
Abstract
Purpose
The extracellular region (EC) of the vascular endothelial growth factor (VEGF) receptor-2 (VEGFR-2) contains seven immunoglobulin-like (Ig-like) domains that are required for specific ligand binding and receptor dimerization. Studies of domain 4–7 deletions and substitutions provided insights into the interaction between receptors in the absence of VEGF. In this study, we investigated the effect of domain 4 in ligand-independent VEGFR-2 dimerization and activation in human vascular endothelial cells and human breast cancer cells.
Methods
To confirm the role of domain 4 in ligand-independent receptor dimerization and activation, two VEGFR-2 fragments with and without domain 4, KFP1 and KFP2, were generated by recombinant DNA technology. We measured the affinity of KFP1 and KFP2 with VEGFR-2, and the roles of KFP1 and FKP2 in dimerization and phosphorylation of VEGFR-2. We also evaluated the effect of KFP1 and FKP2 on cell proliferation and migration in HUVECs and in human breast cancer cells.
Results
We showed that KFP1 did not affect the interaction of VEGFR-2 and VEGF but bound VEGFR-2 in the absence of VEGF. Furthermore, cross-linking and cross-linking immunoblotting demonstrated that KFP1 could form a complex with VEGFR-2, which resulted in VEGFR-2 dimerization in the absence of VEGF. Importantly, we found that the KDR fragment with domain 4 induced phosphorylation of VEGFR-2, as well as phosphorylation of downstream receptor kinases in HUVECs and VEGFR-2-positive breast cancer cells. Consistent with these results, this ligand-independent activation of VEGFR-2 also promoted downstream signaling and cell proliferation and migration.
Conclusions
The domain 4 of VEGFR-2 plays an important role in the interaction between VEGFR receptors in the absence of VEGF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human vascular endothelial growth factor (VEGF), also known as VEGF-A, belongs to the growth factor family. The binding of VEGF to its receptors, VEGFR-1 (Flt-1), VEGFR-2 (KDR), and VEGFR-3 (Flt-4), triggers receptor-associated tyrosine kinase receptor (RTK) activity and regulates the formation of blood and lymphatic vessels. Among these three receptors, VEGFR-2 plays key roles in mediating VEGF-induced endothelial cell proliferation, migration, and angiogenesis. Importantly, VEGR-2 directly regulates the pathogenesis of several tumors including breast cancer [1, 2]. VEGF/VEGFR-2 promotes breast cancer cell proliferation, invasion, and survival in an essential autocrine manner [3–5]. In addition to increased expression in breast cancer cell lines or human tumors, VEGFR-2 expression and activation are related to the disease stage, recurrence, and metastasis, indicating that VEGFR-2 may serve as a specific biomarker in breast cancer [5–8].
VEGFR-2 contains an extracellular (EC) domain composed of seven immunoglobulin-like (Ig-like) domains, a single transmembrane (TM) region, and an intracellular (IC) split tyrosine kinase [9–11]. The extracellular Ig-like domains 2 and 3 of VEGFR-2 have been identified as the ligand-binding region [12–14], and the Ig-like domains 4–7 are required for receptor dimerization [15–18].
It is generally believed that VEGFR-2 is monomeric in the absence of ligand and follows the conventional model of ligand-induced dimerization and activation. However, a recent report suggested that VEGFR-2 can form a dimer in the absence of VEGF and that the dimer is phosphorylated [19]. Although ligand-independent dimerization of VEGFR-2 has not been extensively investigated, previous reports have shown that other growth factor receptors, including PDGFR, EGFR, FGFR, and TrkA, can form dimers in the absence of ligands [20–25]. Furthermore, membrane-proximal regions, such as the Ig-like domain 4, are involved in the ligand-independent dimerization of the PDGFR. The interaction of stem cell factor (SCF) with its receptor KIT also showed that KIT dimerization can be inhibited by a monoclonal antibody targeting the Ig-like domain 4 or through the use of an Ig-like domain 4-deleted receptor, indicating that SCF-induced KIT dimerization depends on the presence of the fourth Ig-like domain [26]. Similarly, the Ig-like domains 4 and 7 of VEGFR-2 are involved in the ligand-independent dimerization and stabilize VEGFR-2 dimers in the absence of VEGF [19, 27–29]. However, the contributions of the three VEGFR-2 domains showed that the EC domain inhibits dimerization, while the TM domain and IC domain enhance dimerization and phosphorylation in the absence of VEGF [30]. In particular, the specific interaction between the IC domains regulates VEGFR-2 dimerization in the absence of ligand. With the deletion of Ig-like domains 4–7, VEGFR-2 activation is observed in the absence of VEGF binding [27].
However, the Ig-like domain 4-mediated VEGFR-2 dimerization is still unclear, and the Ig-like domain 4-mediated ligand-independent receptor activation has yet to be demonstrated in HUVECs and some VEGFR-2-positive tumor cells. Here, we generated recombinant KDR fragments with or without the Ig-like domain 4 and investigated their effects in mediating ligand-independent VEGFR-2 dimerization in human vascular endothelial cells, as well as in human breast cancer cells.
