
Abstract
The redox reactivity of iron is a double-edged sword for cell functions, being either essential or harmful depending on metal concentration and location. Deregulation of iron homeostasis is associated with several clinical conditions, including viral infections. Clinical studies as well as in silico, in vitro and in vivo models show direct effects of several viruses on iron levels. There is support for the strategy of iron chelation as an alternative therapy to inhibit infection and/or viral replication, on the rationale that iron is required for the synthesis of some viral proteins and genes. In addition, abnormal iron levels can affect signaling immune response. However, other studies report different effects of viral infections on iron homeostasis, depending on the class and genotype of the virus, therefore making it difficult to predict whether iron chelation would have any benefit. This review brings general aspects of the relationship between iron homeostasis and the nonspecific immune response to viral infections, along with its relevance to the progress or inhibition of the inflammatory process, in order to elucidate situations in which the use of iron chelators could be efficient as antivirals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron is an essential nutrient for most organisms (Posey and Gherardini 2000; Troxell et al. 2012) and is present in all human cells, given its role on oxygen transport, as well as in ATP and DNA production (Harigae 2018). Thanks to its high redox reactivity, it is important for oxygen transport, ribonucleotide reduction, NADH synthesis, etc. (Halliwell and Gutteridge 2007; Pierce et al. 2003; Sjöberg 1997; Tong and Rouault 2007). However, such reactivity also contributes to oxidative stress when at toxic levels in cells. Thus, both iron overload and deficiency are deleterious (Benite et al. 2002; Grotto 2008; Halliwell and Gutteridge 2007). Viruses exert influence on iron homeostasis in host cells by different mechanisms, according to their classification, given that different genetic materials (DNA or RNA, single-stranded or double-stranded) require different host cell machinery for replication. Cell iron levels can affect the replication of common viruses, such as human immunodeficiency virus (HIV), hepatitis C (HCV) and herpes (Chhabra et al. 2020; Mancinelli et al. 2020). The severity of COVID-19 symptoms is related with ferritin levels in the body, and hyperferritinemia is present in the most severe cases (Colafrancesco et al. 2020; Perricone et al. 2020). Dalamaga et al. (2020) included dysregulation of iron homeostasis (resulting in oxidative stress and inflammatory response) as a possible reason for the systemic effects of COVID-19.
Antiviral drugs may minimize damages caused by viral infection, but they are not always satisfactorily effective due to virus polymorphism (González-Candelas and López-Labrador 2010; Jones et al. 2021; Vahey and Fletcher 2019). Combination of antivirals with other safe substances capable of modulating cell response to infection could overcome this issue. Therefore, this review aims to discuss the relation of some viral infections with iron homeostasis, as well as its relevance to the progress or inhibition of the infection and inflammatory response, in order to elucidate situations in which the use of iron chelators as broader spectrum antivirals may be effective.
Iron homeostasis and dyshomeostasis
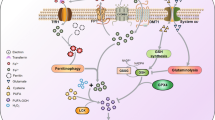
The main biochemical steps involved in iron absorption and mobilization are displayed in Fig. 1 (Andrews 2000; Bruzzese et al. 2023; Coffey and Ganz 2017). Dietary iron has distinct duodenal absorption processes depending upon its speciation. Non-heme iron is normally present as Fe(III), which has to be reduced to Fe(II) by the ferrireductase DCYTB prior to its transport via the divalent metal transport protein (DMT1). The precise transporter of heme iron is currently unknown; heme iron in the cytosol is degraded by heme oxygenase (HO) in order to destroy the iron-porphyrin complex and release Fe(II). This form of “free” iron is either stored in ferritin (FTN, a protein capable of accumulating up to 4500 iron ions) or exported by ferroportin (FPT), the only known iron exporter in vertebrates. Iron is then oxidized to Fe(III) by copper-containing proteins hephaestin (HEPH) or ceruloplasmin (CP). The metal is transported through the circulatory system by transferrin (Tf), a glycoprotein with high affinity for Fe(III). Tf delivers iron to the cell by binding to the transferrin receptor (TfR1 or TfR2), when it is not bound to hemochromatosis protein (HFE). HFE and Tf compete for TfR, whose affinity for Fe-Tf is higher than for Apo-Tf (Salter-Cid et al. 1999; Schmidt et al. 2008; Waheed et al. 2001). The Fe-Tf-TfR complex enters the cell through endosomes, where upon acidification induces reduction of Fe(III) to Fe(II), and ferrous iron is released in the cytosol mediated by DMT1 (Grotto 2008; Halliwell and Gutteridge 2007).

