
Abstract
The application of microfluidic droplet PCR for single-molecule amplification and analysis has recently been extensively studied. Microfluidic droplet technology has the advantages of compartmentalizing reactions into discrete volumes, performing highly parallel reactions in monodisperse droplets, reducing cross-contamination between droplets, eliminating PCR bias and nonspecific amplification, as well as enabling fast amplification with rapid thermocycling. Here, we have reviewed the important technical breakthroughs of microfluidic droplet PCR in the past five years and their applications to single-molecule amplification and analysis, such as high-throughput screening, next generation DNA sequencing, and quantitative detection of rare mutations. Although the utilization of microfluidic droplet single-molecule PCR is still in the early stages, its great potential has already been demonstrated and will provide novel solutions to today’s biomedical engineering challenges in single-molecule amplification and analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The abilities to selectively and sensitively detect single molecules, especially single biomolecules such as DNA and proteins, could lead to enormous breakthroughs in chemistry, biology, medicine and environmental science [1–3]. Compared with ensemble method, single molecule measurements explore the information of time trajectories and status distributions of individual molecules in various systems, which is useful for studying chemical and biological reaction pathways [3–6]. For instance, nucleic acid molecules have been revealed to be responsible for a wide range of cellular functions, such as regulation/silencing of gene expression, structural support for molecular machines, and precise control of cell behaviors [7–9]. The ability to detect gene expression with the sensitivity at the single-molecule level will have considerable value in basic biomedical research, drug discovery and medical diagnostics [7–9]. It is also necessary to distinguish between expression products of related genes, since it is becoming increasingly clear that single nucleotide changes can have significant effects on the biological activity of a gene product and are highly related to various disease developments [10, 11]. One of the most promising technologies for nucleic acid analysis at the single-molecule level is the so-called polymerase chain reaction (PCR), due to its dramatic amplification capability across several orders of magnitude that can generate thousands to millions of copies of a particular DNA sequence from even a single copy [12–15].
Microfluidic technology associated with micro total analysis systems (μTAS), or lab-on-chip, has been developing rapidly with the potential to significantly transform modern chemistry and biology. Microfluidic devices offer many advantages, including low consumption of reagents, high-throughput capabilities, accelerated chemical reactions and heat transfer, and the ability to build cheap and portable devices. One subcategory of microfluidics is droplet-based microfluidics which creates discrete volumes with the use of immiscible phases. The ultrahigh-throughput generation of uniform droplets with nL to pL volume greatly enhances the capability of microfluidics to perform a large number of reactions without increasing device size or complexity. In the past ten years or so, microfluidic droplets have been widely applied to material synthesis, molecular evolution, protein crystallization, drug delivery, as well as single-molecule and single-cell analysis [16–20].
In this review, we will specifically focus on the recent development and application of microfluidic droplets for single-molecule PCR. A brief overview of PCR technology for single-molecule analysis will be presented together with the limitations that have to be addressed. Then, the solutions offered by microfluidic droplets will be given with some recent breakthroughs highlighted in this area. Finally, how this technique can open up new research areas will be examined. More comprehensive reviews on droplet microfluidics can be referred elsewhere [16, 21–24].
Conventional PCR and its limitation on single-molecule analysis
Since the invention of PCR in 1983 by Kary Mullis for DNA amplification [25, 26], PCR has been widely used in biomedical research laboratories, and has revolutionized the life science applications and related areas, such as biological, medical, clinical, and forensic analysis. Due to its dramatic amplification capability across several orders of magnitude that can generate thousands to millions of copies of a particular DNA sequence from even a single copy, it becomes the most promising tool for single-molecule genetic analysis [12–15]. However, to perform single-molecule analysis, conventional PCR often suffers from critical requirements for the experiment parameters in order to achieve the maximum product yield whilst minimizing nonspecific amplification [27]. Moreover, conventional PCR needs to consume a large amount of expensive reagents, and has a preference for amplifying short fragments and producing short chimeric molecules, which is fatal to single-molecule analysis with such low template concentration [13, 15]. The throughput of conventional PCR is also limited by the spatial constraints and the long thermal-cycling time due to the low heating and cooling rates for the relatively large total thermal mass [27].
To overcome all these shortcomings, several attempts have been made to achieve single-molecule amplification capability. Two or more sequential PCRs have been performed, often using nested sets of primers, in order to suppress formation of non-specific products that interfere with PCR and achieve single-molecule sensitivity [28, 29]. However, the requirement of opening vessels between first and second steps is prone to the possibility of cross-contamination.
Later on, it was realized that reaction volume might be a key issue for single-molecule PCR [30]. The TaqMan assay requires near saturating amounts of PCR product to detect enhanced fluorescence, ~1011 product molecules/μL. To reach this concentration of product after 30 cycles in a 10 μL PCR requires at least 103 starting template molecules, i.e.103 × 230/10μL = 1011/μL. With less starting template molecules and more cycle numbers, under special conditions single molecule can be detectable, but this often fails due to the competition between the desired PCR fragments and spurious PCR products, such as primer-dimer. On the other hand, if the PCR volume were reduced to 10 nL, a single template molecule might be sufficient to generate a saturating concentration of PCR product after ~30 cycles, i.e. 1 × 230/10nL = 1011/μL. Kalinina et al. have shown a novel nanoliter scale PCR method using glass capillaries and TaqMan probes for fluorescent detection of amplicons to achieve single-molecule amplification [30]. Meanwhile, stochastic PCR amplification of single DNA template followed by capillary electrophoretic analysis has been realized on an integrated microfluidic device [31]. These micro-devices increase the effective concentration of the initial template DNA by reducing the reactor volume. However, due to the increasing surface-to-volume ratio of these micro-devices, the interaction between the channel surface and the reagent/sample is the major challenge due to induced PCR inhibition and carryover contamination. This problem is particularly serious when working with a single DNA template. Moreover, the particular requirements for the devices and technology limit the throughput of single-molecule detection.
To further reduce the reaction volume without sacrificing PCR efficiency and throughput, single-molecule emulsion PCR was developed [32]. In emulsion PCR, water-in-oil (W/O) emulsion was utilized to generate numerous droplets of reaction mixture in bulk oil phase, capable of performing millions of independent reactions in parallel. This method consists of two steps: the reaction mixture containing the template DNA and PCR reagents is emulsified into the bulk oil phase, which is then followed by the PCR amplification step. For water droplets in the W/O emulsion with diameters about 2 to 10 μm, the volume ranges from 4.2 fL to 0.52 pL and the effective concentration for single DNA molecule in the droplet is around 3.2pM to 0.4nM, which is sufficient to conduct single-molecule PCR. The increased concentration of the initial template DNA can also minimize the interference of the primer-dimer effect and improve the specificity of PCR [33]. Moreover, the hydrophobicity of the oil phase surrounding the droplets can prevent the contact between the surface of the reactor walls and the sample by confining the sample in droplets, thus eliminating the adverse effects due to the large surface-to-volume ratio encountered in microfluidic devices. Although single-molecule amplification could be achieved, the downstream high-throughput manipulation and analysis is quite difficult due to the propensity for loss of monoclonality when the oil phase is removed.