Results
Production of KDR fragments and functional validation
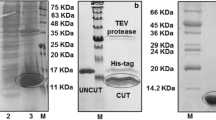
The KDR fragments KFP1 and KFP2 were separately fused to the Fc domain of human IgG1 (Fig. 1a), which is a commonly used strategy to facilitate separation or purification of proteins. KFP1 and KFP2 were purified from the culture supernatant using Protein A affinity chromatography. Purified KFP1 has an apparent molecular mass of ~72 and ~145 kDa as a monomer and a dimer under reducing and non-reducing conditions, respectively. The apparent molecular mass of KFP2 was ~55 and ~110 kDa as a monomer and a dimer (Fig. 1b) under reducing and non-reducing conditions, respectively. To functionally validate purified KFP1 and KFP2, we characterized their binding to VEGF165 using ELISA. As expected, KFP1 displayed a similar binding affinity to KFP2 (Fig. 1c), indicating that the Ig-like domains 2 and 3 mediated ligand binding, and the addition of Ig-like domain 4 does not affect the ligand-binding affinity.

Characterization of the KDR fragment proteins KFP1 and KFP2. a Schematic representation of full-length VEGFR-2/KDR is provided, indicating their seven Ig domains (hexagon), transmembrane regions (black bars), and kinase domain (rectangles). KFP1 contains D2-4 domains of VEGFR-2 fused to the Fc portion of human IgG1. KFP2 is the same construct as KFP1, but the D4 domain has been removed. b KFP1 and KFP2 were subjected to non-reducing and reducing SDS-PAGE, and Western blots were conducted with goat anti-human IgG1 Fc antibody. Sizes of molecular weight markers (kDa) are shown on the left and schematics of KFP1 and KFP2 on the right. c Recombinant VEGF165 was immobilized on microtiter plates and incubated with KFP1 or KFP2. Bound KDR fragment proteins were detected by goat anti-human IgG Fc antibody, and specific binding was calculated as optical density at 450 nm
Ig-like domain 4 mediates VEGFR-2 dimerization in the absence of ligand
To investigate the VEGFR-2 dimerization mediated by Ig-like domain 4, we used cross-linking experiments examining KDR fragment-mediated dimer formation in the absence of VEGF. KFP1 cross-linking revealed two bands corresponding to monomer (145 kDa) and dimer (290 kDa) (Fig. 2a), indicating that the cross-linked KFP1 dimer can be formed in the absence of ligand. However, no dimers were detected in the cross-linked reaction for KFP2 itself (Fig. 2b) and for the interaction between KFP2 and KFP1 (data not shown).

KDR fragment proteins induce the dimerization of VEGFR-2 in the absence of ligand: a self-dimerization of KFP1 with BS3 agent; b elf-dimerization of KFP2 with BS3 agent; c and d dimerization of VEGFR-2 of HUVECs induced by KDR fragment proteins. HUVECs were treated with KFP1 or KFP2 for 2 h. The cells were cross-linked with bis(sulfosuccinimdyl) suberate (BS3), and the cell lysates were immunoblotted with anti-VEGFR-2 antibody (c) or anti-human IgG Fc antibody (d)
In another cross-linked reaction system, the addition of KFP1 to HUVECs resulted in the formation of two cross-linked products that were recognized by anti-VEGFR-2 antibody or anti-human IgG Fc antibody (Fig. 2c and d). Based on the relative molecular mass corresponding to the molecular mass of ~360 and ~580 kDa, respectively, the low-molecular weight cross-linked product should be a VEGFR-2 complex resulting from the receptor–receptor interaction. The high-molecular weight cross-linked product is favored by the KFP1–VEGFR-2 complex formation (Fig. 2c and d, lane 2). Under the same conditions, the cross-linked products were not detected in HUVECs treated by KFP2 (Fig. 2c and d, lane 3). These results indicate that KFP1, a KDR fragment with an Ig-like domain 4, can mediate ligand-independent dimerization of itself, as well as VEGFR-2 dimerization. In contrast, KFP2 without an Ig-like domain 4 did not. Apparently, the Ig-like domain 4 mediated the receptor–receptor interaction.
KDR fragments bind to VEGFR-2
VEGFR-2 dimer formation mediated by KFP1 suggested that the Ig-like domain 4 may bind to VEGFR-2. We therefore examined the binding of KDR fragments to VEGFR-2 in the absence of ligand. Using biolayer interferometry, we first analyzed the binding ability of KDR fragments to membrane VEGFR-2 of HUVECs in the absence of VEGF. As expected, KFP1 could bind to membrane VEGFR-2, whereas the binding of KFP2 was not observed for VEGFR-2 (Fig. 3a and b).

VEGFR-2 binding affinity of KDR fragment proteins. a Biolayer interferometry binding assay to KFP1 or KFP2 and membrane VEGFR-2. KDR fragment proteins were loaded to AHC biosensor probes, and their binding with membrane VEGFR-2 from membrane proteins of HUVECs was detected. b Binding assay of KFP1 and membrane VEGFR-2 by biolayer interferometry. KFP1 was loaded to AHC biosensor probes, and its interaction with the membrane proteins of HUVECs (MP) or membrane proteins of HUVECs depleted of VEGFR-2 by immunoprecipitation with anti-VEGFR-2 antibody (MP (−VEGFR2)) was assessed. c Kinetic affinity measurement of KDR fragment proteins with rhVEGFR-2 using ForteBio. Curves of data indicate the wavelength shift corresponding to phases of association and dissociation. KDR fragment proteins binding to 50 μg/ml biotin–rhVEGFR-2 on an SA biosensor chip. From top, the protein concentrations are 5000 nM (dark blue), 2500 nM (red), 1000 nM (light blue), 500 nM (green), 250 nM (orange), 100 nM (purple), and 50 nM (marine blue). The curves can be used to determine the KD, Kon, and Koff of KDR fragment proteins; see Table 1
To determine the binding affinity of KFP1 for VEGFR-2, biotin-linked recombinant VEGFR-2 (rhVEGFR-2) was used to load streptavidin (SA) biosensor probes, and the interaction with KFP1 or KPF2 was detected in the absence of VEGF. The dynamic association and dissociation curves for the binding of KDR fragments with VEGFR-2 are presented in Fig. 3c. The affinity constants from KDR fragments to VEGFR-2 are listed in Table 1. KFP1 displays a K D of ~123 nM, whereas that of KFP2 was approximately 41200 nM, indicating that KFP1 is capable of binding to VEGFR-2.
