
Abstract
Background
Models that predict the risk of estrogen receptor (ER)-positive breast cancers may improve our ability to target chemoprevention. We investigated the contributions of sex hormones to the discrimination of the Breast Cancer Surveillance Consortium (BCSC) risk model and a polygenic risk score comprised of 83 single nucleotide polymorphisms.
Methods
We conducted a nested case-control study of 110 women with ER-positive breast cancers and 214 matched controls within a mammography screening cohort. Participants were postmenopausal and not on hormonal therapy. The associations of estradiol, estrone, testosterone, and sex hormone binding globulin with ER-positive breast cancer were evaluated using conditional logistic regression. We assessed the individual and combined discrimination of estradiol, the BCSC risk score, and polygenic risk score using the area under the receiver operating characteristic curve (AUROC).
Results
Of the sex hormones assessed, estradiol (OR 3.64, 95% CI 1.64–8.06 for top vs bottom quartile), and to a lesser degree estrone, was most strongly associated with ER-positive breast cancer in unadjusted analysis. The BCSC risk score (OR 1.32, 95% CI 1.00–1.75 per 1% increase) and polygenic risk score (OR 1.58, 95% CI 1.06–2.36 per standard deviation) were also associated with ER-positive cancers. A model containing the BCSC risk score, polygenic risk score, and estradiol levels showed good discrimination for ER-positive cancers (AUROC 0.72, 95% CI 0.65–0.79), representing a significant improvement over the BCSC risk score (AUROC 0.58, 95% CI 0.50–0.65).
Conclusion
Adding estradiol and a polygenic risk score to a clinical risk model improves discrimination for postmenopausal ER-positive breast cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ER-positive cancers represent at least 80% of breast cancers diagnosed in the United States [1]. Preventive medications such as tamoxifen and raloxifene can reduce the risk of estrogen receptor (ER)-positive breast cancers in high-risk women but have potential adverse effects [2]. Uptake is partially limited by the ability to identify high-risk women with the most favorable benefit-harm tradeoff [3, 4].
The United States Preventive Services Task Force (USPSTF) recommends using risk prediction models to identify candidates for chemoprevention but acknowledges that current models have modest discrimination [3]. Validated models such as the Breast Cancer Risk Assessment Tool and Tyrer-Cuzick model incorporate clinical risk factors such as age, race/ethnicity, family history, history of prior breast biopsy, and history of benign breast disease [3, 5,6,7]. The Breast Cancer Surveillance Consortium (BCSC) risk model also includes mammographic breast density, a strong risk factor, and tends to have the highest discrimination of available models [8, 9].
Single nucleotide polymorphisms (SNPs) can improve the performance of clinical risk models. Individual SNPs have modest impacts on risk, but polygenic risk scores (PRS) representing the cumulative effects of multiple SNPs exhibit strong associations with risk that are largely independent from clinical risk factors [10,11,12]. PRS have been shown to improve the discrimination of the BCSC [11, 12] and other [13] risk models. The magnitude of improvement in the area under the receiver operating characteristic curve (AUROC) of these models is modest [11, 12].
Circulating sex hormone levels may improve prediction beyond SNPs and clinical models [7, 14, 15]. Estrone and estradiol have been most robustly associated with breast cancer, particularly ER-positive cancers. Elevated estradiol levels are positively associated with breast cancer risk [14,15,16,17,18,19,20], while undetectable levels are strongly protective against ER-positive cancers [21]. Sex hormone binding globulin (SHBG) binds estradiol, affecting bioavailable levels, and has been inversely associated with risk of invasive breast cancer [14]. The reported effects of testosterone on breast cancer risk have been heterogeneous [14, 18, 19, 22,23,24].
Many prior studies have examined the relationships between individual hormones and breast cancer risk [14,15,16, 18,19,20, 22, 24]. Fewer studies have assessed the combined effects of hormones in risk prediction, particularly in conjunction with risk prediction models. In one study, sex hormones (with the most parsimonious combination being estrone sulfate, testosterone, and prolactin) improved the performance of the Gail and Rosner–Colditz risk models by AUROC of 0.06 and 0.03 (relative improvements of 11 and 6%), respectively [25]. This analysis did not account for breast density or SNPs.
A prediction model for ER-positive breast cancer, which includes sex hormones, SNPs, and clinical risk factors could potentially improve on the discrimination of more restrictive models and help identify women most likely to benefit from chemoprevention by virtue of their risk of ER-positive breast cancer. To investigate this hypothesis, we studied the associations of four sex hormones—estrone, estradiol, testosterone, and SHBG—with ER-positive breast cancer in postmenopausal women. We assessed whether the addition of one or a combination of sex hormones improved the discrimination of the BCSC risk score alone, and the BCSC risk score modified by an 83-SNP PRS.
Methods
Study population
We conducted a nested case-control study [12] within the California Pacific Medical Center (CPMC) Research Institute cohort, comprised of women undergoing screening mammography at the Breast Health Center at CPMC. Between 2004 and 2011, women were asked to provide blood samples for research. Women who provided informed consent for blood collection completed a questionnaire with demographic information and risk factor data, which were collected and pooled by the San Francisco Mammography Registry (SFMR). The protocol was approved by the institutional review boards at CPMC and the University of California, San Francisco.
Blood samples were collected from 19,276 women without a diagnosis of invasive or pre-invasive breast cancer at the time of blood draw. A nested case-control study was performed on 324 participants with blood collected between 9/3/2004 and 11/30/2011. We excluded women self-identified as premenopausal, perimenopausal, or postmenopausal as a direct result of surgery or medical treatments, such as chemotherapy. Women on hormonal therapy or selective estrogen receptor modulators at the time of blood draw were also excluded. Cases were ascertained by linkage to the California Cancer Registry (CCR) and defined as pathologically-confirmed diagnoses of invasive breast cancers with positive/elevated ER expression on immunohistochemical staining. ER-negative cases and those with unavailable ER status were excluded. Women without breast cancer as of last linkage to the CCR on 10/31/2013 were matched as controls in a 2:1 ratio to cases based on age, race/ethnicity, and date of index mammogram. Six cases were matched 1:1 with controls due to missing data, resulting in 110 cases and 214 controls.