To address these problems, a method called BEAMing was developed on the basis of four of its principal components: beads, emulsion, amplification, and magnetics [34, 35]. In BEAMing, the single-molecule emulsion PCR is performed by encapsulating a single primer-immobilized microbead with a single DNA template in individual droplets. The DNA template is amplified on the microbead in the same droplet with thousands of DNA copies coated on the bead, which prevents the possibility of the mixing of DNA products from different droplets after breaking of the emulsion droplets. The use of microbeads is essential to maintain monoclonality of the droplets during downstream processing. Millions beads can be analyzed within minutes using flow cytometry or optical scanning instruments. Such advances have led to the ability to identify and quantify rare mutant genes within large populations and have also enabled the new generation of high-throughput sequencing systems [36, 37]. Ideally, each aqueous droplet should encapsulate one microbead together with similar volumes/quantities of template and reagents to ensure reliable and efficient results. However, the low monodispersity of current techniques for emulsion generation, such as aggressively stirring or agitating, that generates droplets with a diameter distribution of up to 2 orders of magnitude, leads to wide variations in PCR reagents and the probability of encapsulating one microbead in each droplet is low. This leads to low PCR efficiencies, non-uniform amplification, limitation on the length of the DNA amplicons (~250bp), as well as the reduction of the number of parallel reactions which can be successfully performed [38].
General operations of microfluidic droplets
To overcome the aforementioned limitation of conventional PCR for single-molecule analysis, microfluidic techniques for droplet generation becomes an overwhelmingly attractive alternative, which allows a high degree of control over the droplet size and composition in a high-throughput manner. Hence, microfluidic droplet techniques have been increasingly applied to PCR technology. In this section, general information about droplet generation and manipulation will be briefly introduced.
Droplet generation
The high-throughput generation of uniform droplets and the accurate control over the size, shape, morphology and monodispersity of droplets are the prerequisites of entry to this ultrasmall-volume field. Traditional top-down methods of creating droplets by emulsification, such as aggressively stirring or agitating using two immiscible fluids, result in heterogeneous droplets with a broad size distribution. However, for single-molecule and single-cell assays, the sensitivity, accuracy and reproducibility are highly dependent on the manufacture of droplets with high requirements on their monodispersity and homogeneity. Droplets formed by microfluidics are generally monodisperse with less than 1% size variations. Meanwhile, the size of droplets can be precisely tuned by the channel geometry, the surface/interfacial tension, the shear force, liquid velocity, etc. Among the reported droplet-based systems, T-junction and flow-focusing techniques are frequently adopted to continuously generate droplets utilizing the channel geometry.
T-junction
In general, T-junction geometries contain a continuous phase main channel and a disperse phase inlet channel, perpendicular to each other, which looks like the two branches of the “T” (Fig. 1a) [39–41]. A droplet formation cycle starts with the stream of the disperse phase (water) penetrating into the main channel (oil), and a droplet begins to grow. The pressure gradient, the shear force, and the interfacial tension at the fluid-fluid interface distort and elongate the droplet in the downstream direction, until the neck of the disperse phase becomes thin and eventually breaks to release the droplet downstream into the main channel. Then the tip of disperse phase retracts to the end of inlet and the process repeats. Empirically, the size of droplet and its generation process are highly dependent on the capillary number, the flow rates, the viscosity ratio, and the channel geometry [39, 42].

To fully understand this process and achieve fine control over the droplet size, several models have been proposed in recent years to facilitate the optimization and predication of droplet size. For example, Steijn et al. presented a theoretical model to control the droplet size by the geometry of a junction with fixed flow rates [43]. Chiu and colleagues performed a systematic experimental study to validate this model and improve it by highlighting the importance of physical fluid properties [44]. These and other models [42, 45, 46] greatly advance our understanding and predictive capability over the droplet size, which is important for the accuracy and reproducibility of single-molecule and single-cell applications using microfluidics.
Flow-focusing
In the flow-focusing configuration, as shown in Fig. 1b, a liquid flows in a central channel and a second immiscible liquid flows through two symmetric perpendicular channels. The two liquid phases are then impelled to flow through a small orifice that is located downstream of the inlet channels. The outer fluid applies pressure and viscous stresses that drive the inner fluid into a narrow strand, which then breaks into droplets inside or downstream of the orifice [47, 48]. The design employs symmetric shearing force by the continuous phase which enables more stable and controllable generation of droplets [49]. Again, the droplet size, monodispersity, and droplet generation frequency are highly dependent on flow rate, liquid properties, and device geometry. Yobas et al. found that the size of droplets can be decreased by increasing flow rates of the continuous phase with the increase of generation frequency [50]. Another important factor is interfacial tension. As illustrated by Peng et al., interfacial tension can be expected to decrease with a decrease in droplet size and generation rate and monodisperse droplets of a different size could be obtained by simply adjusting the concentration of surfactant [51]. Many variations of basic flow-focusing design and the channel fabrication have been developed to facilitate more complex applications. For example, in Fig. 1c, Takeuchi et al. fabricated a microfluidic axisymmetric flow-focusing device that confines droplets in the central axis of a microfluidic channel and avoids contact of the droplets with the walls of the outlet channel, thus preventing the damage resulting from shearing, adhesion or wetting at the walls [52]. Utada et al. designed a coaxial microcapillary fluidic device to generate double emulsions that contained a single internal droplet in a core-shell geometry [53].
Droplet manipulation
Once the droplets are formed, further controls are then required to split the droplets for scaling up parallel reactions, fuse droplets with different reagents, mix reagents inside droplets, as well as analyze the content of droplets and sort them for desired properties. With the growth in this field, various technologies have been developed to facilitate the precise operation and manipulation of droplets on demand. These controls can be achieved either passively or actively.
Droplet fission
Since each droplet can serve as a vessel for reagents, by splitting a single droplet into two or more droplets, droplet size can be reduced, the content concentrations of daughter droplets can be controlled, and the experimental capacity can be easily scaled up. It can be achieved by passive or active methods. Passive methods mainly rely on the shear forces created by channel geometry to split the droplets at certain positions, such as T-junction and branching channels (Fig. 2a) [54–56]. For instance, Hatch et al. designed a 256 droplet-splitter that can generate over 1-million, momodisperse, 50 pL droplets in 2-7 minutes for PCR, thus greatly increasing the throughput and dynamic-range of digital PCR assays [56]. In contrast, active methods may rely on external power or electrical control of the splitting mechanism. Shown in Fig. 2b, by charging the oil-water interface, precise control of droplet splitting can be realized by electrostatic force [57]. Electrowetting on dielectric (EWOD) is also a powerful technology to actively divide a droplet into smaller droplets [58].

Droplet manipulation. (a) Droplet fission by T-junction (adapted from Ref [55]). (b) Droplet fission by electrostatic force (adapted from Ref [57]). (c) Droplet fusion by channel geometry (adapted from Ref [54]). (d) Droplet fusion by electrical field (adapted from Ref [57]). (e) Mixing within droplet inside winding channels (adapted from Ref [59]). (f) Bumpy serpentine mixer for fast mixing in droplet (adapted from Ref [68]). (g) Gravity-driven droplet sorting (adapted from Ref [73]). (h) Droplet sorting by DEP (adapted from Ref [74])
Droplet fusion
Time controlled coalescence of droplets is important for performing reactions within droplets (Fig. 2c–d). It has been achieved by a variety of methods including: matching the frequency of two sets of droplets in order to merge within the main channel, geometry design to control flow rates, channel obstruction, channel expansion, and selective hydrophilic treatment of a portion of a microchannel [54, 59–62]. It can also been achieved using EWOD, dielectrophoresis (DEP) and other electrically-controlled methods [57, 63–65]. The merging can take place between droplets with either the same or different sizes and with two droplets or several droplets [54, 57].