Ig-like domain 4 mediates the phosphorylation of VEGFR-2 and activation of downstream kinases in the absence of ligand
To investigate whether Ig-like domain 4 mediates the phosphorylation of VEGFR-2 as well as the activation of the downstream kinases, serum-starved HUVECs were treated with 1.5 μM KFP1 in the absence of VEGF for 0–120 min. VEGFR-2 phosphorylation was determined by a Phosphorylation Assay Kit with a phosphotyrosine-specific antibody. As shown in Fig. 4, KFP1 induced phosphorylation of VEGFR-2 in the absence of VEGF. The phosphorylation of VEGFR-2 was observed after ~15 min, peaking within 60 min (Fig. 4a). The VEGFR-2 phosphorylation was blocked by a specific antibody against the VEGFR-2 ectodomain (Fig. 4b). KFP1-mediated VEGFR-2 phosphorylation also indicated the activation of downstream kinases in the absence of ligand. We therefore tested two kinases: phospholipase Cγ (PLCγ) and the extracellular signal-regulated kinase (ERK1/2). As shown in Fig. 4c, d, serum-starved HUVECs were treated with 1.5 μM KFP1 in the absence of VEGF for 60 and 120 min, and significantly increased phosphorylation of PLCγ and ERK1/2 was observed, indicating that the downstream kinase activity of VEGFR-2 was also mediated by Ig-like domain 4.

Phosphorylation of VEGFR-2 and activation of downstream kinases in HUVECs induced by KDR fragment proteins. a HUVECs were serum-starved for 16 h and stimulated for the indicated times with 1.5 μM KDR fragment proteins. b Serum-starved HUVECs were stimulated for 1 h with the indicated concentrations of KFP1 in the presence or absence of specific antibody against the VEGFR-2 ectodomain and then used in VEGFR-2-phosphorylated and total ELISAs. c HUVECs were stimulated with 1.5 μM KFP1 for 1 h following a serum starvation for 16 h. PLCγ kinase activity was determined by Western blot analysis. d HUVECs were serum-starved for 16 h and stimulated with 1.5 μM KFP1 for 2 h, and phosphorylated and total ERKs were determined by Western blot analysis. *p < 0.05, compared with control “0”; # p < 0.05, compared with group control “−mAB”
Ig-like domain 4-mediated HUVEC proliferation and migration in the absence of ligand
Endothelial cell proliferation and migration play a central role in angiogenesis, and the VEGFR-2-mediated signaling pathway has an important function. We next examined whether Ig-like domain 4 mediates HUVEC proliferation and migration. HUVEC viability was measured by the CCK-8 method. In the absence of VEGF, KFP1 dose-dependently stimulated HUVEC proliferation, whereas KFP2 had no effect on HUVEC proliferation (Fig. 5a). In addition, HUVEC mobility was assessed in a modified Boyden chamber assay. Following KFP1 treatment, HUVEC migration significantly increased, but KFP2 had no effect on migration (Fig. 5c). More importantly, the cell proliferation and migration stimulated by KFP1 in the absence of ligand were blocked by a specific antibody against the VEGFR-2 ectodomain (Fig. 5b and d).

KDR fragment proteins induce the proliferation and migration of endothelial cells in the absence of VEGF. a HUVECs were serum-starved for 16 h and then treated for 96 h with various concentrations of KDR fragment proteins. b Serum-starved HUVECs were stimulated with 1.5 μM KFP1 in the presence or absence of specific antibody against VEGFR-2 ectodomain. Cell proliferation was measured by the CCK-8 method and normalized to PBS controls. c HUVECs were placed in the upper compartment of the Boyden chamber and allowed to migrate towards basal media containing 0.1% FBS with or without a series of KDR fragment proteins. The percentage of total migration was calculated as (FSample − FBlank)/(FBasal − FBlank) × 100, where FBasal is the fluorescence in the absence of KDR fragment proteins, FSample is the fluorescence in the presence of KDR fragment proteins, and FBlank is the fluorescence with no cells. d HUVEC migration was assessed in the absence and presence of specific antibody against VEGFR-2 ectodomain with 1.5 μM KFP1. Fold migration was calculated as the ratio FSample/FBasal. Data are the mean ± SEM from three independent experiments. *p < 0.05, vs. PBS control “0”; # p < 0.05, vs. group control “−mAB”
Ig-like domain 4-mediated ligand-independent activation promotes proliferation, migration, and invasion in VEGFR-2-positive breast cancer cells
VEGFR-2 is not exclusively expressed in vascular endothelial cells; it is prominently expressed in breast cancer cells, including estrogen receptor-positive or receptor-negative breast cancer. To determine the role of KFP1 with Ig-like domain 4 on the proliferation of breast cancer cells, we first tested the basal level of VEGFR-2 expression in six cultured cell lines. We showed that three lines, MDA-MB-231, MDA-MB-435, and HCC1937, had high VEGFR-2 expression (in Supplementary material). We used these lines to test the effect of KFP1 on cell proliferation in VEGFR-2-positive cell lines and showed that KFP1 significantly stimulated cell proliferation at 48 h in the absence of VEGF (p < 0.05) (Fig. 6a). In contrast, cell proliferation was not observed in three VEGFR-2-negative cell lines (Fig. 6b). In VEGFR-2-positive breast cancer cell lines, the proliferation stimulated by KFP1 was blocked by a specific antibody against the VEGFR-2 ectodomain (Fig. 6c).