Sex hormone measurement
Blood samples for sex hormone measurements were collected at the time of consent. Whole blood was centrifuged within 15 min of venipuncture and serum was aliquoted into 1 mL tubes stored at −80°C. Samples were shipped on dry ice by overnight courier to Mayo Medical Laboratory (Rochester, MN) for hormone measurement. Liquid chromatography–mass spectrometry (LC–MS, Agilent Technologies, Santa Clara, CA) was used to measure estrone, estradiol, and testosterone levels. Prior to LC–MS, estradiol and estrone were extracted with methylene chloride and underwent derivatization with dansyl chloride followed by high-pressure liquid chromatography. SHBG was measured using a Siemens chemiluminescent assay (Siemens Healthcare Diagnostics, Deerfield, IL). The inter- and intra-assay coefficients of variation for each assay are shown in Supplementary Table S1. Women with estradiol levels over 25 pg/ml were excluded from the analysis given the possibility that extreme values were due to unreported or incorrectly ascertained exogenous hormone usage or menopausal status. The free estradiol index was calculated by dividing the total estradiol level by the SHBG level and multiplying by 100.
SNP genotyping and calculation of polygenic risk score
Whole blood was sent to the Genomics Core at the University of Minnesota for DNA extraction. A total of 113 cases and 113 matched controls randomly selected from the overall dataset were genotyped using an OncoArray platform (Illumina, San Diego, CA), resulting in 1:1 matching within the genotyped subgroup. We included 83 SNPs (Supplementary Table S2) selected based on review of the genome-wide association study (GWAS) catalog and published associations with invasive breast cancer in Caucasian, Asian, or Hispanic populations, as previously described [12]. The PRS was calculated using a previously described method using published odds ratios and allele frequencies [12]. In brief, it is the composite likelihood ratio (LR) of breast cancer representing the individual effects of each SNP assuming that the SNPs are inherited independently (in linkage equilibrium) and that there are no interactions between them.
BCSC model
The BCSC risk model provides estimates of 5-year absolute risk using age, race/ethnicity, presence of a first-degree relative with breast cancer, history of breast biopsy, and breast density [8, 9]. Community radiologists participating in the SFMR assessed breast density on the index mammogram (acquired 1997–2011) according to the Breast imaging reporting and data system (BI-RADS) fourth edition or earlier categories: almost entirely fat (a), scattered fibroglandular densities (b), heterogeneously dense (c), and extremely dense (d) [26]. Although an updated version of the BCSC model has been published [9], we used version 1.0 [8] to allow the calculation of risk estimates for women older than 74 years.
Statistical analysis
Demographic data and risk factors were compared between cases and controls using the chi-squared test for categorical measures and the unpaired t test for body mass index. The median hormone levels across cases and controls were compared using the non-parametric k-sample equality of medians test. Statistical tests were two-sided, with α = 0.05.
To examine the effects of individual hormones on ER-positive breast cancer risk, we performed univariate conditional logistic regression. Hormone levels were categorized into quartiles based on their respective distributions in controls. We compared point estimates across quartiles using tests of linear trend.
To examine the relative contributions of sex hormone levels, the BCSC risk score, and PRS to ER-positive risk, we performed conditional logistic regression using univariate and multivariable models within the subset of women (n = 218, i.e., 109 matched case-control pairs) with complete genotype, hormone, density, and clinical data. Hormone levels were log2-transformed so that a one-unit increase represented a doubling of levels. The PRS was standardized according to the mean and standard deviation of the PRS in controls. The 5-year risk estimate generated by BCSC version 1.0 was used as a continuous variable in logistic regression models.
Area under the receiver operating characteristic curve (AUROC) was used to compare discrimination. To avoid the bias introduced when the same data are used to both fit and evaluate the model, we performed tenfold cross-validation to confirm the internal validity of the model. Briefly, we split the dataset into ten equally-sized groups containing randomly-selected case-control pairs, used 9/10 as a training dataset to fit a regression model, then used the fitted model to generate predicted probabilities in the remaining 1/10. We repeated this process ten times so that each 1/10 of the dataset was used once as a validation set, and then used the aggregate predicted probabilities to calculate the final cross-validated AUC and 95% CI using a stratified bootstrap approach (n = 1000 replications) accounting for matched case-control clusters. Reported AUROCs represent cross-validated estimates. We compared AUROCs using a Wald test with bootstrap variance and covariance estimates.
Primary statistical analysis was performed using STATA 14.1 (StataCorp, College Station, TX, USA). The PRS was generated using R (R Foundation, Vienna, Austria), and the BCSC risk estimate was generated using SAS Version 9.3 (SAS Institute, Cary, NC, USA).
Results
We identified 110 postmenopausal women with ER-positive, invasive breast cancers and 214 matched controls. Approximately 80% of cases and controls were Caucasian/White by self-report (Table 1). Cases were more likely to have a positive family history of breast cancer, prior breast biopsy, and higher body mass index, although the absolute difference was 1 kg/m2. The distribution of breast density was generally similar between cases and controls, with most women having scattered fibroglandular densities (BI-RADS b) or heterogeneously dense (BI-RADS c) breasts. Fatty breasts (BI-RADS a) were more common in cases, while extremely dense breasts (BI-RADS d) were more common in controls. However, the overall difference in density did not reach statistical significance (p = 0.12).
