al. (2011). Determination of 17q gain in patients with neuroblastoma by analysis of circulating DNA. Pediatric Blood & Cancer, 56(5), 757–761. https://doi.org/10.1002/pbc.22816.
Yagyu, S., Iehara, T., Gotoh, T., Miyachi, M., Katsumi, Y., Kikuchi, K., et al. (2011). Preoperative analysis of 11q loss using circulating tumor-released DNA in serum: a novel diagnostic tool for therapy stratification of neuroblastoma. Cancer Letters, 309(2), 185–189. https://doi.org/10.1016/j.canlet.2011.05.032.
Misawa, A., Tanaka, S., Yagyu, S., Tsuchiya, K., Iehara, T., Sugimoto, T., et al. (2009). RASSF1A hypermethylation in pretreatment serum DNA of neuroblastoma patients: a prognostic marker. British Journal of Cancer, 100(2), 399–404. https://doi.org/10.1038/sj.bjc.6604887.
Yagyu, S., Gotoh, T., Iehara, T., Miyachi, M., Katsumi, Y., Tsubai-Shimizu, S., et al. (2008). Circulating methylated-DCR2 gene in serum as an indicator of prognosis and therapeutic efficacy in patients with MYCN nonamplified neuroblastoma. Clinical Cancer Research, 14(21), 7011–7019. https://doi.org/10.1158/1078-0432.CCR-08-1249.
Hayashi, M., Zhu, P., McCarty, G., Meyer, C. F., Pratilas, C. A., Levin, A., et al. (2017). Size-based detection of sarcoma circulating tumor cells and cell clusters. Oncotarget, 8(45), 78965–78977. https://doi.org/10.18632/oncotarget.20697.
Barris, D. M., Weiner, S. B., Dubin, R. A., Fremed, M., Zhang, X., Piperdi, S., et al. (2018). Detection of circulating tumor DNA in patients with osteosarcoma. Oncotarget, 9(16), 12695–12704. https://doi.org/10.18632/oncotarget.24268.
Shulman, D. S., Klega, K., Imamovic-Tuco, A., Clapp, A., Nag, A., Thorner, A. R., et al. (2018). Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group. British Journal of Cancer, 119(5), 615–621. https://doi.org/10.1038/s41416-018-0212-9.
Allen-Rhoades, W., Kurenbekova, L., Satterfield, L., Parikh, N., Fuja, D., Shuck, R. L., et al. (2015). Cross-species identification of a plasma microRNA signature for detection, therapeutic monitoring, and prognosis in osteosarcoma. Cancer Medicine, 4(7), 977–988. https://doi.org/10.1002/cam4.438.
Ma, W., Zhang, X., Chai, J., Chen, P., Ren, P., & Gong, M. (2014). Circulating miR-148a is a significant diagnostic and prognostic biomarker for patients with osteosarcoma. Tumour Biology, 35(12), 12467–12472. https://doi.org/10.1007/s13277-014-2565-x.
Schleiermacher, G., Peter, M., Oberlin, O., Philip, T., Rubie, H., Mechinaud, F., et al. (2003). Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. Journal of Clinical Oncology, 21(1), 85–91. https://doi.org/10.1200/JCO.2003.03.006.
Hayashi, M., Chu, D., Meyer, C. F., Llosa, N. J., McCarty, G., Morris, C. D., et al. (2016). Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer, 122(19), 3015–3023. https://doi.org/10.1002/cncr.30144.
Krumbholz, M., Hellberg, J., Steif, B., Bauerle, T., Gillmann, C., Fritscher, T., et al. (2016). Genomic EWSR1 Fusion Sequence as Highly Sensitive and Dynamic Plasma Tumor Marker in Ewing Sarcoma. Clinical Cancer Research, 22(17), 4356–4365. https://doi.org/10.1158/1078-0432.CCR-15-3028.
Allegretti, M., Casini, B., Mandoj, C., Benini, S., Alberti, L., Novello, M., et al. (2018). Precision diagnostics of Ewing's sarcoma by liquid biopsy: circulating EWS-FLI1 fusion transcripts. Therapeutic Advances in Medical Oncology, 10, 1758835918774337. https://doi.org/10.1177/1758835918774337.