KDR fragment proteins induce the proliferation, migration, and invasion of breast cancer cells in the absence of VEGF. VEGFR-2-positive (a) or VEGFR-2-negative (b) breast cancer cells were treated for 48 h with various concentrations of KDR fragment proteins after being serum-starved for 16 h. Serum-starved VEGFR-2-positive breast cancer cells were stimulated with 1.5 μM KFP1 in the presence or absence of specific antibody against VEGFR-2 ectodomain (c). Cell proliferation was measured by the CCK-8 method and normalized to PBS controls. VEGFR-2-positive (d) or VEGFR-2-negative (e) breast cancer cells were placed in the upper compartment of the Boyden chamber and allowed to migrate towards the basal media containing 0.1% FBS with 1.5 μM KDR fragment proteins for 24 h, and the migrated cells were shown by indirect immunofluorescence and quantified with fluorescence. The percentage of total migration was calculated as (FSample − FBlank)/(FBasal − FBlank) × 100, where FBasal is the fluorescence in the absence of KDR fragment proteins, FSample is the fluorescence in the presence of KDR fragment proteins, and FBlank is the fluorescence with no cells. VEGFR-2-positive (g) or VEGFR-2-negative (f) breast cancer cells were placed in the upper compartment of the invasion chamber and allowed to invade towards the basal media containing 0.1% FBS with 1.5 μM KDR fragment proteins for 24 h, and the invaded cells were shown by indirect immunofluorescence and quantified with fluorescence. The percentage of total invasion was calculated as (FSample − FBlank)/(FBasal − FBlank) × 100, where FBasal is the fluorescence in the absence of KDR fragment proteins, FSample is the fluorescence in the presence of KDR fragment proteins, and FBlank is the fluorescence with no cells. Values and error bars represent the value and standard error of the mean from three independent experiments. *p < 0.05, vs. PBS control “0”; # p < 0.05, vs. group control “−mAB”
We next tested the effect of KFP1 on cell migration in VEGFR-2-positive cell lines. Consistent with the effect of KFP1 on cell proliferation, KFP1 significantly increased VEGFR-2-positive cell migration at 24 h in the absence of VEGF (p < 0.05). However, no cell migration was observed in VEGFR-2-negative cell lines (Fig. 6d and e).
Finally, we tested the effect of KFP1 on cell invasion in VEGFR-2-positive cell lines. As expected, KFP1 also increased VEGFR-2-positive cell invasion at 24 h in the absence of VEGF (p < 0.05). No cell invasion was observed in VEGFR-2-negative cell lines (Fig. 6f and g).
Discussion
Several research teams have shown that the Ig-like domains 4 and 7 of VEGFR-2 are involved in the ligand-independent dimerization via stabilization of VEGFR-2 dimers in the absence of VEGF [19, 28]. Our results also clearly demonstrated that the Ig-like domain 4 is required for the ligand-independent dimerization and activation. We generated two KDR fragments, KFP1 with Ig-like domain 4 and KFP2 without Ig-like domain 4, to investigate the ligand-independent VEGFR-2 activation. Activation was due to a direct interaction of Ig-like domain 4 with VEGFR-2.
First, we confirmed that domain 4 mediates VEGFR-2 dimerization in the absence of ligand and that this depends on domain 4 binding with VEGFR-2. In the absence of VEGF165, KFP1 was shown to bind either membrane VEGFR-2 or rhVEGFR-2, and the binding affinity of KFP2 to VEGFR-2 was not observed. In the cross-linked reaction system with HUVECs, a complex of KFP1–VEGFR-2 was formed in the absence of VEGF165 (Fig. 2c and d, lane 2), and the cross-linked products were not detected in HUVECs treated by KFP2 (Fig. 2c and d, lane 3). Unexpectedly, KFP1 self-cross-linking revealed dimer formation in the absence of VEGF165, indicating that the Ig-like domain 4 is likely to be the determinant factor in the process of dimer formation. This hypothesis is supported by electron microscopy analysis showing that VEGFR-2 ECD monomers are always held together at both ends and form a predimerized unliganded domain, and the crossover events of the ECD monomers often take place in the Ig-like domain 4 [30].
Second, we confirmed that domain 4 mediates the activation of VEGFR-2 and its downstream molecules in the absence of ligand. In cultured HUVECs, KFP1 induces phosphorylation of VEGFR-2 in the absence of VEGF165, and the phosphorylation stimulated by KFP1 is observed within 15 min. In the same experimental conditions, KFP1 also induced phosphorylation of the downstream molecules ERK1/2 and PLCγ in HUVECs. In contrast, we did not observe any phosphorylation of VEGFR-2 and its downstream molecules after treatment with KFP2, indicating that domain 4 is required for the ligand-independent activation of VEGFR-2 in HUVECs. Surprisingly, the domain 4-mediated ligand-independent VEGFR-2 activation can also promote HUVEC proliferation and migration; the domain 4-mediated HUVEC proliferation and migration were decreased by antibodies against the VEGFR-2 ectodomain. Proliferation and migration of endothelial cells in response to VEGF play an important role in angiogenesis associated with pathologies such as atherosclerosis, diabetes, and tumor development [31–35]. Many studies have shown that VEGF induces sustained ERK1/2 or PLCγ activation in malignant tumor cells and results in cell proliferation and migration [36, 37]. However, it is unclear whether Ig-like domain 4-mediated HUVEC proliferation and migration is observed in tumor cells.