Cases had higher median estrone and estradiol levels while controls had higher SHBG (Table 1). Cases and controls had similar testosterone levels. The distributions of all hormone levels were right-skewed, while log-transformed levels approximated a normal distribution (Supplementary Figure S1).
We examined the associations between individual sex hormones and ER-positive breast cancer using univariate logistic regression (Table 2). Estradiol had the strongest association with ER-positive breast cancer, with increasing levels corresponding to higher risk. The associations reached statistical significance in the highest (OR 3.64, 95% CI 1.64–8.06) and second-highest (OR 3.12, 95% CI 1.41–6.90) quartiles relative to the bottom quartile, and followed a linear trend. Free estradiol index showed a similar trend. Estrone levels above 14 pg/ml were associated with an approximately twofold increase in risk. SHBG was inversely associated with risk, though no quartile associations reached statistical significance. The second-highest quartile of testosterone was associated with elevated risk (OR 2.55, 95% CI 1.23–5.26) but a null effect could not be excluded for the other quartiles.
The individual and combined contributions of the BCSC risk score, PRS, and estradiol levels to risk prediction were evaluated in a subset of 218 women representing 109 cases and 109 matched controls who were genotyped in addition to having hormone levels and breast density measured. We selected estradiol because it was most robustly associated with ER-positive breast cancer in univariate logistic regression.
In univariate analysis, estradiol was associated with ER-positive cancer, as were the PRS and BCSC risk score (Table 3, column 1). In a model containing the BCSC risk score and estradiol levels, the OR per doubling of estradiol levels slightly increased from 1.57 (95% CI 1.13–2.19) to 1.79 (95% CI 1.24–2.58). The ORs and confidence intervals per 1% increase in the BCSC risk score and per doubling of estradiol modestly increased when combined in a joint model (Table 3, column 2), but remained similar when the PRS was added to the model (Table 3, column 3). Adjustment for BMI slightly attenuated the associations between estradiol and ER-positive cancer but did not have a substantial effect on the BCSC risk score or the PRS (Supplementary Table S3).
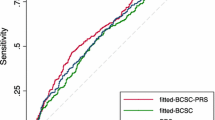
We compared the discrimination of the above models using ROC curve analysis generated by tenfold cross-validation (Table 4). AUROCs for estradiol and the PRS were higher than that of the BCSC risk score, though differences did not reach statistical significance. Adding estradiol to the BCSC risk score resulted in modestly improved discrimination from AUROC 0.58 (95% CI 0.50–0.65) to 0.67 (95% CI 0.57–0.71), p = 0.02, while the AUROC increase with adding PRS to the BCSC risk score was of borderline statistical significance. The combination of the BCSC risk score, PRS, and estradiol levels had the highest discrimination out of all models, with AUROC 0.72 (95% CI 0.65–0.79), representing an AUROC increase of 0.14 (24%) relative to the BCSC risk score alone, p < 0.001. Furthermore, the model including BCSC risk score, PRS, and estradiol had improved discrimination relative to the BCSC-PRS (p = 0.02) and BCSC-estradiol models (p = 0.01). The ROC curves for the BCSC risk score alone and with the addition of the PRS and/or estradiol levels are shown in Fig. 1. We confirmed that inclusion of estrone, testosterone, or SHBG to models containing the BCSC risk score and estradiol levels did not further improve model performance (Supplementary Table S4).

Receiver operating characteristic curves for estrogen receptor-positive breast cancers are shown for the Breast Cancer Surveillance Consortium (BCSC) model alone and in combination with estradiol level, polygenic risk score (PRS), or both. The dashed reference line corresponds to an area under the curve of 0.5
We further examined the physiologic relationship between estradiol and ER-positive breast cancer using a cubic splines model (Figure S2). The risk of ER-positive breast cancers increased linearly with rising estradiol levels until a threshold of approximately 5 pg/ml, beyond which there was no further risk elevation associated with higher estradiol levels. The precision of our OR estimate was limited in the upper range of estradiol levels due to scarcity of data.
Discussion
The addition of estradiol levels and the PRS to the BCSC model improved its performance, with the combined model having an AUROC of 0.72 (95% CI 0.65–0.79) for ER-positive cancers, representing a statistically significant improvement from the BCSC risk score, both alone and in combination with estradiol or an 83-SNP polygenic risk score. The sample size and retrospective nature of the analysis limits our ability to definitively assess discrimination and calibration, particularly the precision of our AUROC point estimates. Nevertheless, our results contribute to the growing body of literature showing that the addition of circulating measures such as SNPs and hormone levels can improve the performance of clinical risk models.
While the PRS has been shown to improve the discrimination of the BCSC model in several reports [11, 12], these analyses focused on invasive breast cancer overall. Differential effects of clinical risk factors [27], breast density [28, 29], and SNPs [30, 31] with ER subtype have been reported. Neither the BCSC model nor PRS are fitted to receptor subtype, but these models are likely well-fitted to ER-positive cancers given that at least 80% of breast cancers diagnosed in the United States are ER-positive [27], with the proportion rising with older age [1]. In our study, it is possible that both the PRS and estradiol levels contributed subtype-specific prediction, suggesting that measurement of both biomarkers may be of clinical value.
Our study is the first to investigate the combined contributions of clinical risk factors, breast density, SNPs (as represented by a PRS), and sex hormones to breast cancer risk prediction. The most comparable study examined the contributions of seven sex hormones to the Gail and Rosner–Colditz risk models [25]. The authors used stepwise regression to identify a subset of hormones (estrone, testosterone, and prolactin) that maximized predictive power of the respective models. Direct comparison of the AUROCs in our study to those reported by Twroger et al. [25] is limited by differences in study design, statistical analysis, and performance of risk models evaluated in each study. Neither the Gail nor Rosner–Colditz models incorporate breast density, and the study [25] did not evaluate the effects of genetic variants.