Mixing in droplets
The control of mixing is an essential prerequisite for carrying out and studying the kinetics of biological and chemical reactions. Rapid and full mixing of reagents is necessary to decrease reaction time and improve the reaction efficiency. Chaotic advection is a useful technique to achieve complete mixing in microfluidics [66]. It relies on repeated folding and stretching of the two fluids inside the droplet and is created mainly by channel geometries, such as the use of winding channels (Fig. 2e) [59, 67]. The addition of bumpy mixer or small protrusions can further increase the circulation within droplets, and even viscous biological samples have been reported to be mixed well within milliseconds (Fig. 2f) [68, 69]. Regarding active mixing, electric control can also be applied for mixing inside droplets, which allows flexible manipulation of individual droplets [70, 71].
Droplet sorting
A more challenging operation is droplet sorting, which can analyze and transport a droplet individually out of a population. Many microfluidic sorters have been developed, using mechanical, optical, magnetic, electro-osmotic, electrophoretic, and DEP actuations. For example, channel geometry has been employed to sort droplets passively by size, and droplets with a size difference as small as 4 μm can be differentiated [72]. As shown in Fig. 2g, Huh et al. developed the μSOHSA—microfluidic sorting device with hydrodynamic separation amplification—which is able to sort different sizes of particles based on gravity induced sorting and channel geometry [73]. Baret et al. developed a highly efficient microfluidic sorting system that actively sorts droplets using DEP based on their fluorescence signal at rate up to 2000 droplets/s, named fluorescence-activated droplet sorting (FADS) (Fig. 2h) [74]. It could be used to sort bacteria, cells, viruses, or even single genes expressed in vitro.
In addition to the methods mentioned above, more techniques have been designed to be incorporated with droplet-based microfluidic systems, such as droplet trapping [75–78], incubation [79–81], and presentation to an analytical instrument [17, 82–84]. Meanwhile, in order to carry out complex biological experiments and produce automated droplet microfluidic systems, module integration is a major challenge to be addressed. Many integrated devices have been developed to enable multiple droplet operations, from droplet generation to final analysis all on chip, thus realizing laboratory miniaturization in droplets. More detailed reviews can be found elsewhere [21, 23, 24].
Microfluidic droplet PCR
Microfluidic techniques for droplet generation allow a high degree of control over the droplet size and composition with particularly good monodispersity, at the rates of up to several thousand per second. Due to the high surface-to-volume ratios at the microscale, heat and mass transfer times are shorter, facilitating fast reaction time. Similar to the W/O emulsion, the contact between the surface of channels and the sample inside droplets has been eliminated by the surrounding immiscible fluid. Meanwhile, droplet microfluidics allows the independent control over each droplet, such as mixing, transporting and analysis. Since identical droplets are produced at a very high frequency within one experiment, massively parallel processing and experimentation can be easily achieved in ultra-short time and with confident reproducibility.
Hence, such techniques are well suited to emulsion PCR. Microfluidic droplet techniques have been increasingly applied to PCR due to the inherent advantages of compartmentalizing reactions into discrete volumes, performing highly parallel reactions in monodisperse droplets, reducing cross-contamination between droplets, eliminating PCR bias and nonspecific amplification, as well as enabling fast amplification with rapid thermocycling. Herein, a summary of reaction conditions used in different microfluidic droplet PCR systems will be presented, followed by specific examples of advanced microfluidic droplet techniques for single-molecule PCR.
Reaction conditions in different microfluidic droplet PCR protocols
In general, microfluidic droplet PCR can be classified into two categories based on whether the amplification is performed on-chip or off-chip. For on-chip amplification, either the whole chip has to go through thermocycles for amplifications [85, 86], or the specific geometry designs have been made such that droplets can go through different temperature zones inside the chip [87, 88]. These highly integrated devices will facilitate the development of lab-on-a-chip toolkits as well as the future evolution of chip-based instrumentation. However, the control over amplification conditions on-chip is still very limited with simplified procedures. To guarantee the PCR amplification efficiency for sensitive detection, most of microfluidic droplet PCR is still performed using off-chip amplification. In some other cases, such as if the droplets contain microbeads [38, 89] or require other materials [90, 91], off-chip amplification will be more compatible. For off-chip amplification, uniform droplets are generated on chip, collected in a PCR tube, and amplified inside a PCR machine. Afterwards, the droplets can be reinjected into the microfluidic device for on-chip analysis. Or, the emulsion will be broken and the products can be collected for off-chip analysis, such as gel electrophoresis, sequencing, etc.
The thermostability of droplets is very important for their successful application and becomes a prerequisite of W/O droplet vesicles. Droplet stability depends on a number of factors such as oil phase and surfactants in oil/aqueous phases. Especially, these materials also need to be compatible with PCR conditions. Other factors that need to be considered include the droplet generation method, the generation frequency, droplet volume as well as amplicon size. Table 1, has summarized the experimental conditions used in different microfluidic droplet PCR protocols.
Bead-free on-chip emulsion PCR in microfluidic droplets
The first successful example of combining emulsion PCR with microfluidic droplets was reported by Beer et al., where an on-chip digital microfluidic real-time PCR instrument was developed for generating monodisperse microdroplet reactors in a single channel chamber, thermal cycling for amplification reaction, and monitoring amplification real-time in the individual picoliter droplets [85]. This system utilized a shearing cross-flow T-junction in a silicon device to generate W/O microdroplets (Fig. 3a). Highly monodisperse droplets were generated at the rate 1 kHz with diameter around 24-31μm at different experimental conditions and with a variation of less than 1μm. Figure 3b schematically illustrates their integrated instrument with droplet generator, thermal cycler and fluorescence detector. In particular, an off-chip valving system can stop the droplet generation before performing thermal cycling and real-time monitoring on-chip. The six order of magnitude reduction of reactor size allowed detection of a single-molecule DNA at significantly reduced cycle thresholds with high-throughput and low reagent usage to achieve single copy PCR amplification (Fig. 3c). Subsequently, they developed a similar system for on-chip single-copy real-time Reverse-Transcription PCR in monodisperse picoliter droplets [86]. Hence, single-molecule PCR throughput is significantly increased, and DNA, RNA and viruses from a complex environment could be isolated and detected for single-molecule or single-cell genetic analysis and gene-profiling applications.