VEGFR-2 is not exclusively expressed in vascular endothelial cells; there is also a prominent expression in some cancer cells [38–45]. In breast cancer, VEGF and VEGFR-2 are co-expressed on the breast tumor epithelia [46], and increased VEGF or VEGFR2 expression on breast tumor cells correlates with decreased survival [47]. In addition, VEGFR-2 was significantly increased in HER2+ breast cancer [48]. Overexpression of VEGFR-2 may provide a model of ligand-independent receptor activation in breast cancer cells. Finally, similar results were also observed in human VEGFR2-positive breast cancer cells. KFP1 with domain 4 stimulated cell proliferation and migration in the absence of VEGF165. In contrast, cell proliferation and migration was not observed in three VEGFR-2-negative breast cancer cell lines. Additionally, in VEGFR-2-positive breast cancer cell lines, the proliferation and migration stimulated by KFP1 was also blocked by a specific antibody against the VEGFR-2 ectodomain. Although KFP1 mediates proliferation and migration in cultured VEGFR-2-positive breast cancer cells, whether molecules with domain 4 have a negative effect on VEGFR-2-positive tumor remains to be determined. However, the interaction of KFP1 with VEGF165 showed that the binding affinity of KFP1 is unaffected by the addition of the Ig-like domain 4 (Fig. 1c), and even the dynamic dissociation from VEGF165 is slower than that of KFP2. This delayed dissociation may be beneficial for the design of pharmaceutical molecules. In fact, a new pharmaceutical molecule with domain 4, conbercept (KH902), has an inhibitory effect on VEGF-mediated HUVEC proliferation and migration and antiangiogenic effects in age-related macular degeneration [49–51] and proliferative diabetic retinopathy [52, 53].
In conclusion, we showed that the Ig-like domain 4 is indispensable for VEGFR-2 dimerization and activation. In addition to ligand-independent VEGFR-2 activation, receptor–receptor interactions by the Ig-like domain 4 likely occur in the presence of ligand. Our data shed new light on the function of domain 4 in ligand-independent VEGFR-2 activation. Furthermore, we showed that domain 4-induced VEGFR-2 dimerization promotes downstream signaling and function in VEGFR-2-positive cells.
Materials and methods
Cell lines and cell culture
The breast cancer cell lines MCF-7 (THB-22), T-47D (THB-133), ZR-75-30 (CRL-1504), MDA-MB-435 (THB-129), MDA-MB-231 (THB-26), and HCC1937 (CRL-2336) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). These cells were grown in RPMI 1640 (Thermo Fisher Scientific formerly Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Primary human umbilical vein endothelial cells (HUVECs) were ordered from Cascade (Carlsbad, CA, USA) and cultured in endothelial cell medium (ECM, ScienCell, San Diego, CA, USA) with endothelial cell growth supplements (ECGS) and 5% fetal calf serum (FBS), as provided by the manufacturers.
Reagents and antibodies
Streptavidin (SA) and Anti-human IgG Fc Capture (AHC) biosensor tips were purchased from Pall Life Sciences (New York City, NY, USA). ELISA reagents and kits were obtained from R&D SYSTEMS (In Minneapolis, MN, USA). Bis-(sulfosuccinimidyl) suberate (BS3) was purchased from Thermo Fisher Scientific. Anti-VEGFR-2 antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) and R&D SYSTEMS. Anti-ERK or PLCγ antibodies were ordered from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The BioCoat Cell Migration Chamber Plate was purchased from BD Biosciences (Franklin Lakes, NJ, USA). The cell invasion Chamber Plate was obtained from Merck Millipore (Boston, MA, USA).
KDR fragment protein production
The KDR protein fragments used in this study are as follows: KFP1 was created by fusing the second, third, and fourth domains of VEGFR-2/KDR to the constant region (Fc) of human IgG1; KFP2 was engineered by fusing the second and third domains of VEGFR-2/KDR to the constant region (Fc) of human IgG1. The gene constructs, protein expression, and purification are described in the Supplementary materials.
Binding kinetics and affinity assays
Biolayer Interferometry (BLI), a label-free technology, was used for measuring the binding kinetics of protein fragments to VEGFR-2. Kinetics measurements were monitored in real time on a ForteBio Octet QKe instrument (Pall, NY, USA) equipped with Streptavidin (SA) biosensor tips. Assays were performed at 30 °C in solid black 96-well plates. Recombinant human VEGFR-2 Fc chimera protein (R&D System, MN, USA) in PBS buffer and NHS-LCLC-biotin were mixed at a mass concentration of 1:1 for 1 h at room temperature, and then excess NHS-LCLC-biotin was dialyzed using a PD-10 desalination column. Biotin–VEGFR-2 was used to load SA biosensor probes, which were then used to measure the association with KDR fragments (50–5000 nM); complexes were then allowed to dissociate in PBS buffer. Data analysis and curve fitting were carried out using Octet software, version 7.0. Experimental data were fitted with the binding equations describing a 1:1 interaction and plotted to produce a calibration curve to determine the kinetic parameters. Furthermore, binding affinity of KDR fragments with membrane proteins was analyzed by BLI. KDR fragment proteins were used to load AHC biosensor probes, which were then used to measure the association with membrane proteins of HUVECs, which showed high expression levels of VEGFR-2/KDR protein.