Our findings around the association of the individual sex hormones with breast cancer risk are qualitatively consistent with previous studies. Estradiol has been robustly associated with risk. In our study, estradiol had the strongest association with ER-positive breast cancer in univariate analyses, and the results of our cubic splines analysis suggested a threshold effect. Estrone has also been associated with breast cancer risk in numerous studies [14, 19, 20], consistent with our results. Our results suggest that SHBG (which modulates estradiol’s physiologic effects) provides a small improvement in prediction, but our analysis was not powered to definitively assess this observation. Testosterone did not meaningfully contribute to risk prediction in our dataset, which is unsurprising given the heterogeneous results in the literature [14, 18, 19, 22,23,24].
Our findings should be interpreted in light of several considerations. First, the discrimination of the BCSC risk score in our dataset was lower than in the previous studies, where the AUROCs ranged from 0.65 to 0.66 [8, 9]. The lower AUROC we observed was likely due to matching based on age and race/ethnicity (both strong predictors of invasive breast cancer in the BCSC model). Additionally, the distribution of breast density did not differ significantly between cases and controls, possibly due to a combination of chance and the restriction of our analysis to postmenopausal women with ER-positive breast cancers, a group where the association with breast density is attenuated with increasing age [27]. The discrimination of the BCSC risk score would likely be higher in larger, unmatched studies.
Importantly, the sample size of our study limited the precision of our point estimates and comparisons. In particular, the AUROC for the PRS was higher than previously reported, though the latter studies’ AUROCs ranged from 0.60 to 0.62, which overlaps with our confidence intervals [10, 11]. The AUROC for the PRS in combination with the BCSC risk score was slightly lower than that of the PRS alone possibly due to sample splitting for cross-validation, and relative insensitivity of the AUROC for improvements in discrimination with the addition of risk factors [32]. Our AUROC comparisons must also be interpreted in light of an underlying assumption of the Wald test, that the differences in AUROC estimators follows a normal distribution. Due to our small sample size, this assumption may not have been satisfied [33].
Additionally, the case-control design limited our ability to assess calibration. Careful evaluation of model calibration in independent, preferably unmatched, datasets is essential prior to adoption of such a tool into clinical practice. Lastly, our population was predominantly white, creating the need for further assessment of discrimination and calibration in a multiracial, multiethnic population.
Our statistical approach differed from those described by others. We did not adjust our main analysis for additional covariates beyond those included in the BCSC model. Previously published studies have conditioned on such variables as parity, number of live births, age at menarche, and time since menopause [14]. These analytical approaches tend to isolate the causal effect of the hormonal pathway by controlling for potential mediators, likely leading to a conservative estimate of the hormone’s effect. In contrast, our goal was to investigate the net predictive power of sex hormones, which encompasses the effects of mediating and confounding pathways. We examined hormones without adjustment for covariates to determine whether they improved the prediction of our models. For example, we did not adjust for BMI because the relationship between obesity and breast cancer risk is primarily mediated through its effect on circulating estrogen levels [34,35,36]. Supporting this assumption, adjusting for BMI in secondary analyses minimally attenuated the association between estradiol and ER-positive breast cancer.
We chose ER-positive cancers as our endpoint given the direct causal relationship between estradiol (and other sex hormones) with these cancers. In the clinical setting, our results are therefore most applicable to chemoprevention. Guidelines empirically recommend using risk prediction models to identify high-risk women eligible for chemoprevention [3, 37], although such models are not specific to particular subtypes of breast cancer. Selective estrogen receptor modulators (SERMs) and aromatase inhibitors have established efficacy in preventing the development of ER-positive cancers in high-risk women. Improving risk prediction for ER-positive cancers may improve our ability to identify women who may specifically benefit from chemoprevention, although this approach merits further evaluation in specimens stored from randomized trials.
The threshold effect of estradiol is consistent with prior studies showing that high-risk women with undetectable estradiol levels were not at elevated risk of developing breast cancers, and did not benefit from chemoprevention with raloxifene, a selective estrogen receptor modulator [21]. Although our splines analysis is purely exploratory and limited by sample size, future studies could further investigate whether a threshold relationship exists and attempt to identify an estradiol level cutoff that could be used alone, or in conjunction with risk models, to risk-stratify women for ER-positive cancer.
Our results suggest that the addition of estradiol to clinical risk factors, breast density, and a PRS may improve the prediction of ER-positive breast cancer in postmenopausal women. This combination of predictors may improve the identification of postmenopausal women who are most likely to benefit from chemoprevention.