As most iron in the body is involved in oxygen transport in red blood cells, it is not surprising that iron homeostasis is affected by oxygen levels. At low O2 levels, cells produce transcription factors, such as the hypoxia-inducible factor (HIFs), of which three isoforms are known. HIFs regulate genes associated with the expression of iron homeostasis proteins, such as ferroportin, transferrin receptor, heme-oxygenase 1 and erythropoietin (EPO) (Shah and Xie 2014).
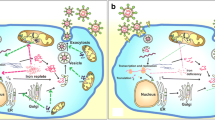
The balance between intra and extracellular iron is orchestrated by hepcidin (HPN), which induces FPT endocytosis and degradation. HPN production in liver cells via bone morphogenetic protein (BMP) is triggered by high iron levels and is suppressed by erythroferrone (ERFE), whose synthesis is stimulated by erythropoietin (EPO) secretion from kidneys when iron is demanded for hemoglobin synthesis (Antunes and Canziani 2016; Barupala et al. 2016; Ganz 2019; Singh 2018). Iron is recycled when senescent erythrocytes are phagocytosed by macrophages, then it is exported by FPT or stored by FTN (Grotto 2008). When increase in HPN levels results in iron overload, TfR translation is inhibited and FTN translation is stimulated by iron regulatory elements and proteins (IRE and IRP, respectively) (Fig. 2). IREs are non-coding stretches of messenger RNA (mRNA), found at the 5' end of FTN-mRNA or 3' end of TfR-mRNA. In the first case, the IRP-IRE binding inhibits mRNA translation and, consequently, ferritin expression. On the other hand, the interaction between an IRP and the IRE of the TfR-mRNA stabilizes the molecule, enabling the production of TfR (Halliwell and Gutteridge 2007; Kiihn and Hentze 1992; Schalinske et al. 1997). Iron regulatory proteins (IRP1 and IRP2) have a higher affinity for IRE when iron availability is low. Otherwise, IRP2 undergoes degradation and IRP1 converts to aconitase. This enzyme contains an iron-sulfur cluster cofactor that catalyzes the reversible isomerization of citrate, important both to the Krebs cycle and fatty acid metabolism (Tong and Rouault 2007).

Translation of FTN and TfR modulated by IRP (adapted Kiihn and Hentze 1992)
Iron dyshomeostasis may be due to (i) lack, or (ii) overload of the metal. On the one hand, iron deficiency anemia (IDA) is the world’s most common disease, affecting an estimated one billion people worldwide, especially women and children in developing countries. IDA is mainly associated to low income and lack of access to high quality nutrition (World Health Organization (WHO) 2023). On the other hand, iron overload diseases (IOD) occur when there is either an inherited disorder of iron absorption (such as in hereditary hemochromatosis) or after a treatment of a chronic condition (such as in multiple blood transfusions in thalassemia) (Bruzzese et al. 2023). IOD are specially challenging since there is no dedicated mechanism of excretion of excess iron, therefore the buildup of a redox-active, essential metal outside the biochemical compartments designed to store it may give rise to a series of dysfunctions. The rigorous regulation of iron homeostasis can therefore modulate defense mechanisms against pathogens by: (i) controlling production of reactive species via Fenton and Haber–Weiss reactions, and (ii) avoiding iron sequestration by invading microorganisms. In this perspective, the regulation of iron homeostasis plays a fundamental role in the immune system.
Iron and immune system
Iron deficiency can impair the immune system, turning macrophages inefficient for initial immune response due to decreased production of reactive oxygen and nitrogen species (respectively, ROS and RNS) (Dickson and Zhou 2020). Although the precise mechanism is debated, there is consensus that iron affects immune response, since ROS and RNS act simultaneously in the direct attack on antigens and in the signaling mechanisms for cytokines production and release (Andrés et al. 2022; Yang et al. 2013). Cytokines are produced in endosomes of phagocytic cells, such as neutrophils and macrophages. Both of them secrete lactoferrin (LCF), a natural iron chelator, whose high affinity for Fe(III) inhibits uncontrolled production of reactive species and prevents the use of iron by invading microorganisms (Cruvinel et al. 2010; Ni et al. 2022; Ward et al. 2011).
Macrophages participate in the immune system as Antigen-Presenting Cells (APCs). They recognize Pathogen Associated Molecular Patterns (PAMPs) through Toll-Like Receptors (TLRs), digest antigens and present the peptide residues to immature T lymphocytes through a protein complex known as the Major Histocompatibility Complex (MHC). The MHC induces production of mature T lymphocytes: CD4 (helper T cells) or CD8 (cytotoxic T cells), according to the type of protein group on its surface. CD8 induces infected cells to death, while CD4 helps B lymphocytes to produce specific antibodies. When MHC is activated by an infection, the synthesis of HFE can also be affected, hindering the absorption of iron by TfR (Janeway et al. 2001). Proper macrophage activation also depends on the cluster of differentiation 71 (CD71), which is the transferrin receptor 1 (TfR1). CD71 is responsible for activating T lymphocytes via iron endocytosis through an interleukin 2 (IL-2)-dependent pathway (Candelaria et al. 2021).