Eguchi-Ishimae, M., Tezuka, M., Kokeguchi, T., Nagai, K., Moritani, K., Yonezawa, S., et al. (2019). Early detection of the PAX3-FOXO1 fusion gene in circulating tumor-derived DNA in a case of alveolar rhabdomyosarcoma. Genes, Chromosomes & Cancer, 58(8), 521–529. https://doi.org/10.1002/gcc.22734.
Miyachi, M., Tsuchiya, K., Yoshida, H., Yagyu, S., Kikuchi, K., Misawa, A., et al. (2010). Circulating muscle-specific microRNA, miR-206, as a potential diagnostic marker for rhabdomyosarcoma. Biochemical and Biophysical Research Communications, 400(1), 89–93. https://doi.org/10.1016/j.bbrc.2010.08.015.
Jimenez, I., Chicard, M., Colmet-Daage, L., Clement, N., Danzon, A., Lapouble, E., et al. (2019). Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. International Journal of Cancer, 144(1), 68–79. https://doi.org/10.1002/ijc.31620.
Murray, M. J., Raby, K. L., Saini, H. K., Bailey, S., Wool, S. V., Tunnacliffe, J. M., et al. (2015). Solid tumors of childhood display specific serum microRNA profiles. Cancer Epidemiology, Biomarkers & Prevention, 24(2), 350–360. https://doi.org/10.1158/1055-9965.EPI-14-0669.
Ludwig, N., Nourkami-Tutdibi, N., Backes, C., Lenhof, H. P., Graf, N., Keller, A., et al. (2015). Circulating serum miRNAs as potential biomarkers for nephroblastoma. Pediatric Blood & Cancer, 62(8), 1360–1367. https://doi.org/10.1002/pbc.25481.
Schmitt, J., Backes, C., Nourkami-Tutdibi, N., Leidinger, P., Deutscher, S., Beier, M., et al. (2012). Treatment-independent miRNA signature in blood of Wilms tumor patients. BMC Genomics, 13, 379. https://doi.org/10.1186/1471-2164-13-379.
Treger, T. D., Chagtai, T., Butcher, R., Cresswell, G. D., Al-Saadi, R., Brok, J., et al. (2018). Somatic TP53 Mutations Are Detectable in Circulating Tumor DNA from Children with Anaplastic Wilms Tumors. Translational Oncology, 11(6), 1301–1306. https://doi.org/10.1016/j.tranon.2018.08.006.
Charlton, J., Williams, R. D., Weeks, M., Sebire, N. J., Popov, S., Vujanic, G., et al. (2014). Methylome analysis identifies a Wilms tumor epigenetic biomarker detectable in blood. Genome Biology, 15(8), 434. https://doi.org/10.1186/s13059-014-0434-y.
Liu, W., Chen, S., & Liu, B. (2016). Diagnostic and prognostic values of serum exosomal microRNA-21 in children with hepatoblastoma: a Chinese population-based study. Pediatric Surgery International, 32(11), 1059–1065. https://doi.org/10.1007/s00383-016-3960-8.
Jiao, C., Jiao, X., Zhu, A., Ge, J., & Xu, X. (2017). Exosomal miR-34s panel as potential novel diagnostic and prognostic biomarker in patients with hepatoblastoma. Journal of Pediatric Surgery, 52(4), 618–624. https://doi.org/10.1016/j.jpedsurg.2016.09.070.
Gerson, J. M., Schlesinger, H. R., Sereni, P., Moorhead, P. S., & Hummeler, K. (1977). Isolation and characterization of a neuroblastoma cell line from peripheral blood in a patient with disseminated disease. Cancer, 39(6), 2508–2512. https://doi.org/10.1002/1097-0142(197706)39:6<2508::aid-cncr2820390630>3.0.co;2-x.
Pelkey, T. J., Frierson Jr., H. F., & Bruns, D. E. (1996). Molecular and immunological detection of circulating tumor cells and micrometastases from solid tumors. Clinical Chemistry, 42(9), 1369–1381.
Kreissman, S. G., Seeger, R. C., Matthay, K. K., London, W. B., Sposto, R., Grupp, S. A., et al. (2013). Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. The Lancet Oncology, 14(10), 999–1008. https://doi.org/10.1016/S1470-2045(13)70309-7.
Miyajima, Y., Kato, K., Numata, S., Kudo, K., & Horibe, K. (1995). Detection of neuroblastoma cells in bone marrow and peripheral blood at diagnosis by the reverse transcriptase-polymerase chain reaction for tyrosine hydroxylase mRNA. Cancer, 75(11), 2757–2761. https://doi.org/10.1002/1097-0142(19950601)75:11<2757::aid-cncr2820751120>3.0.co;2-s.