Abbreviations
- AUROC:
-
Area under the receiver operating characteristic curve
- BCSC:
-
Breast Cancer Surveillance Consortium
- BI-RADS:
-
Breast Imaging Reporting and Data System
- BMI:
-
Body mass index
- CCR:
-
California Cancer Registry
- CPMC:
-
California Pacific Medical Center
- GWAS:
-
Genome-wide association study
- LR:
-
Likelihood ratio
- OR:
-
Odds ratio
- PRS:
-
Polygenic risk score
- SFMR:
-
San Francisco Mammography Registry
- SHBG:
-
Sex hormone binding globulin
- SNPs:
-
Single nucleotide polymorphisms
- USPSTF:
-
United States Preventive Services Task Force
References
Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LA, Cronin KA (2014) US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst 106(5):dju055. doi:10.1093/jnci/dju055
Moyer VA (2013) Medications to decrease the risk for breast cancer in women: recommendations from the U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 159(10):698–708. doi:10.7326/0003-4819-159-10-201311190-00717
Nelson HD, Smith MEB, Griffin JC, Fu R (2013) Use of medications to reduce risk for primary breast cancer: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 158(8):604–614. doi:10.7326/0003-4819-158-8-201304160-00005
Burns RB, Schonberg MA, Tung NM, Libman H (2016) Should we offer medication to reduce breast cancer risk? Grand rounds discussion from Beth Israel Deaconess Medical Center should we offer medication to reduce breast cancer risk? Ann Intern Med 165(3):194–204. doi:10.7326/M16-0940
Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C, Mulvihill JJ (1989) Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 81(24):1879–1886
Tyrer J, Duffy SW, Cuzick J (2004) A breast cancer prediction model incorporating familial and personal risk factors. Stat Med 23(7):1111–1130. doi:10.1002/sim.1668
Cummings SR, Tice JA, Bauer S, Browner WS, Cuzick J, Ziv E, Vogel V, Shepherd J, Vachon C, Smith-Bindman R, Kerlikowske K (2009) Prevention of breast cancer in postmenopausal women: approaches to estimating and reducing risk. J Natl Cancer Inst 101(6):384–398. doi:10.1093/jnci/djp018
Tice JA, Cummings SR, Smith-Bindman R, Ichikawa L, Barlow WE, Kerlikowske K (2008) Using clinical factors and mammographic breast density to estimate breast cancer risk: development and validation of a new predictive model. Ann Intern Med 148(5):337–347
Tice JA, Miglioretti DL, Li CS, Vachon CM, Gard CC, Kerlikowske K (2015) Breast density and benign breast disease: risk assessment to identify women at high risk of breast cancer. J Clin Oncol 33(28):3137–3143. doi:10.1200/jco.2015.60.8869
Mavaddat N, Pharoah PDP, Michailidou K, Tyrer J, Brook MN, Bolla MK, Wang Q, Dennis J, Dunning AM, Shah M, Luben R, Brown J, Bojesen SE, Nordestgaard BG, Nielsen SF, Flyger H, Czene K, Darabi H, Eriksson M, Peto J, dos Santos-Silva I, Dudbridge F, Johnson N, Schmidt MK, Broeks A, Verhoef S, Rutgers EJ, Swerdlow A, Ashworth A, Orr N, Schoemaker MJ, Figueroa J, Chanock SJ, Brinton L, Lissowska J, Couch FJ, Olson JE, Vachon C, Pankratz VS, Lambrechts D, Wildiers H, Van Ongeval C, van Limbergen E, Kristensen V, Grenaker Alnæs G, Nord S, Borresen-Dale A-L, Nevanlinna H, Muranen TA, Aittomäki K, Blomqvist C, Chang-Claude J, Rudolph A, Seibold P, Flesch-Janys D, Fasching PA, Haeberle L, Ekici AB, Beckmann MW, Burwinkel B, Marme F, Schneeweiss A, Sohn C, Trentham-Dietz A, Newcomb P, Titus L, Egan KM, Hunter DJ, Lindstrom S, Tamimi RM, Kraft P, Rahman N, Turnbull C, Renwick A, Seal S, Li J, Liu J, Humphreys K, Benitez J, Pilar Zamora M, Arias Perez JI, Menéndez P, Jakubowska A, Lubinski J, Jaworska-Bieniek K, Durda K, Bogdanova NV, Antonenkova NN, Dörk T, Anton-Culver H, Neuhausen SL, Ziogas A, Bernstein L, Devilee P, Tollenaar RAEM, Seynaeve C, van Asperen CJ, Cox A, Cross SS, Reed MWR, Khusnutdinova E, Bermisheva M, Prokofyeva D, Takhirova Z, Meindl A, Schmutzler RK, Sutter C, Yang R, Schürmann P, Bremer M, Christiansen H, Park-Simon T-W, Hillemanns P, Guénel P, Truong T, Menegaux F, Sanchez M, Radice P, Peterlongo P, Manoukian S, Pensotti V, Hopper JL, Tsimiklis H, Apicella C, Southey MC, Brauch H, Brüning T, Ko Y-D, Sigurdson AJ, Doody MM, Hamann U, Torres D, Ulmer H-U, Försti A, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Andrulis IL, Knight JA, Glendon G, Marie Mulligan A, Chenevix-Trench G, Balleine R, Giles GG, Milne RL, McLean C, Lindblom A, Margolin S, Haiman CA, Henderson BE, Schumacher F, Le Marchand L, Eilber U, Wang-Gohrke S, Hooning MJ, Hollestelle A, van den Ouweland AMW, Koppert LB, Carpenter J, Clarke C, Scott R, Mannermaa A, Kataja V, Kosma V-M, Hartikainen JM, Brenner H, Arndt V, Stegmaier C, Karina Dieffenbach A, Winqvist R, Pylkäs K, Jukkola-Vuorinen A, Grip M, Offit K, Vijai J, Robson M, Rau-Murthy R, Dwek M, Swann R, Annie Perkins K, Goldberg MS, Labrèche F, Dumont M, Eccles DM, Tapper WJ, Rafiq S, John EM, Whittemore AS, Slager S, Yannoukakos D, Toland AE, Yao S, Zheng W, Halverson SL, González-Neira A, Pita G, Rosario Alonso M, Álvarez N, Herrero D, Tessier DC, Vincent D, Bacot F, Luccarini C, Baynes C, Ahmed S, Maranian M, Healey CS, Simard J, Hall P, Easton DF, Garcia-Closas M (2015) Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst. doi:10.1093/jnci/djv036