On-chip, real-time, single-molecule PCR in picoliter droplet reactors. (a) Overall channel and flow configuration. (b) Schematic of the integrated instrument for real-time PCR in droplets. (c) Real-time PCR data from picoliter droplets at an estimated 7, ~0.06, and 0 copies of genomic DNA per single droplet, respectively. Droplets were identified from the bright-field image and then monitored at each cycle to generate real-time fluorescence curves. (Adapted from Ref [85])
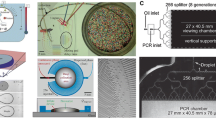
However, the set-ups presented above have involved thermal cycling of the entire device, thus limiting cycling rate and throughput. To further improve the throughput of single-molecule droplet PCR, several types of chip design have been proposed. Kiss et al. presented a continuous-flow-based real-time PCR system, in which a range of template concentrations could be detected and the results agreed closely with the frequencies predicted by Poisson statistics (Fig. 4) [87]. Millions of picoliter droplets containing PCR reagents and DNA templates were generated by a flow-focusing design at a rate of 500/s (Fig. 4b). Then droplets were conveyed through the designed chip by the continuous-flow of oil, passing alternative denaturation and annealing zones controlled by static heaters below the chip. This format avoids temperature cycling of the entire chip and leads to more rapid (55s per cycle) and efficient PCR amplification. By placing channel restrictions called “neckdowns” periodically throughout the chip where droplets can only go through one by one, the amplification process within individual droplets at specific channel locations can be monitored (Fig. 4c). As a result, amplification of a 245-bp product can be detected and quantified in 35 min with 35 cycles at starting template concentrations as low as 1 template molecule/167 droplets. This represents the first successful example of continuous-flow microfluidic PCR chip for high-throughput, single-molecule DNA amplification and quantification.

Image of continuous flow droplet PCR chip. (a) Schematic of the overall flow configuration. Pink-shaded zones of the chip were set at 95°C, and nonshaded zones were at 67°C. The yellow regions for the interrogation neckdowns and the corresponding cycle numbers are indicated on the left. The nozzle is highlighted in red, and the oil extractor (OE) is in blue. (b) Optical image of droplet generation at the nozzle. (c) Optical image of uniform droplets in the downstream channel and flowing through one of the neckdowns. (Adapted from Ref [87])
Meanwhile, Schaerli et al. reported another high-throughput microfluidic device for continuous-flow PCR of single-copy DNA in water-in-oil nanoliter droplets [88]. As shown in Fig. 5, they designed a radial pattern featured with a central hot zone for DNA denaturation and a lower temperature periphery for DNA annealing and extension. Droplets were formed at a T-junction and flowed through alternating temperature zones in a continuous-flow of oil. Fluorescence lifetime imaging (FLIM) of Rhodamine B was used to measure the temperature inside the droplets to provide precise temperature control for successful DNA amplification by PCR. Here product analysis was completed off-line by gel electrophoresis, real-time PCR and sequencing. Amplification of 85-bp products from as low as 0.3 template/droplet was achieved in 17 min for 34 cycles, while an attempt for longer, 505bp fragments resulted in lower efficiency. These two examples demonstrated high-throughput amplification from single molecule DNA in droplets that is paving the way for microfluidic digital PCR.

Design of the continuous-flow radial PCR device for single-molecule DNA amplification. The device contains an oil inlet (a) that joins two aqueous inlet channels (B1 and B2) to form droplets at a T-junction (c). The droplets pass through the inner circles (500-μm wide channels) in the hot zone (d) to ensure initial denaturation and travel on to the periphery in 200-μm wide channels where annealing and extension occur (e). The droplets then flow back to the center where the DNA is denatured and a new cycle begins. Finally, the droplets exit the device after 34 cycles (f). All channels are 75 μm deep. The positions of the underlying copper rod and the Peltier module are indicated with orange and blue areas, respectively. (Adapted from Ref [88])
Bead-based emulsion PCR in microfluidic droplets
From simple emulsion PCR to the more popular BEAMing method, control over downstream high-throughput manipulation and analysis was greatly enhanced. Therefore, since the development of emulsion PCR in microfluidic droplets, researchers have been seeking to combine BEAMing with microfluidic droplets for both more precise volume control, easier manipulation and more importantly, flexible downstream processing and analysis. Several breakthroughs have been made recent years.
The bigest difficulty for incorporating beads into microfluidic droplets lies in the microbead sedimentation, i.e. preventing bead settlement within the syringe. Kumaresan et al. successfully solved this problem by designing a hybrid glass-PDMS-glass microdroplet generator (μDG) integrated with a three-valve diaphragm micropump that provides uniform droplet size, controlled generation frequency, and effective microbead transportation and encapsulation (Fig. 6) [38]. A high-throughput single-copy genetic amplification (SCGA) process was developed based on this design. Individual DNA molecules or cells together with primer functionalized microbeads were encapsulated in uniform PCR mix droplets. After bulk PCR amplification, the droplets were broken and the beads were collected for downstream analysis via flow cytometry. For the first time, the researchers have shown the generation of 100 amol of >600 bp PCR amplicons on individual beads starting from a single template, which are ideally suited for next-generation de novo sequencing applications, as well as high-throughput genetic analysis of single cells.

Single copy genetic amplification (SCGA). a Monodisperse droplets containing target DNA or cells, beads and the PCR reagent (blue) are formed in a carrier oil (yellow) at the cross-injector and routed into a tube for temperature cycling. b Each functional PCR droplet contains a bead covalently labeled with the reverse primer, dye-labeled forward primer, and a single target copy. Subsequent steps of PCR generate dye-labeled double-stranded product on the bead surface. c Layout of the device, showing the PCR solution inlet, the two oil inlets, and the droplet outlet ports(red).A three layer(glass-PDMS-glass) pneumatically controlled micropump is integrated on-chip to deliver PCR reagent. d Comparison of PCR yields for 380 (∼175 amol/bead), 624 (∼155 amol/bead), and 1139 bp (∼10 amol/bead) amplicons from a starting pUC18 template concentration of 10 molecules per droplet. e Flow cytometry analysis of beads carrying a 624 bp product amplified from 1 template per droplet (upper) and 0 template per droplet (lower). (Adapted from Ref [38])
The droplet generation frequency of the μDG reported above was only about 6Hz. To further improve the throughput and enable detection of extremely low-frequency events in a vast population, the single-channel μDG was scaled up using microfabricated emulsion generator array (MEGA) devices containing 4, 32, and 96 channels with a flexible capability of generating up to 3.4 million nanoliter-volume droplets per hour [89]. As shown in Fig. 7, on chip micropumps provided sufficient power to drive the generation of multiple droplets in parallel, and the symmetrical design ensured even fluidic transport. A multiplex single-cell PCR was demonstrated to efficiently amplify multiple target genes specific to different cell types. The large size of microbeads (34 μm) used allows multiple, different forward primers to be conjugated, and the PCR mix contains reverse primers each labeled with a unique fluorescent dye. Individual cells together with microbeads were encapsulated in uniform PCR mix droplets. The droplets were collected and thermally cycled in parallel. Afterwards, the beads were recovered from the emulsion and rapidly analyzed by multicolor flow cytometry. The method was able to identify and quantify a pathogen-to-background ratio in the order of 1/105, demonstrating that multiplex MEGA is an ultrahigh-throughput microfluidic platform for large-scale quantitative genotypic studies at single-molecule and single-cell levels.