Miyajima, Y., Horibe, K., Fukuda, M., Matsumoto, K., Numata, S., Mori, H., et al. (1996). Sequential detection of tumor cells in the peripheral blood and bone marrow of patients with stage IV neuroblastoma by the reverse transcription-polymerase chain reaction for tyrosine hydroxylase mRNA. Cancer, 77(6), 1214–1219. https://doi.org/10.1002/(sici)1097-0142(19960315)77:6<1214::aid-cncr31>3.0.co;2-2.
Uemura, S., Ishida, T., Thwin, K. K. M., Yamamoto, N., Tamura, A., Kishimoto, K., et al. (2019). Dynamics of Minimal Residual Disease in Neuroblastoma Patients. Frontiers in Oncology, 9, 455. https://doi.org/10.3389/fonc.2019.00455.
Viprey, V. F., Corrias, M. V., Kagedal, B., Oltra, S., Swerts, K., Vicha, A., et al. (2007). Standardisation of operating procedures for the detection of minimal disease by QRT-PCR in children with neuroblastoma: quality assurance on behalf of SIOPEN-R-NET. European Journal of Cancer, 43(2), 341–350. https://doi.org/10.1016/j.ejca.2006.08.007.
Schleiermacher, G., Mosseri, V., London, W. B., Maris, J. M., Brodeur, G. M., Attiyeh, E., et al. (2012). Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. British Journal of Cancer, 107(8), 1418–1422. https://doi.org/10.1038/bjc.2012.375.
Pinto, N., Mayfield, J. R., Raca, G., Applebaum, M. A., Chlenski, A., Sukhanova, M., et al. (2016). Segmental Chromosomal Aberrations in Localized Neuroblastoma Can be Detected in Formalin-Fixed Paraffin-Embedded Tissue Samples and Are Associated With Recurrence. Pediatric Blood & Cancer, 63(6), 1019–1023. https://doi.org/10.1002/pbc.25934.
Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E., & Bishop, J. M. (1984). Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science, 224(4653), 1121–1124.
Pugh, T. J., Morozova, O., Attiyeh, E. F., Asgharzadeh, S., Wei, J. S., Auclair, D., et al. (2013). The genetic landscape of high-risk neuroblastoma. Nature Genetics, 45(3), 279–284. https://doi.org/10.1038/ng.2529.
Durinck, K., & Speleman, F. (2018). Epigenetic regulation of neuroblastoma development. Cell and Tissue Research, 372(2), 309–324. https://doi.org/10.1007/s00441-017-2773-y.
Brodeur, G. M., & Bagatell, R. (2014). Mechanisms of neuroblastoma regression. Nature Reviews. Clinical Oncology, 11(12), 704–713. https://doi.org/10.1038/nrclinonc.2014.168.
Applebaum, M. A., Barr, E. K., Karpus, J., Nie, J., Zhang, Z., Armstrong, A. E., et al. (2019). 5-Hydroxymethylcytosine Profiles Are Prognostic of Outcome in Neuroblastoma and Reveal Transcriptional Networks That Correlate With Tumor Phenotype. JCO Precision Oncology, (3), 1–12. https://doi.org/10.1200/PO.18.00402.
van Groningen, T., Koster, J., Valentijn, L. J., Zwijnenburg, D. A., Akogul, N., Hasselt, N. E., et al. (2017). Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nature Genetics, 49(8), 1261–1266. https://doi.org/10.1038/ng.3899.
Ostler, K. R., Yang, Q., Looney, T. J., Zhang, L., Vasanthakumar, A., Tian, Y., et al. (2012). Truncated DNMT3B isoform DNMT3B7 suppresses growth, induces differentiation, and alters DNA methylation in human neuroblastoma. Cancer Research, 72(18), 4714–4723. https://doi.org/10.1158/0008-5472.CAN-12-0886.
Yang, Q., Kiernan, C. M., Tian, Y., Salwen, H. R., Chlenski, A., Brumback, B. A., et al. (2007). Methylation of CASP8, DCR2, and HIN-1 in neuroblastoma is associated with poor outcome. Clinical Cancer Research, 13(11), 3191–3197. https://doi.org/10.1158/1078-0432.CCR-06-2846.