Vachon CM, Pankratz VS, Scott CG, Haeberle L, Ziv E, Jensen MR, Brandt KR, Whaley DH, Olson JE, Heusinger K, Hack CC, Jud SM, Beckmann MW, Schulz-Wendtland R, Tice JA, Norman AD, Cunningham JM, Purrington KS, Easton DF, Sellers TA, Kerlikowske K, Fasching PA, Couch FJ (2015) The contributions of breast density and common genetic variation to breast cancer risk. J Natl Cancer Inst 107(5):dju397. doi:10.1093/jnci/dju397
Shieh Y, Hu D, Ma L, Huntsman S, Gard CC, Leung JW, Tice JA, Vachon CM, Cummings SR, Kerlikowske K, Ziv E (2016) Breast cancer risk prediction using a clinical risk model and polygenic risk score. Breast Cancer Res Treat. doi:10.1007/s10549-016-3953-2
Dite GS, MacInnis RJ, Bickerstaffe A, Dowty JG, Allman R, Apicella C, Milne RL, Tsimiklis H, Phillips KA, Giles GG, Terry MB, Southey MC, Hopper JL (2016) Breast cancer risk prediction using clinical models and 77 independent risk-associated SNPs for women aged under 50 years: Australian Breast Cancer Family Registry. Cancer Epidemiol Biomark Prev 25(2):359–365. doi:10.1158/1055-9965.epi-15-0838
Key T, Appleby P, Barnes I, Reeves G (2002) Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst 94(8):606–616
Endogenous Hormones Breast Cancer Collaborative Group, Key TJ, Appleby PN, Reeves GK, Roddam AW, Helzlsouer KJ, Alberg AJ, Rollison DE, Dorgan JF, Brinton LA, Overvad K, Kaaks R, Trichopoulou A, Clavel-Chapelon F, Panico S, Duell EJ, Peeters PH, Rinaldi S, Fentiman IS, Dowsett M, Manjer J, Lenner P, Hallmans G, Baglietto L, English DR, Giles GG, Hopper JL, Severi G, Morris HA, Hankinson SE, Tworoger SS, Koenig K, Zeleniuch-Jacquotte A, Arslan AA, Toniolo P, Shore RE, Krogh V, Micheli A, Berrino F, Barrett-Connor E, Laughlin GA, Kabuto M, Akiba S, Stevens RG, Neriishi K, Land CE, Cauley JA, Lui LY, Cummings SR, Gunter MJ, Rohan TE, Strickler HD (2011) Circulating sex hormones and breast cancer risk factors in postmenopausal women: reanalysis of 13 studies. Br J Cancer 105(5):709–722. doi:10.1038/bjc.2011.254
Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE (2004) Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst 96(24):1856–1865. doi:10.1093/jnci/djh336
Woolcott CG, Shvetsov YB, Stanczyk FZ, Wilkens LR, White KK, Caberto C, Henderson BE, Le Marchand L, Kolonel LN, Goodman MT (2010) Plasma sex hormone concentrations and breast cancer risk in an ethnically diverse population of postmenopausal women: the Multiethnic Cohort Study. Endocr Relat Cancer 17(1):125–134. doi:10.1677/ERC-09-0211
Zeleniuch-Jacquotte A, Shore RE, Koenig KL, Akhmedkhanov A, Afanasyeva Y, Kato I, Kim MY, Rinaldi S, Kaaks R, Toniolo P (2004) Postmenopausal levels of oestrogen, androgen, and SHBG and breast cancer: long-term results of a prospective study. Br J Cancer 90(1):153–159. doi:10.1038/sj.bjc.6601517
Baglietto L, Severi G, English DR, Krishnan K, Hopper JL, McLean C, Morris HA, Tilley WD, Giles GG (2010) Circulating steroid hormone levels and risk of breast cancer for postmenopausal women. Cancer Epidemiol Biomark Prev 19(2):492–502. doi:10.1158/1055-9965.EPI-09-0532
Eliassen AH, Missmer SA, Tworoger SS, Spiegelman D, Barbieri RL, Dowsett M, Hankinson SE (2006) Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J Natl Cancer Inst 98(19):1406–1415. doi:10.1093/jnci/djj376
Cummings SR, Duong T, Kenyon E, Cauley JA, Whitehead M, Krueger KA (2002) Serum estradiol level and risk of breast cancer during treatment with raloxifene. JAMA 287(2):216–220
Cummings SR, Lee JS, Lui LY, Stone K, Ljung BM, Cauleys JA (2005) Sex hormones, risk factors, and risk of estrogen receptor-positive breast cancer in older women: a long-term prospective study. Cancer Epidemiol Biomark Prev 14(5):1047–1051. doi:10.1158/1055-9965.EPI-04-0375
Beattie MS, Costantino JP, Cummings SR, Wickerham DL, Vogel VG, Dowsett M, Folkerd EJ, Willett WC, Wolmark N, Hankinson SE (2006) Endogenous sex hormones, breast cancer risk, and tamoxifen response: an ancillary study in the NSABP Breast Cancer Prevention Trial (P-1). J Natl Cancer Inst 98(2):110–115. doi:10.1093/jnci/djj011
Farhat GN, Cummings SR, Chlebowski RT, Parimi N, Cauley JA, Rohan TE, Huang AJ, Vitolins M, Hubbell FA, Manson JE, Cochrane BB, Lane DS, Lee JS (2011) Sex hormone levels and risks of estrogen receptor-negative and estrogen receptor-positive breast cancers. J Natl Cancer Inst 103(7):562–570. doi:10.1093/jnci/djr031
Tworoger SS, Zhang X, Eliassen AH, Qian J, Colditz GA, Willett WC, Rosner BA, Kraft P, Hankinson SE (2014) Inclusion of endogenous hormone levels in risk prediction models of postmenopausal breast cancer. J Clin Oncol 32(28):3111–3117. doi:10.1200/jco.2014.56.1068