To act in immune response, macrophages need to be activated. This can be triggered by inflammatory processes in two different ways: classically (M1) or alternatively (M2). Each one produces a different phenotype (Table 1). Macrophage polarization depends on several factors, such as PAMPs and type of cytokines released, among other effects caused by signaling species. For example, both iron overload and deficiency induce M1 polarization, while adequate concentrations of this nutrient induce M2 polarization (Agoro et al. 2018; Ni et al. 2022).
Macrophage interaction with PAMPs activates two distinct pathways: (i) the HIF-α, which induces hypoferremia (Ward et al. 2011); and (ii) the Nuclear Factor kappa B (NF-κB), a protein complex that stimulates gene expression of inflammatory cytokines (Kawasaki and Kawai 2014). Pro-inflammatory interleukin 12 (IL-12) stimulates natural killer (NK) cells as well as the production of interferons gamma (IFN-γ), increasing the microbicidal potential of macrophages (Machado et al. 2004). IL-6 induces expression of Acute Phase Proteins (APP) type II, such as ferritin and hepcidin (Georgopoulou et al. 2014; Liu and Ahearn 2005; Liu et al. 2020). In case of acute inflammation, FTN can be produced independently of iron levels, acting as an immunosuppressant (Chen et al. 2005; Colafrancesco et al. 2020; Ruddell et al. 2009) and stimulating more classical activation of macrophages (Ni et al. 2022). Increased hepcidin levels promote iron intracellular accumulation, resulting in ferritin excess (hyperferritinemia) and iron deficiency anemia (IDA) (Castro et al. 2019).
Abnormally low iron levels (hypoferremia) is common in populations in which iron deficiency is prevalent and the burden of infections is high. It can impair humoral response to vaccines and, as a result, boosts death rate from preventable diseases (Peace and O’Neill 2021; Preston et al. 2021; Stoffel et al. 2020). Finally, enzymes containing iron–sulfur (Fe–S) clusters can modulate the immune system through viral particle recognition, which activates the expression of pro-inflammatory cytokines and the IFN-stimulating gene. Generally, in Fe–S clusters, iron is hexacoordinated by cysteines and a labile water molecule, which exposes the metal to solvents and allow it to react with ROS and RNS, destroying the protein. Inactivation of this type of enzyme can inhibit viral replication, since viruses use the host's enzymatic machinery, including enzymes containing Fe–S clusters, to replicate (Castro et al. 2019; Ebrahimi et al. 2022).
Iron and viral infection
General overview
Gene replication and protein synthesis are crucial processes for all kinds of living organisms and viruses, and iron plays an essential role in both. Therefore, competition for iron between parasite and host is a determining factor in parasitic diseases. Limitation of iron availability is one of the main defense strategies of the host, but some viruses can actively modulate iron levels of the infected organism, through different mechanisms, according to virus composition and host.
Given the complexity and sensibility of iron homeostasis, it is not surprising that there are several pathways for its regulation during inflammation. One of them is lactoferrin excretion by macrophages and neutrophils (Telek et al. 2022). Literature describes LCF antiviral effect against Herpes Simplex Virus (HSV), Human Papillomavirus (HPV), Rotavirus, Cytomegalovirus (CMV), Hepatitis B and C (HBV and HCV), Human Immunodeficiency Virus (HIV) and Severe Acute Respiratory Syndrome Coronavirus 1 and 2 (SARS-CoV-1 and SARS-CoV-2) (Campione et al. 2021; Mancinelli et al. 2020). Although in vitro studies suggest that LCF can prevent SARS-CoV-2 infection, it cannot treat it (Campione et al. 2021), and according to an in silico study, instead of iron chelation, the interaction between LCF and the coronavirus Spike (S) protein is the major mechanism of inhibition (Kell et al. 2020). If iron chelation is relevant to its antiviral activity, it is interesting to note that LCF affinity constant for iron is ~ 1020, and the most common bacterial siderophores present affinity constants of ~ 1030 (Baker and Baker 2004). Hence, using LCF as an antiviral could provide favorable conditions for opportunistic organisms such as bacteria. Kung et al. (2022) demonstrated that Long Chain Acyl-CoA Synthetase 4 (ACSL4) is involved in the production of viral replication organelles in some enteroviruses infections. This enzyme is involved in lipid peroxidation associated with ferroptosis. Enteroviruses, such as meningitis and poliomyelitis virus, are non-enveloped positive single-stranded (ss)RNA (Li et al. 2020). The negative-sense ssRNA avian influenza virus (H5N1) can change membrane potential of mitochondria and disrupt lysosome permeability, liberating iron and proteases capable of degrading ferritin into the cytosol (Wang et al. 2022). Macrophages from Herpes-infected mice displayed increased pro-inflammatory cytokines production and dysregulated iron recycling (Biron et al. 2014). It is worth mentioning that herpes is a double-stranded (ds)DNA (Wei et al. 2012). Therefore, it requires iron-dependent ribonucleotide reductase (RNR), the only known enzyme that catalyze reduction of ribonucleotide (NTP) to deoxyribonucleotide (dNTP), essential for DNA synthesis (Sjöberg 1997; Torrents 2014). Other DNA viruses that can subvert iron homeostasis are Canine and Feline Parvovirus (respectively, CPV and FPV). Both can use CD71 transferrin receptor to get access to the cell (Drakesmith and Prentice 2008). Among the viruses capable of actively affect iron homeostasis, three that stand out will be discussed in more detail below.