Lu, Z., Tian, Y., Salwen, H. R., Chlenski, A., Godley, L. A., Raj, J. U., et al. (2013). Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anti-Cancer Drugs, 24(5), 484–493. https://doi.org/10.1097/CAD.0b013e32835ffdbb.
Decock, A., Ongenaert, M., Van Criekinge, W., Speleman, F., & Vandesompele, J. (2016). DNA methylation profiling of primary neuroblastoma tumors using methyl-CpG-binding domain sequencing. Scientific Data, 3, 160004. https://doi.org/10.1038/sdata.2016.4.
Olsson, M., Beck, S., Kogner, P., Martinsson, T., & Caren, H. (2016). Genome-wide methylation profiling identifies novel methylated genes in neuroblastoma tumors. Epigenetics, 11(1), 74–84. https://doi.org/10.1080/15592294.2016.1138195.
Decock, A., Ongenaert, M., Cannoodt, R., Verniers, K., De Wilde, B., Laureys, G., et al. (2016). Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget, 7(2), 1960–1972. https://doi.org/10.18632/oncotarget.6477.
Applebaum, M., Barr, E., Karpus, J., West-Szymanski, D., Zhang, W., Salwen, H., et al. 5-Hydroxymethylcytosine (5HMC) Profiles of Cell-Free DNA (CFDNA): novel Liquid Biopsy Biomarkers for Children with Neuroblastoma. In Pediatric blood & cancer, 2019 (Vol. 66, pp. S318–S318). Hoboken: Wiley.
Burningham, Z., Hashibe, M., Spector, L., & Schiffman, J. D. (2012). The epidemiology of sarcoma. Clinical Sarcoma Research, 2(1), 14. https://doi.org/10.1186/2045-3329-2-14.
Reed, D. R., Hayashi, M., Wagner, L., Binitie, O., Steppan, D. A., Brohl, A. S., et al. (2017). Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer, 123(12), 2206–2218. https://doi.org/10.1002/cncr.30589.
McBride, D. J., Orpana, A. K., Sotiriou, C., Joensuu, H., Stephens, P. J., Mudie, L. J., et al. (2010). Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes, Chromosomes & Cancer, 49(11), 1062–1069. https://doi.org/10.1002/gcc.20815.
Vo, K. T., Edwards, J. V., Epling, C. L., Sinclair, E., Hawkins, D. S., Grier, H. E., et al. (2016). Impact of Two Measures of Micrometastatic Disease on Clinical Outcomes in Patients with Newly Diagnosed Ewing Sarcoma: a Report from the Children's Oncology Group. Clinical Cancer Research, 22(14), 3643–3650. https://doi.org/10.1158/1078-0432.CCR-15-2516.
Irtan, S., Ehrlich, P. F., & Pritchard-Jones, K. (2016). Wilms tumor: "State-of-the-art" update, 2016. Seminars in Pediatric Surgery, 25(5), 250–256. https://doi.org/10.1053/j.sempedsurg.2016.09.003.
Brioude, F., Lacoste, A., Netchine, I., Vazquez, M. P., Auber, F., Audry, G., et al. (2013). Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Hormone Research in Pædiatrics, 80(6), 457–465. https://doi.org/10.1159/000355544.
Bisogno, G., De Salvo, G. L., Bergeron, C., Gallego Melcon, S., Merks, J. H., Kelsey, A., et al. (2019). Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology, 20(11), 1566–1575. https://doi.org/10.1016/S1470-2045(19)30617-5.
Parikh, A. R., Leshchiner, I., Elagina, L., Goyal, L., Levovitz, C., Siravegna, G., et al. (2019). Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nature Medicine, 25(9), 1415–1421. https://doi.org/10.1038/s41591-019-0561-9.
Funding
MAA is supported by the NIH Grant Number and K08CA226237. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
CH is a shareholder of Shanghai Epican Genetech Co. Ltd. that licensed 5hmC-Seal from The University of Chicago. CH is a scientific founder and scientific advisory board member of Accent Therapeutics, Inc.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Daniel A. Weiser, Diana C. West-Szymanski and Ellen Fraint Denotes co-first authors
Rights and permissions
About this article

Cite this article
Weiser, D.A., West-Szymanski, D.C., Fraint, E. et al. Progress toward liquid biopsies in pediatric solid tumors. Cancer Metastasis Rev 38, 553–571 (2019). https://doi.org/10.1007/s10555-019-09825-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-019-09825-1