Sickles EA, Orsi CJ, Bassett LW (2013) Mammography. ACR BI-RADS atlas, breast imaging reporting and data system. American College of Radiology, Reston
Kerlikowske K, Gard CC, Tice JA, Ziv E, Cummings SR, Miglioretti DL (2017) Risk factors that increase risk of estrogen receptor-positive and -negative breast cancer. J Natl Cancer Inst 109(5):djw276. doi:10.1093/jnci/djw276
Bertrand KA, Scott CG, Tamimi RM, Jensen MR, Pankratz VS, Norman AD, Visscher DW, Couch FJ, Shepherd J, Chen YY, Fan B, Wu FF, Ma L, Beck AH, Cummings SR, Kerlikowske K, Vachon CM (2015) Dense and nondense mammographic area and risk of breast cancer by age and tumor characteristics. Cancer Epidemiol Biomark Prev 24(5):798–809. doi:10.1158/1055-9965.epi-14-1136
Bertrand KA, Tamimi RM, Scott CG, Jensen MR, Pankratz V, Visscher D, Norman A, Couch F, Shepherd J, Fan B, Chen YY, Ma L, Beck AH, Cummings SR, Kerlikowske K, Vachon CM (2013) Mammographic density and risk of breast cancer by age and tumor characteristics. Breast Cancer Res 15(6):R104. doi:10.1186/bcr3570
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, Turnbull C, Rahman N, Fletcher O, Peto J, Gibson L, Dos Santos Silva I, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Czene K, Irwanto A, Liu J, Waisfisz Q, Meijers-Heijboer H, Adank M, van der Luijt RB, Hein R, Dahmen N, Beckman L, Meindl A, Schmutzler RK, Muller-Myhsok B, Lichtner P, Hopper JL, Southey MC, Makalic E, Schmidt DF, Uitterlinden AG, Hofman A, Hunter DJ, Chanock SJ, Vincent D, Bacot F, Tessier DC, Canisius S, Wessels LF, Haiman CA, Shah M, Luben R, Brown J, Luccarini C, Schoof N, Humphreys K, Li J, Nordestgaard BG, Nielsen SF, Flyger H, Couch FJ, Wang X, Vachon C, Stevens KN, Lambrechts D, Moisse M, Paridaens R, Christiaens MR, Rudolph A, Nickels S, Flesch-Janys D, Johnson N, Aitken Z, Aaltonen K, Heikkinen T, Broeks A, Veer LJ, van der Schoot CE, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Zamora MP, Perez JI, Pita G, Alonso MR, Cox A, Brock IW, Cross SS, Reed MW, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Henderson BE, Schumacher F, Le Marchand L, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Lindblom A, Margolin S, Hooning MJ, Hollestelle A, van den Ouweland AM, Jager A, Bui QM, Stone J, Dite GS, Apicella C, Tsimiklis H, Giles GG, Severi G, Baglietto L, Fasching PA, Haeberle L, Ekici AB, Beckmann MW, Brenner H, Muller H, Arndt V, Stegmaier C, Swerdlow A, Ashworth A, Orr N, Jones M, Figueroa J, Lissowska J, Brinton L, Goldberg MS, Labreche F, Dumont M, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Bruning T, Radice P, Peterlongo P, Manoukian S, Bonanni B, Devilee P, Tollenaar RA, Seynaeve C, van Asperen CJ, Jakubowska A, Lubinski J, Jaworska K, Durda K, Mannermaa A, Kataja V, Kosma VM, Hartikainen JM, Bogdanova NV, Antonenkova NN, Dork T, Kristensen VN, Anton-Culver H, Slager S, Toland AE, Edge S, Fostira F, Kang D, Yoo KY, Noh DY, Matsuo K, Ito H, Iwata H, Sueta A, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Shu XO, Lu W, Gao YT, Cai H, Teo SH, Yip CH, Phuah SY, Cornes BK, Hartman M, Miao H, Lim WY, Sng JH, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Shen CY, Hsiung CN, Wu PE, Ding SL, Sangrajrang S, Gaborieau V, Brennan P, McKay J, Blot WJ, Signorello LB, Cai Q, Zheng W, Deming-Halverson S, Shrubsole M, Long J, Simard J, Garcia-Closas M, Pharoah PD, Chenevix-Trench G, Dunning AM, Benitez J, Easton DF (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45(4):353–361. doi:10.1038/ng.2563 361e351-352
Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Maranian MJ, Bolla MK, Wang Q, Shah M, Perkins BJ, Czene K, Eriksson M, Darabi H, Brand JS, Bojesen SE, Nordestgaard BG, Flyger H, Nielsen SF, Rahman N, Turnbull C, Fletcher O, Peto J, Gibson L, dos Santos-Silva I, Chang-Claude J, Flesch-Janys D, Rudolph A, Eilber U, Behrens S, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Khan S, Aaltonen K, Ahsan H, Kibriya MG, Whittemore AS, John EM, Malone KE, Gammon MD, Santella RM, Ursin G, Makalic E, Schmidt DF, Casey G, Hunter DJ, Gapstur SM, Gaudet MM, Diver WR, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Berg CD, Chanock SJ, Figueroa J, Hoover RN, Lambrechts D, Neven P, Wildiers H, van Limbergen E, Schmidt MK, Broeks A, Verhoef S, Cornelissen S, Couch FJ, Olson JE, Hallberg E, Vachon C, Waisfisz Q, Meijers-Heijboer H, Adank MA, van der Luijt RB, Li J, Liu J, Humphreys K, Kang D, Choi JY, Park SK, Yoo KY, Matsuo K, Ito H, Iwata H, Tajima K, Guenel P, Truong T, Mulot C, Sanchez M, Burwinkel B, Marme F, Surowy H, Sohn C, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Gonzalez-Neira A, Benitez J, Zamora MP, Perez JI, Shu XO, Lu W, Gao YT, Cai H, Cox A, Cross SS, Reed MW, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Lindblom A, Margolin S, Teo SH, Yip CH, Taib NA, Tan GH, Hooning MJ, Hollestelle A, Martens JW, Collee JM, Blot W, Signorello LB, Cai Q, Hopper JL, Southey MC, Tsimiklis H, Apicella C, Shen CY, Hsiung CN, Wu PE, Hou MF, Kristensen VN, Nord