Microfluidic emulsion generator array (MEGA) devices. (a) Layout of a glass/PDMS/glass hybrid four-channel MEGA device with a pneumatically controlled three-valve micropump integrated to drive four nozzles for droplet generation. (b) Design of a 32-channel MEGA device using an array of eight identical micropumps to operate 32 nozzles simultaneously. c Layout of 96-channel MEGA on a 4 in. wafer composed of a single ring pump and 96 droplet generators. Inset: close-ups of a single repeating unit composed of four T-shaped nozzles (left) and the pump structure schematically showing three pairs of coaxial ring-shaped valves and displacement trenches (right). (d) Exploded view of the complete four-layer 96-channel MEGA device and the plexiglass assembly module used to infuse oil and to collect the generated emulsion. (e-f) E. coli O157 detection using a 96-channel MEGA shows the measured O157 ratios consistent with the inputs: (e) 0.94/103 vs 1/103(10 cpd) and (f) 0.85/104 vs 1/104 (100cpd). (Adapted from Ref [89])
Agarose droplet microfluidics for single-molecule emulsion PCR
The ability of primer functionalized microbeads that can link with the PCR amplicons generated from a single DNA molecule or a single cell is critical for efficient high-throughput downstream manipulation and analysis. However, the use of microbeads also leads to many problems, including poor PCR efficiency, (i.e. 40% for microbead ePCR [38]) and short product length due to the steric hindrance effect and charge repulsion when PCR is performed on a solid surface. Moreover, according to the Poisson statistics, the encapsulation of diluted molecules and microbeads into droplets at random leads to a large number of void droplets including empty droplets or droplets with only either molecule or microbead, which is wasteful and negates the speed and efficiency afforded by ePCR.
To address these challenging problems and still preserve the monoclonal nature of the DNA product in each droplet, we introduced agarose as a trapping matrix to replace conventional primer functionalized microbeads [90]. Illustrated in Fig. 8, the agarose-in-oil droplets generated by a flow-focusing design contain agarose solution, PCR mix and DNA templates. Agarose has a unique thermoresponsive sol-gel switching property. It remains in liquid phase at all PCR temperatures, such that PCR can take place with high efficiency. After off-chip PCR amplification, the solution form of the agarose droplet can be switched to the solid gel phase by simply cooling the solution below gelling point of agarose. Since PCR forward primer is conjugated to agarose, amplicons can physically attach to the agarose matrix after PCR. As a result, DNA products amplified in the droplet can retain their monoclonality even after the oil phase is removed and afford flexible downstream processing and analysis, such as flow cytometry, sequencing, or long term storage etc. This method does not require coencapulation of primer labeled microbeads, allows high-throughput generation of uniform droplets (~500Hz), and enables high PCR efficiency (~95%), making it a promising platform for many single copy genetic studies.

Agarose droplet microfluidics for single-molecule emulsion PCR. (a) Schematics of the agarose emulsion droplet microfluidic method for single copy genetic analysis. Statistically diluted templates are encapsulated into uniform nanolitre agarose-in-oil droplets, which are then thermally cycled for PCR amplification. Following ePCR, the droplets are cooled to gelate to agarose beads for downstream genetic analysis. (b-c) Fluorescence microscope images of agarose beads after amplification from template concentrations of (b) 1.5 and (c) 0 copy/bead. d Percentage of microbeads carrying PCR product. The theoretical value (blue) was calculated according to Poisson distribution and the observed value (green) was the statistic result according to the experimental data. (Adapted from Ref [90])
Applications of microfluidic droplet PCR
Recent technical advances in microfluidic droplet PCR have proven this technique to be an important and capable tool for the execution of many new biological applications and assays, especially its great advantages in single-molecule amplification and analysis. Herein, we cover three key applications in this field using the aforementioned techniques.
High-throughput screening
For decades, biotechnologists have kept exploiting various technologies for high-throughput screening of genes and proteins to obtain novel functionalities from a large population library [95–97]. One particular technology is so-called SELEX (systematic evolution of ligands by exponential enrichment) for generation of aptamers, which are single-stranded DNA or RNA sequences that can bind to their target molecules with high affinity and specificity [98–102]. The SELEX process involves progressive enrichment of aptamers sequences by 8-30 rounds of partitioning and amplification from a starting library containing 1014 to 1016 molecules [98–100]. After several rounds of enrichment, normally the enriched DNA library has to be cloned into plasmids, which are then transfected into bacteria. Bacteria are then grown and colonies are picked and sequenced in large quantities to obtain aptamer candidates. After bioinformatic analysis, possible candidates are then chemically synthesized, and their binding affinities are measured individually. Such a process is time-consuming, labor-intensive, inefficient and expensive.
To address these problems, very recently, we have developed a novel method for efficiently screening aptamers from a complex single-stranded DNA (ssDNA) library by employing single-molecule emulsion PCR based on the technique just mentioned, agarose droplet microfluidics [103]. As the working-flow shown in Fig. 9a, ssDNA of pre-enriched library against Shp2 protein, a cancer biomarker, was statistically diluted and encapsulated into individual uniform agarose droplets using a flow-focusing method for droplet PCR to generate clonal agarose beads. Only the bright clonal beads containing DNA were picked out (Fig. 9c). The binding ability of amplified ssDNA from each clonal bead was then screened via high-throughput fluorescence flow cytometry (Fig. 9d-e). Only the amplified ssDNA with high binding affinity and high selectivity were chosen as aptamers and sequenced, or they can be directly used for downstream biomedical applications. Compared to conventional cloning-sequencing-synthesis-screening work flow, this method takes the advantage of the compartmentalization of microfluidic droplets and allows rapid molecular evolution of individual DNA sequences from an enriched library prior to knowing their exact sequence information, which makes the whole process more rapid, efficient and cost-effective. This approach could also be further applied to other molecular evolution technologies including mRNA display, phage display etc.

Aptamer screening by agarose droplet microfluidic technology. a Schematic working-flow. Single DNA sequences of an enriched library obtained by traditional SELEX are encapsulated individually into agarose droplets for high-throughput single copy DNA amplification. The resulting agarose droplets are cooled to become agarose beads and stained with SYBR Green in order to pick out high fluorescent beads containing DNA colonies. Binding affinity of DNA in each fluorescent bead against target molecule are screened and aptamers are identified. (b-c) Microscope images of agarose beads after PCR amplification and SYBR Green staining at DNA template concentration of 0 (b) and 0.3 copies/droplet (c). (d-e) Binding assay of DNA in clonal beads with Shp2 protein (d) and GST protein (e) monitored by flow cytometry. Compared with unselected library, 10th library exhibited clear fluorescence intensity shift. Among 30 positive clones, 3 possible aptamers (15th, 18th and 21st positive clone) displayed obvious fluorescence shift with target protein Shp2 (d) and did not show observable fluorescence shift with control protein GST (e). (Adapted from Ref [103])
Next generation DNA sequencing
DNA sequencing has been widely applied to numerous fields, such as diagnostics, biotechnology, forensic biology and biological systematics [7, 104, 105]. The Human Genome Project was successfully completed in 2003. However, to further make it applicable for the development of medicines and the healthcare of individuals, high accuracy, long-range contiguity, high-throughput as well as low cost are still the goals for researchers to develop next-generation sequencing techniques. Eliminating the conventional cloning-based DNA library preparation is the first step for next-generation sequencing. BEAMing based microemulsion technology has emerged as a rapid, low-cost alternative [34, 35]. However, it suffers from non-uniform amplification with a limitation on the DNA amplicon’s length (~250 bp), mainly due to the small and variable volume of the emulsion. Kumaresan et al. developed a high-throughput SCGA process, as explained above, and demonstrated its application for DNA sequencing [38]. Uniform droplets with tunable 2-5 nL volumes were generated and encapsulated with microbeads of 34 μm in diameter. Compared to the small beads and small droplets used in the BEAMing technique, the large surface area of the microbeads allows sufficient forward primers to be conjugated resulting in 4.4 fmol of primers per bead, and the reverse primers and other PCR reagents in the large droplets are ~10-fold excess required for the efficient generation of PCR amplicons [89]. As shown in Fig. 6d-e, approximately 175 amol of 380 bp DNA product was generated on each bead, ~150amol of 624 bp DNA product per bead, and ~10 amol of 1139 bp DNA product per bead. ~100 amol of a 624 bp product from a single bead was sufficient to be directly sequenced by attomole-scale Sanger sequencing [106], demonstrating the feasibility of SCGA to generate sufficient amounts of long DNA on beads from a single template molecule to enable next generation Sanger and pyrosequencing [37, 107–109].