Cross-linking and cross-linking immunoblotting
Cross-linking of KDR fragments with itself was conducted by (BS3), a water-soluble, homobifunctional N-hydroxysuccinimide ester, in a conjugation buffer containing 20 mM sodium phosphate, pH 7.5, with 0.15 M NaCl. BS3 was directly added at a final concentration of 2 mM. The reaction was quenched using 1 M Tris–HCl, pH 7.5, at a final concentration of 20 mM and incubated for 15 min at room temperature. The cross-linked reaction mixtures were verified by SDS polyacrylamide gel electrophoresis.
HUVECs were cultured in 150-mm dishes to subconfluency and serum-starved overnight. After treatment with KDR fragments for 30 min at 4 °C and then 2 h at 37 °C, the cells were collected in PBS. BS3 was added to a final concentration of 2.5 mM and incubated on ice for 2 h. The quenching solution was then added and incubated for 15 min on ice. VEGFR-2 dimerization was analyzed by immunoblotting.
Immunoprecipitation and Western blotting
Cells were then lysed for 2 h in ice-cold buffer (20 mM Tris–HCl, pH 7.5, 50 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 100 mM sodium fluoride, 1 mM activated sodium orthovanadate, 10 μg/ml aprotinin, and 1 μM pepstatin A). VEGFR-2 was immunoprecipitated and targeted in Western blots with a VEGFR-2-specific antibody. Proteins were visualized, and relative band intensities were measured on an imaging system (Chemi Doc™ XRS+, Bio-Rad, CA, USA).
Receptor activation assay
HUVECs were stimulated with different concentrations of KDR fragment proteins, alone or preincubated with the anti-VEGFR-2 ectodomain antibody at 37 °C for 2 h, and the samples were used in total and phospho-VEGFR-2 ELISAs as per the manufacturer’s instructions.
Cell proliferation assay
HUVECs were resuspended at 3 × 104 cells/ml in ECM with ECGS and FBS supplementation, and 100 μl of the cell suspension was seeded in each well of a 96-well tissue culture plate and incubated for 24 h. Serial dilutions of KDR fragment proteins, alone or preincubated with anti-VEGFR-2 ectodomain antibody at 37 °C for 2 h, were then added to HUVECs and continuously incubated at 37 °C with 5% CO2 for 4 days. At the end of incubation, 20 μl of CCK-8 (Dojindo, Kyushu, Japan) was added to each well, and the plate was incubated for an additional 4 h under the same conditions. Then the plate was read at 450 nm on a plate reader.
Four thousand breast cancer cells were plated per well of a 96-well tissue culture plate, serum-starved for 16 h, and challenged for 48 h with 1.5 μM of the KDR fragment proteins KFP1 and KFP2, or were preincubated with the anti-VEGFR-2 ectodomain antibody at 37 °C for 2 h. Then the cell viability was assayed using the CCK-8 method.
Cell migration and invasion Assays
Endothelial cell migration was assessed using a modified Boyden chamber (BD FluroBlok™ 96-well BioCoat angiogenesis system: Endothelial cell migration (ECM)), according to the manufacturer’s suggested protocol. Briefly, serum-starved HUVECs were thawed and resuspended at 3.3 × 105 cells/ml, and an aliquot of resuspended cells (~50,000 cells/well) was placed in the upper well of an ECM plate. Serial dilutions of KDR fragment proteins, with or without anti-VEGFR-2 ectodomain antibody following a 2-h incubation at 37 °C, were placed in the lower well. The ECM plate was incubated at 37 °C with 5% CO2 for 24 h to allow cells from the upper well to migrate through the FluroBlok™ membrane towards the lower well. The migrated cells were stained with the fluorescent dye Calcein AM (Anaspec, Freemont, CA, USA) for 1.5 h in a 37 °C/5% CO2 incubator. Fluorescence emission was measured at 517 nm with excitation at 494 nm in a bottom-reading fluorescent plate reader.
Then 8.5 × 104 cells of serum-starved breast cancer cells were placed in the insert of a BioCoat Cell Migration Chamber Plate, and 150 μl of 1.5 μM KDR fragment proteins were added to the lower plate. After incubating for 24 h at 37 °C, the migrated cells were detected by Calcein AM and validated with Smart Flare human VEGF-Cy3 (Life Technologies, CA, USA).
Finally, 1.2 × 105 cells of serum-starved breast cancer cells were loaded into the ECMatrix-coated membrane chamber of a cell invasion Chamber Plate (Merk, Darmstadt, Germany), and 150 μl of 1.5 μM KDR fragment proteins were added to the wells of feeder trays. After incubating for 20 h at 37 °C, the invaded cells in feeder tray were detected by CyQuant GR Dye, incubated with DAPI, and visualized by a fluorescence microscope.
Statistics
Statistical analysis was performed using SPSS version 16.0 software. All data are presented as the mean ± standard deviation. Differences among groups were determined with one-way ANOVA. Comparisons were considered to be statistically significant if p < 0.05.