S, Alnaes GI, Giles GG, Milne RL, McLean C, Canzian F, Trichopoulos D, Peeters P, Lund E, Sund M, Khaw KT, Gunter MJ, Palli D, Mortensen LM, Dossus L, Huerta JM, Meindl A, Schmutzler RK, Sutter C, Yang R, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Hartman M, Miao H, Chia KS, Chan CW, Fasching PA, Hein A, Beckmann MW, Haeberle L, Brenner H, Dieffenbach AK, Arndt V, Stegmaier C, Ashworth A, Orr N, Schoemaker MJ, Swerdlow AJ, Brinton L, Garcia-Closas M, Zheng W, Halverson SL, Shrubsole M, Long J, Goldberg MS, Labreche F, Dumont M, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Bruning T, Radice P, Peterlongo P, Manoukian S, Bernard L, Bogdanova NV, Dork T, Mannermaa A, Kataja V, Kosma VM, Hartikainen JM, Devilee P, Tollenaar RA, Seynaeve C, Van Asperen CJ, Jakubowska A, Lubinski J, Jaworska K, Huzarski T, Sangrajrang S, Gaborieau V, Brennan P, McKay J, Slager S, Toland AE, Ambrosone CB, Yannoukakos D, Kabisch M, Torres D, Neuhausen SL, Anton-Culver H, Luccarini C, Baynes C, Ahmed S, Healey CS, Tessier DC, Vincent D, Bacot F, Pita G, Alonso MR, Alvarez N, Herrero D, Simard J, Pharoah PP, Kraft P, Dunning AM, Chenevix-Trench G, Hall P, Easton DF (2015) Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet 47(4):373–380. doi:10.1038/ng.3242
Pepe MS, Kerr KF, Longton G, Wang Z (2013) Testing for improvement in prediction model performance. Stat Med 32(9):1467–1482. doi:10.1002/sim.5727
Demler OV, Pencina MJ, D’Agostino RB (2012) Misuse of DeLong test to compare AUCs for nested models. Stat Med 31(23):2577–2587. doi:10.1002/sim.5328
Anderson GL, Neuhouser ML (2012) Obesity and the risk for premenopausal and postmenopausal breast cancer. Cancer Prev Res 5(4):515–521. doi:10.1158/1940-6207.capr-12-0091
Cecchini RS, Costantino JP, Cauley JA, Cronin WM, Wickerham DL, Land SR, Weissfeld JL, Wolmark N (2012) Body mass index and the risk for developing invasive breast cancer among high-risk women in NSABP P-1 and STAR breast cancer prevention trials. Cancer Prev Res 5(4):583–592. doi:10.1158/1940-6207.capr-11-0482
Neuhouser ML, Aragaki AK, Prentice RL, Manson JE, Chlebowski R, Carty CL, Ochs-Balcom HM, Thomson CA, Caan BJ, Tinker LF, Urrutia RP, Knudtson J, Anderson GL (2015) Overweight, obesity, and postmenopausal invasive breast cancer risk: a secondary analysis of the women’s health initiative randomized clinical trials. JAMA Oncol 1(5):611–621. doi:10.1001/jamaoncol.2015.1546
Visvanathan K, Hurley P, Bantug E, Brown P, Col NF, Cuzick J, Davidson NE, Decensi A, Fabian C, Ford L, Garber J, Katapodi M, Kramer B, Morrow M, Parker B, Runowicz C, Vogel VG 3rd, Wade JL, Lippman SM (2013) Use of pharmacologic interventions for breast cancer risk reduction: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 31(23):2942–2962. doi:10.1200/jco.2013.49.3122
Acknowledgements
We are grateful to Charles E. McCulloch, PhD for his input on statistical methods. The collection of cancer data was supported in part by the California Cancer Registry. For a full description, please see: http://breastscreening.cancer.gov/work/acknowledgement.html. We thank the participating women, mammography facilities, and radiologists for the data they have provided for this study.
Funding
This work was supported by the NCI-funded Breast Cancer Surveillance Consortium grant P01 CA154292. E. Ziv also received support from the National Cancer Institute under grant K24 CA169004. S.R. Cummings also received support for the collection of blood specimens from the DaCosta Fund for the Prevention of Breast Cancer, the Clinical Research in Clinical Care (CRCLE) funds provided by the California Pacific Medical Center, and by a grant from the Eli Lilly Foundation. Y. Shieh was supported by a National Research Service Award through the National Institutes of Health T32 HP19025. Data collection for this work was supported by the National Cancer Institute-funded Breast Cancer Surveillance Consortium (HHSN261201100031C). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Specifically, each registry in the Breast Cancer Surveillance Consortium and the Statistical Coordinating Center (SCC) have received institutional review board approval for either active or passive consenting processes or a waiver of consent to enroll participants, link data, and perform analytic studies. All procedures are Health Insurance Portability and Accountability Act (HIPAA) compliant and all registries and the SCC have received a Federal Certificate of Confidentiality and other protection for the identities of women, physicians, and facilities who are subjects of this research.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shieh, Y., Hu, D., Ma, L. et al. Joint relative risks for estrogen receptor-positive breast cancer from a clinical model, polygenic risk score, and sex hormones. Breast Cancer Res Treat 166, 603–612 (2017). https://doi.org/10.1007/s10549-017-4430-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4430-2