Quantitative and sensitive detection of rare mutations
Genetic alterations, such as deletion, point mutation, and rearrangement, play an essential role in the development and progression of cancers, which can serve as biomarkers for cancer diagnostics [10, 110]. Mutations within tumor cells are released by the cells into clinical samples, such as blood, lymph, and urine, where the mutation detection has to be performed in a background of a large excess of non-mutated DNA from normal cells. Thus a simple, sensitive and quantitative method to measure the ratio of mutant to wide-type genes in clinical samples is highly desired.
The detection of low-frequency genetic variations requires high analysis throughput in order to obtain the statistically significant population for the target. Zeng et al. applied their high-throughput microfabricated emulsion generator array (MEGA) devices containing 4, 32 and 96 channels to detect and quantify both wild type and mutant/pathogenic cells [89]. Escherichia coli bacterial cells were taken as a model to detect toxic E. coli O157 cells in a high background of normal K12 cells. Microbeads functionalized with multiple forward primers targeting specific genes from different cell types were used for solid-phase PCR in droplets. By increasing the ratio of cells per droplet (cpd) from 0.2 cpd to 10 cpd or even 100 cpd, the number of droplets/beads that must be processed was reduced by 50-fold or 500-fold. As shown in Fig. 7e, to detect E. coli O157 cells at a frequency of 1/103, 9492 events were recorded with 10 cpd with the detection of 88 positive events, giving an output fraction of 0.94/103. To further improve the detection capability, 100 cpd was applied to detection a frequency of 1/104 with the output fraction of 0.85/104 (Fig. 7f). Here, 96-channels were operated at ~7 Hz to generate droplets encapsulating up to 104 cells within 5 min. The limit of detection was determined by further challenging the system with lower pathogetic ratios with the result of 3/105 and a 99% confidence. Their results demonstrated the capability of using ultrahigh-throughput microfluidic platforms for large-scale quantitative genotypic studies of complex biological systems at the single-molecule and single-cell level.
Pekin et al. performed work using another design of microfluidic droplet PCR for the highly sensitive detection of mutated DNA in a quantitative manner within complex mixtures of DNA [94]. In Fig. 10, using a microfluidic system, single target DNA molecules were compartmentalized in microdroplets with PCR mix and clinically validated fluorogenic TaqMan probes specific for mutated and wide-type KRAS, which generated green and red fluorescence signals, respectively. The droplets were collected in a tube and thermocycled for PCR amplification. Afterwards, the droplets were reinjected onto a microfluidic chip and the green and red fluorescent signals of each droplet were analyzed on chip upon laser excitation. Thus, the ratio of mutant to wild-type alleles can be quantitatively determined from the ratio of red and green fluorescence. By single molecule compartmentalization in picoliter droplets and fast fluorescence analysis on chip, this technique enabled the determination of mutants in several cancer cell lines and quantification of DNA down to 1 mutant KRAS gene in 200 000 wild-type KRAS genes when analyzing ~106 fluorescent droplets. The sensitivity could be further increased if a greater number of droplets can be analyzed. Furthermore, simultaneous detection and quantification of multiple mutations in a single experiment were also demonstrated by one-to-one fusion of droplets containing gDNA with any one of the several different types of droplets, each containing a TaqMan probe specific for a different KRAS mutation or wild-type KRAS, and an optical code. Very recently, Hindson et al. reported another droplet digital PCR system and also showed sensitive detection of mutant DNA in a 100 000-fold excess of wild-type background [111]. They further demonstrated absolute quantitation of circulating fetal and maternal DNA from cell-free plasma. Thus, the high sensitivity and quantitation of such procedures could allow many other applications in complex biological samples, such as detection of infectious disease, identification of alleles, and analysis of transcription.

Microfluidic droplet PCR for rare mutations detection. (a-c) Overview of the system. a An aqueous phase containing the gDNA, PCR reagents and TaqMan probes specific for the wild-type and mutant genes is emulsified within a microfluidic device. (b) The emulsion is collected in a PDMS sealed tubeand thermocycled. (c) The emulsion is then reinjected onto a microfluidic chip, the droplets are spaced by oil, and the fluorescent signal of each droplet is analyzed. (d-f) Fluorescence confocal microscopy analysis of thermocycled droplets. gDNA extracted from homozygous cell-lines bearing wild-type KRAS alleles (SW48) (d, red), mutant KRAS alleles (SW620) (e, green), and a heterozygous cell-line bearing both mutant and wild-type KRAS alleles (H358) (f red and green) were analyzed by fluorescence confocal microscopy. (Adapted from Ref [94])
Conclusion and outlook
Microfluidic droplet technology has dramatically changed conventional PCR for DNA amplification, especially single DNA molecule amplification. Microfluidic techniques allow a high degree of control over the droplet size and composition, as well as independent control over each droplet, such as mixing, transporting and analysis. Secondly, due to the high surface-to-volume ratios at the microscale, it can greatly reduce the heat and mass transfer times and facilitate fast PCR processing, which is critical for instant medical diagnosis and in-field detection. Thirdly, the contact between the surface of channels and the sample inside droplets has been eliminated by the surrounding immiscible fluid, thus reducing cross-contamination between droplets, minimizing the possibility of template loss and eliminating PCR bias. Moreover, since identical droplets are produced at a very high frequency within one experiment, massively parallel processing and experimentation can be easily achieved in ultra-short timescales and with confident reproducibility and quantitative single-molecule analysis. Although utilization of microfluidic droplet for PCR is still in the early stages, its great potential for becoming the next generation of single-molecule PCR amplification has already been demonstrated. The high fidelity manipulation of droplets in microfluidic channels, the ability to perform single molecule DNA amplification, and the high throughput workflow with high accuracy using minute sample quantities will certainly drive its further development and applications across branches of experimental sciences. For instance, droplet microfluidics has great advances for in vitro directed evolution by increasing the size of the libraries and the throughput of the screening. Microbead cloning has provided the basis for novel high-throughput genome sequencing technologies. By taking the advantage of droplet microfluidics, it is possible to develop powerful automated instruments, thus further accelerating the speed and reducing the cost of sequencing. Moreover, the compartmentalization of single molecules and even single cells offered by droplet microfluidics enables its applications in rare mutation detection and single-cell analyses of large populations, such as mutant genetic analysis, pathogen detection, and disease cell capture. It might also have a great potential on exploring single-cell functional genomics, proteomics and metabolomics. However, several technical concerns need to be solved for its future development, such as the stability of droplets for downstream manipulation, the molecular interactions on the interface and outside the droplets, the techniques for droplet detection and sorting. Future technology development is still required to provide more sophisticated and more versatile functionality, such as system integration, device design optimization, device manufacture, and system automation.