Abbreviations
- VEGF:
-
Vascular endothelial growth factor
- VEGFR-2:
-
Vascular endothelial growth factor receptor-2
- KDR:
-
Kinase insert domain receptor
- RTK:
-
Receptor tyrosine kinase
- ERK:
-
Extracellular signal-regulated kinase
- PLCγ:
-
Phospholipase Cγ
- PDGFR:
-
Platelet-derived growth factor receptor
- EGFR:
-
Epidermal growth factor receptor
- FGFR:
-
Fibroblast growth factor receptor
- SCFR:
-
Stem cell factor receptor
- ECM:
-
Endothelial cell medium
- ELISA:
-
Enzyme-linked immunosorbent assay
References
Hicklin DJ, Ellis LM (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 23(5):1011–1027
Guo S, Colbert LS, Fuller M, Zhang Y, Gonzalez-Perez RR (2010) Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophy Acta 1806(1):108–121
Bachelder RE, Wendt MA, Mercurio AM (2002) Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res 62(24):7203–7206
Nakopoulou L, Stefanaki K, Panayotopoulou E, Giannopoulou I, Athanassiadou P, Gakiopoulou-Givalou H, Louvrou A (2002) Expression of the vascular endothelial growth factor receptor-2/Flk-1 in breast carcinomas: correlation with proliferation. Hum Pathol 33:863–870
Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, Mercurio AM (2001) Vascular endothelial growth factor is an autocrine survival factor for Neuropilin-expressing breast carcinoma cells. Cancer Res 61(15):5736–5740
Ryden L, Linderholm B, Nielsen NH, Emdin S, Jonsson PE, Landberg G (2003) Tumor specific VEGF-A and VEGFR2/KDR protein are co-expressed in breast cancer. Breast Cancer Res Treat 82(3):147–154
Koukourakis MI, Limberis V, Tentes I, Kontomanolis E, Kortsaris A, Sivridis E, Giatromanolaki A (2011) Serum VEGF levels and tissue activation of VEGFR2/KDR receptors in patients with breast and gynecologic cancer. Cytokine 53(3):370–375
Ryden L, Jirstrom K, Halund M, Stal O, Ferno M (2010) Epidermal growth factor receptor and vascular endothelial growth factor receptor 2 are specific biomarkers in triple-negative breast cancer. Results from a controlled randomized trial with long-term follow-up. Breast Cancer Res Treat 120(2):491–498
Yarden Y, Escobedo JA, Kuang WJ, Yang-Feng TL, Daniel TO, Tremble PM, Chen EY, Ando ME, Harkins RN, Francke U (1986) Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature 323(6085):226–232
Koch S, Claesson-Welsh L (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2(7):a006502
Claesson-Welsh L, Heldin CH (1989) Platelet-derived growth factor. Three isodorms that bind to two distinct cell surface receptors. Acta Oncol 28(3):331–334
Shinkai A, Ito M, Anazawa H, Yamaguchi S, Shitara K, Shibuya M (1998) Mapping of the site involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J Biol Chem 273(47):31283–31288
Fuh G, Li B, Crowley C, Cunningham B, Wells JA (1998) Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J Biol Chem 273(18):11197–11204
Leppanen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, Lankinen H, Goldman A, Ballmer-Hofer K, Alitalo K (2010) Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc Natl Acad Sci USA 107(6):2425–2430
Dosch DD, Ballmer-Hofer K (2010) Transmembrane domain-mediated orientation of receptor monomers in active VEGFR-2 dimers. FASEB J 24(1):32–38
Yang Y, Xie P, Opatowsky Y, Schlessinger J (2010) Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc Natl Acad Sci USA 107(5):1906–1911
Kisko K, Brozzo MS, Missimer J, Schleier T, Menzel A, Leppanen VM, Alitalo K, Walzthoeni T, Aebersold R, Ballmer-Hofer K (2011) Structural analysis of vascular endothelial growth factor receptor-2/ligand complexes by small-angle X-ray solution scattering. FASEB J 25(9):2980–2986
Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J (2007) Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 130(2):323–334
Sarabipour S, Ballmer-Hofer K, Hristova K (2016) VEGFR-2 conformational switch in response to ligand binding. Elife 5:e13876
Herren B, Rooney B, Weyer KA, Iberg N, Schmid G, Pech M (1993) Dimerization of extracellular domains of platelet-derived growth factor receptors. A revised model of receptor-ligand interaction. J Biol Chem 268(20):15088–15095
Naithani S, Chookajorn T, Ripoll DR, Nasrallah JB (2007) Structural modules for receptor dimerization in the S-locus receptor kinase extracellular domain. Proc Natl Acad Sci USA 104(29):12211–12217
Schlessinger J (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110(6):669–672
Heldin CH (1995) Dimerization of cell surface receptors in signal transduction. Cell 80(2):213–223
Omura T, Miyazawa K, Ostman A, Heidin CH (1997) Identification of a 190-kDa vascular endothelial growth factor 165 cell surface binding protein on a human glioma cell line. J Biol Chem 272(37):23317–23322
Omura T, Heldin CH, Ostman A (1997) Immunoglobulin-like domain 4 receptor-receptor interactions contribute to platelet-derived growth factor-induced receptor dimerization. J Biol Chem 272(9):12676–12682
Blechman JM, Lev S, Barg J, Eisenstein M, Vaks B, Vogel Z, Givol D, Yarden Y (1995) The fourth immunoglobulin domain of the stem cell factor receptor couples ligand binding to signal transduction. Cell 80(1):103–113
Tao Q, Backer MV, Backer JM, Terman BI (2001) Kinase insert domain receptor (KDR) extracellular immunoglobulin-like domains 4–7 contain structural features that block receptor dimerization and vascular endothelial growth factor-induced signaling. J Biol Chem 276(24):21916–21923
King C, Stoneman M, Raicu V, Hristova K (2016) Fully quantified spectral imaging reveals in vivo membrane protein interactions. Integr Biol (Camb) 8(2):216–229