References
Xie XS, Yu J, Yang WY (2006) Science 312:228–230
Sims CE, Allbritton NL (2007) Lab Chip 7:423–440
Li GW, Xie XS (2011) Nature 475:308–315
Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T (1995) Nature 374:555–559
Wang MD, Schnitzer MJ, Yin H, Landick R, Gelles J, Block SM (1998) Science 282:902–907
Weiss S (1999) Science 283:1676–1683
Check E (2005) Nature 437:1084–1086
Raser JM, O'Shea EK (2005) Science 309:2010–2013
Misteli T, Soutoglou E (2009) Nat Rev Mol Cell Biol 10:243–254
Stratton MR, Campbell PJ, Futreal PA (2009) Nature 458:719–724
Burkhart DL, Sage J (2008) Nat Rev Cancer 8:671–682
Jeffreys AJ, Wilson V, Neumann R, Keyte J (1988) Nucleic Acids Res 16:10953–10971
Jeffreys AJ, Neumann R, Wilson V (1990) Cell 60:473–485
Li HH, Gyllensten UB, Cui XF, Saiki RK, Erlich HA, Arnheim N (1988) Nature 335:414–417
Ruano G, Kidd KK, Stephens JC (1990) Proc Natl Acad Sci U S A 87:6296–6300
Song H, Chen DL, Ismagilov RF (2006) Angew Chem Int Ed Engl 45:7336–7356
Dittrich PS, Manz A (2006) Nat Rev Drug Discovery 5:210–218
Price AK, Culbertson CT (2007) Anal Chem 79:2614–2621
Chiu DT, Lorenz RM (2009) Acc Chem Res 42:649–658
Lindstrom S, Andersson-Svahn H (2010) Lab Chip 10:3363–3372
Teh SY, Lin R, Hung LH, Lee AP (2008) Lab Chip 8:198–220
Huebner A, Sharma S, Srisa-Art M, Hollfelder F, Edel JB, Demello AJ (2008) Lab Chip 8:1244–1254
Theberge AB, Courtois F, Schaerli Y, Fischlechner M, Abell C, Hollfelder F, Huck WTS (2010) Angew Chem Int Edit 49:5846–5868
Baroud CN, Gallaire F, Dangla R (2010) Lab Chip 10:2032–2045
Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Science 230:1350–1354
Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H (1986) Cold Spring Harbor Symp Quant Biol 51:263–273
Zhang C, Da X (2010) Chem Rev (Washington, DC, U S) 110:4910–4947
Kemp DJ, Smith DB, Foote SJ, Samaras N, Peterson MG (1989) Proc Natl Acad Sci U S A 86:2423–2427
Porterjordan K, Rosenberg EI, Keiser JF, Gross JD, Ross AM, Nasim S, Garrett CT (1990) J Med Virol 30:85–91
Kalinina O, Lebedeva I, Brown J, Silver J (1997) Nucleic Acids Res 25:1999–2004
Lagally ET, Medintz I, Mathies RA (2001) Anal Chem 73:565–570
Nakano M, Komatsu J, Matsuura S, Takashima K, Katsura S, Mizuno A (2003) J Biotechnol 102:117–124
Shao KK, Ding WF, Wang F, Li HQ, Ma D, Wang HM (2011) PLoS One 6
Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B (2003) Proc Natl Acad Sci U S A 100:8817–8822
Diehl F, Li M, He YP, Kinzler KW, Vogelstein B, Dressman D (2006) Nat Methods 3:551–559
Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM (2005) Science 309:1728–1732
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Nature 437:376–380
Kumaresan P, Yang CJ, Cronier SA, Blazej RG, Mathies RA (2008) Anal Chem 80:3522–3529
Thorsen T, Roberts RW, Arnold FH, Quake SR (2001) Phys Rev Lett 86:4163–4166
Nisisako T, Torii T, Higuchi T (2002) Lab Chip 2:24–26
Zagnoni M, Anderson J, Cooper JM (2010) Langmuir 26:9416–9422
Gupta A, Kumar R (2010) Microfluid Nanofluid 8:799–812
van Steijn V, Kleijn CR, Kreutzer MT (2010) Lab Chip 10:2513–2518
Schneider T, Burnham DR, VanOrden J, Chiu DT (2011) Lab Chip 11:2055–2059
Sivasamy J, Wong TN, Nguyen NT, Kao LTH (2011) Microfluid Nanofluid 11:1–10
Liu HH, Zhang YH (2009) J Appl Phys 106
Anna SL, Bontoux N, Stone HA (2003) Appl Phys Lett 82:364–366
Ward T, Faivre M, Abkarian M, Stone HA (2005) Electrophoresis 26:3716–3724
Nie ZH, Seo MS, Xu SQ, Lewis PC, Mok M, Kumacheva E, Whitesides GM, Garstecki P, Stone HA (2008) Microfluid Nanofluid 5:585–594
Yobas L, Martens S, Ong WL, Ranganathan N (2006) Lab Chip 6:1073–1079
Peng L, Yang M, Guo SS, Liu W, Zhao XZ (2011) Biomed Microdevices 13:559–564
Takeuchi S, Garstecki P, Weibel DB, Whitesides GM (2005) Adv Mater (Weinheim, Ger) 17:1067
Utada AS, Lorenceau E, Link DR, Kaplan PD, Stone HA, Weitz DA (2005) Science 308:537–541
Tan YC, Fisher JS, Lee AI, Cristini V, Lee AP (2004) Lab Chip 4:292–298
Link DR, Anna SL, Weitz DA, Stone HA (2004) Phys Rev Lett 92:054503
Hatch AC, Fisher JS, Tovar AR, Hsieh AT, Lin R, Pentoney SL, Yang DL, Lee AP (2011) Lab Chip 11:3838–3845
Link DR, Grasland-Mongrain E, Duri A, Sarrazin F, Cheng Z, Cristobal G, Marquez M, Weitz DA (2006) Angew Chem Int Ed Engl 45:2556–2560
Cho SK, Moon HJ, Kim CJ (2003) J Microelectromech S 12:70–80
Song H, Tice JD, Ismagilov RF (2003) Angew Chem Int Edit 42:768–772
Hung LH, Choi KM, Tseng WY, Tan YC, Shea KJ, Lee AP (2006) Lab Chip 6:174–178
Kohler JM, Henkel T, Grodrian A, Kirner T, Roth M, Martin K, Metze J (2004) Chem Eng J (Lausanne) 101:201–216
Fidalgo LM, Abell C, Huck WTS (2007) Lab Chip 7:984–986
Mazutis L, Araghi AF, Miller OJ, Baret JC, Frenz L, Janoshazi A, Taly V, Miller BJ, Hutchison JB, Link D, Griffiths AD, Ryckelynck M (2009) Anal Chem 81:4813–4821
Tresset G, Takeuchi S (2005) Anal Chem 77:2795–2801
Tan WH, Takeuchi S (2006) Lab Chip 6:757–763
Stroock AD, Dertinger SKW, Ajdari A, Mezic I, Stone HA, Whitesides GM (2002) Science 295:647–651
Bringer MR, Gerdts CJ, Song H, Tice JD, Ismagilov RF (2004) Philos T Roy Soc A 362:1087–1104
Liau A, Karnik R, Majumdar A, Cate JHD (2005) Anal Chem 77:7618–7625
Cabral JT, Hudson SD (2006) Lab Chip 6:427–436
Paik P, Pamula VK, Pollack MG, Fair RB (2003) Lab Chip 3:28–33
Chatterjee D, Hetayothin B, Wheeler AR, King DJ, Garrell RL (2006) Lab Chip 6:199–206
Tan YC, Ho YL, Lee AP (2008) Microfluid Nanofluid 4:343–348
Huh D, Bahng JH, Ling YB, Wei HH, Kripfgans OD, Fowlkes JB, Grotberg JB, Takayama S (2007) Anal Chem 79:1369–1376
Baret JC, Miller OJ, Taly V, Ryckelynck M, El-Harrak A, Frenz L, Rick C, Samuels ML, Hutchison JB, Agresti JJ, Link DR, Weitz DA, Griffiths AD (2009) Lab Chip 9:1850–1858
Boukellal H, Selimovic S, Jia YW, Cristobal G, Fraden S (2009) Lab Chip 9:331–338
Edgar JS, Milne G, Zhao YQ, Pabbati CP, Lim DSW, Chiu DT (2009) Angew Chem Int Edit 48:2719–2722
Wang W, Yang C, Li CM (2009) Lab Chip 9:1504–1506
Bai YP, He XM, Liu DS, Patil SN, Bratton D, Huebner A, Hollfelder F, Abell C, Huck WTS (2010) Lab Chip 10:1281–1285
Huebner A, Bratton D, Whyte G, Yang M, deMello AJ, Abell C, Hollfelder F (2009) Lab Chip 9:692–698
Frenz L, Blank K, Brouzes E, Griffiths AD (2009) Lab Chip 9:1344–1348
Mary P, Abate AR, Agresti JJ, Weitz DA (2011) Biomicrofluidics 5
Hettiarachchi K, Talu E, Longo ML, Dayton PA, Lee AP (2007) Lab Chip 7:463–468
Malic L, Veres T, Tabrizian M (2009) Lab Chip 9:473–475
Marz A, Henkel T, Cialla D, Schmitt M, Popp J (2011) Lab Chip 11:3584–3592
Beer NR, Hindson BJ, Wheeler EK, Hall SB, Rose KA, Kennedy IM, Colston BW (2007) Anal Chem 79:8471–8475
Beer NR, Wheeler EK, Lee-Houghton L, Watkins N, Nasarabadi S, Hebert N, Leung P, Arnold DW, Bailey CG, Colston BW (2008) Anal Chem 80:1854–1858
Kiss MM, Ortoleva-Donnelly L, Beer NR, Warner J, Bailey CG, Colston BW, Rothberg JM, Link DR, Leamon JH (2008) Anal Chem 80:8975–8981
Schaerli Y, Wootton RC, Robinson T, Stein V, Dunsby C, Neil MAA, French PMW, deMello AJ, Abell C, Hollfelder F (2009) Anal Chem 81:302–306
Zeng Y, Novak R, Shuga J, Smith MT, Mathies RA (2010) Anal Chem 82:3183–3190
Leng XF, Zhang WH, Wang CM, Cui LA, Yang CJ (2010) Lab Chip 10:2841–2843
Novak R, Zeng Y, Shuga J, Venugopalan G, Fletcher DA, Smith MT, Mathies RA (2011) Angew Chem Int Edit 50:390–395
Hua ZS, Rouse JL, Eckhardt AE, Srinivasan V, Pamula VK, Schell WA, Benton JL, Mitchell TG, Pollack MG (2010) Anal Chem 82:2310–2316
Zhong Q, Bhattacharya S, Kotsopoulos S, Olson J, Taly V, Griffiths AD, Link DR, Larson JW (2011) Lab Chip 11:2167–2174
Pekin D, Skhiri Y, Baret JC, Le Corre D, Mazutis L, Ben Salem C, Millot F, El Harrak A, Hutchison JB, Larson JW, Link DR, Laurent-Puig P, Griffiths AD, Taly V (2011) Lab Chip 11:2156–2166
Reetz MT, Jaeger KE (1999) Biocatalysis - from Discovery to Application 200:31–57
Pluckthun A, Schaffitzel C, Hanes J, Jermutus L (2001) Advances in Protein Chemistry 55(55):367–403
Paegel BM (2010) Curr Opin Chem Biol 14:568–573
Ellington AD, Szostak JW (1990) Nature 346:818–822
Tuerk C, Gold L (1990) Science 249:505–510
Robertson DL, Joyce GF (1990) Nature 344:467–468
Nutiu R, Li Y (2005) Angew Chemie Int Ed 44:1061–1065
Fang X, Tan W (2010) Acc Chem Res 43:48–57
Zhang WY, Zhang WH, Liu ZY, Li C, Zhu Z, Yang CJ (2012) Anal Chem 84:350–355
Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, Vamathevan J, Miranda M, Anderson IJ, Fraser JA, Allen JE, Bosdet IE, Brent MR, Chiu R, Doering TL, Dontin MJ, D'Souza CA, Fox DS, Grinberg V, Fu JM, Fukushima M, Haas BJ, Huang JC, Janbon G, Jones SJM, Koo HL, Krzywinski MI, Kwon-Chung JK, Lengeler KB, Maiti R, Marra MA, Marra RE, Mathewson CA, Mitchell TG, Pertea M, Riggs FR, Salzberg SL, Schein JE, Shvartsbeyn A, Shin H, Shumway M, Specht CA, Suh BB, Tenney A, Utterback TR, Wickes BL, Wortman JR, Wye NH, Kronstad JW, Lodge JK, Heitman J, Davis RW, Fraser CM, Hyman RW (2005) Science 307:1321–1324
Zhao SY, Shetty J, Hou LH, Delcher A, Zhu BL, Osoegawa K, de Jong P, Nierman WC, Strausberg RL, Fraser CM (2004) Genome Res 14:1851–1860
Blazej RG, Kumaresan P, Cronier SA, Mathies RA (2007) Anal Chem 79:4499–4506
Ronaghi M, Uhlen M, Nyren P (1998) Science 281:363–365
Goldberg SMD, Johnson J, Busam D, Feldblyum T, Ferriera S, Friedman R, Halpern A, Khouri H, Kravitz SA, Lauro FM, Li K, Rogers YH, Strausberg R, Sutton G, Tallon L, Thomas T, Venter E, Frazier M, Venter JC (2006) Proc Natl Acad Sci U S A 103:11240–11245
Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM (2008) Nature 452:U872–U875
Vogelstein B, Kinzler KW (2004) Nat Med (N Y, NY, U S) 10:789–799
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang SL, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW (2011) Anal Chem 83:8604–8610
Acknowledgment
This work is supported by the National Basic Research Program of China (Grant 2010CB732402), National Scientific Foundation of China (Grants 21075104 and 20805038), and the Natural Science Foundation of Fujian Province for Distinguished Young Scholars (Grant 2010 J06004)
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Young Investigators in Analytical and Bioanalytical Science with guest editors S. Daunert, J. Bettmer, T. Hasegawa, Q. Wang and Y. Wei.
Rights and permissions
About this article
Cite this article
Zhu, Z., Jenkins, G., Zhang, W. et al. Single-molecule emulsion PCR in microfluidic droplets. Anal Bioanal Chem 403, 2127–2143 (2012). https://doi.org/10.1007/s00216-012-5914-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-5914-x