Hyde CA, Giese A, Stuttfeld E, Abram Saliba J, Villemagne D, Schleier T, Bina HK, Ballmer-Hofer K (2012) Targeting extracellular domain D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor sites. Mol Cell Biol 32(19):3802–3813
Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer-Hofer K (2007) Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nat Struct Mol Biol 14(3):249–250
Giatromanolaki A, Koukourakis MI, Sivridis E, Chlouverakis G, Vourvouhaki E, Turley H, Harris AL, Gatter KC (2007) Activated VEGFR2/KDR pathway in tumour cells and tumour associated vessels of colorectal cancer. Eur J Clin Invest 37(11):878–886
Straume O, Akslen LA (2003) Increased expression of VEGF-receptors (FLT-1, KDR, NRP-1) and thrombospondin-1 is associated with glomeruloid microvascular proliferation, an aggressive angiogenic phenotype, in malignant melanoma. Angiogenesis 6(4):295–301
Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM (1995) Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res 55(18):3964–3968
Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, Casley DJ, Bach LA, Kelly DJ, Gilbert RE (1999) Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 48(11):2229–2239
Urschel K, Garlichs CD, Daniel WG, Cicha I (2011) VEGFR2 signalling contributes to increased endothelial susceptibility to TNF-α under chronic non-uniform shear stress. Atherosclerosis 219(2):499–509
Svensson S, Jirstrom K, Ryden L, Roos G, Emdin S, Ostrowski MC, Landberg G (2005) ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene 24(27):4370–4379
Wang Z, Gluck S, Zhang L, Moran MF (1998) Requirement for phospholipase C-gamma1 enzymatic activity in growth factor-induced mitogenesis. Mol Cell Biol 18(1):590–597
Higgins KJ, Abdelrahim M, Liu S, Yoon K, Safe S (2006) Regulation of vascular endothelial growth factor receptor-2 expression in pancreatic cancer cells by Sp proteins. Biochem Biophys Res Commun 345(1):292–301
Guo S, Colbert LS, Fuller M, Zhang Y, Gonzalez-Perez RR (2010) Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta 1806(1):108–121
Weigand M, Hantel P, Kreienberg R, Waltenberger J (2005) Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis 8(3):197–204
Gonzalez RR, Cherfils S, Escobar M, Yoo JH, Carino C, Styer AK, Sullivan BT, Sakamoto H, Olawaiye A, Serikawa T, Lynch MP, Rueda BR (2006) Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2). J Biol Chem 281(36):26320–26328
Spannuth WA, Nick AM, Jennings NB, Armaiz-Pena GN, Mangala LS, Danes CG, Lin YG, Merritt WM, Thaker PH, Kamat AA, Han LY, Tonra JR, Coleman RL, Ellis LM, Stood AK (2009) Functional significance of VEGR-2 on ovarian cancer cells. Int J Cancer 124(5):1045–1053
Sato H, Takeda Y (2009) VEGFR2 expression and relationship between tumor neovascularization and histologic characteristics in oral squamous cell carcinoma. J Oral Sci 51(4):551–557
Gockel I, Moehler M, Frerichs K, Drescher D, Trinh TT, Duenschede F, Borschitz T, Schimanski K, Biesterfeld S, Herzer K, Galle PR, Lang H, Junginger T, Schimanski CC (2008) Co-expression of receptor tyrosine kinases in esophageal adenocarcinoma and squamous cell cancer. Oncol Rep 20(4):845–850
Badalian G, Derecskei K, Szendroi A, Szendroi M, Timar J (2007) EGFR and VEGFR2 protein expressions in bone metastases of clear cell renal cancer. Anticancer Res 27(2):889–894
Ryden L, Linderholm B, Nielsen NH, Emdin S, Jonsson PE, Landberg G (2003) Tumor specific VEGF-A and VEGFR2/KDR protein are co-expression in breast cancer. Breast Cancer Res Treat 82(3):147–154
Ghosh S, Sullivan CA, Zerkowski MP, Molinaro AM, Rimm DL, Camp RL, Chung GG (2008) High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol 39(12):1835–1843
Fertig EJ, Lee E, Pandey NB, Popel AS (2015) Analysis of gene expression of secreted factors associated with breast cancer metastases in breast cancer subtypes. Sci Rep 15(5):12133. doi:10.1038/srep12133
Zhang M, Zhang J, Yan M, Li H, Yang C, Yu D (2008) Recombinant anti-vascular endothelial growth factor fusion protein efficiently suppresses choroidal neovascularization in monkeys. Mol Vis 14:37–49
Zhang M, Yu D, Yang C, Xia Q, Li W, Liu B, Li H (2009) The pharmacology study of a new recombinant human VEGF receptor-fc fusion protein on experimental choroidal neovascularization. Pharm Res 26(1):204–210
Nguyen TT, Guymer R (2015) Conbercept (KH-902) for the treatment of neovascular age-related macular degeneration. Expert Rev Clin Pharmacol 8(5):541–548
Huang J, Li X, Li M, Li S, Xiao W, Chen X, Cai M, Wu Q, Luo D, Tang S, Luo Y (2012) Effects of intravitreal injection of KH902, a vascular endothelial growth factor receptor decoy, on the retinas of streptozotocin-induced diabetic rats. Diabetes Obes Metab 14(7):644–653
Su L, Ren X, Wei H, Zhao L, Zhang X, Liu J, Su C, Tan L, Li X (2016) Intravitreal Conbercept (KH902) for surgical treatment of severe proliferative diabetic retinopathy. Rentina 36(5):938–943
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare no actual, potential, or perceived conflict of interest that would prejudice the impartiality of the article.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, S., Gao, X., Fu, W. et al. Immunoglobulin-like domain 4-mediated ligand-independent dimerization triggers VEGFR-2 activation in HUVECs and VEGFR2-positive breast cancer cells. Breast Cancer Res Treat 163, 423–434 (2017). https://doi.org/10.1007/s10549-017-4189-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4189-5