Human immunodeficiency virus (HIV)
Hijacking host’s RNR is far from being a DNA virus exclusivity, since retrovirus reverse transcription also requires dNTPs. This is the case of HIV, known for infecting T cells and macrophages, in addition to promoting CD4-lymphocyte depletion, compromising the immune system (Merino et al. 2017; Naif 2013). The mechanisms by which HIV affects iron homeostasis are not fully understood, but infection symptoms include an increased expression of hepcidin proportional to the decrease of CD4 count in seropositive patients (Armitage et al. 2014; Drakesmith and Prentice 2008; Garrido-Rodríguez et al. 2022; Wisaksana et al. 2013). Armitage et al. (2014) found significantly higher levels of hepcidin in both acute and chronic phases of HIV infection, either in untreated and in antiretroviral treatment (ART) patients. In their study, HPN rise was not associated with its inducer IL-6, suggesting HIV can affect hepcidin expression through other pathways. However, in contradiction to this, Szymczak et al. (2022) found no significant differences in hepcidin expression between ART and control groups.
Despite the lack of consensus about HIV infection and its effects on iron homeostasis, researchers agree that blocking iron exit from infected cells contributes to development of Iron Deficiency Anemia (IDA) and tuberculosis (TB) in untreated patients. Clinical trials in patients with tuberculosis associated with HIV infection showed that intracellular iron accumulation caused anemia due to chronic disease (ACD) concomitantly with IDA, which was treated with iron supplementation, when HPN patient levels were lower than 20 nmol mL−1 (Kerkhoff et al. 2016).
In line with this observation, Esan et al. (2013) observed an increase in CD4 cells and lower propensity to develop acquired immune deficiency syndrome (AIDS) in anemic HIV-infected children receiving iron supplementation. However, the incidence of malaria escalated. Since anemia in HIV carriers is a consequence of a blockade in iron exodus from cells rather than an actual lack of iron in the body, iron supplementation may favor the proliferation of opportunistic pathogens, such as malaria Plasmodium. Moreover, an ex vivo inhibition of HIV in peripheral blood mononuclear cells (PBMCs) obtained from patients with sickle cell disease (SCD) was linked to the increased expression of ferroportin and reduced intracellular iron levels after treatment with hemin (Kumari et al. 2016). One must consider that iron deficiency is implicit in SCD, unlike in healthy PBMCs.
Hepatitis C Virus (HCV)
Even more intriguing is the iron homeostasis dysregulation in Hepatitis C Virus (HCV) infection. There are, at least, six different genotypes and multiple sub-genotypes for HCV (Mancinelli et al. 2020). Each one is a distinct enveloped positive RNA that codifies a single polyprotein, which is processed by proteases in structural and nonstructural viral proteins, among which are those that interact with regulators of iron homeostasis (Gawlik and Gallay 2014). These polyproteins are (i) the structural Core protein, crucial to viral capsid construction, which can bind to RNA and, thanks to its hydrophobic C-terminal domain, interacts with lipids; (ii) the NS5A nonstructural protein, essential for viral replication and assembly, which is a metalloprotein containing zinc as cofactor (Dimitriadis et al. 2021); and (iii) the NS5B nonstructural protein, a RNA-dependent RNA-polymerase (RdRp), which is inhibited by iron ions (Fillebeen et al. 2007).
Increase of hepatic iron levels is the most relevant effect of HCV on iron homeostasis, but there is no consensus to whether iron overload induces or inhibits progress of infection, since contradictory findings indicate either promotion or suppression of HCV replication in iron overload conditions (Zou and Sun 2017). Studies in vitro and in vivo suggest that HCV induces up-regulation of hepcidin through IL-6 stimulation, in response to the Core protein expression (Foka et al. 2014). These studies were performed using only the Core protein gene of HCV-1a or HCV-1b genotype transfected into HepG2 and Huh7 cells or into mice, instead of complete genome. However, Miyachi et al. (2011) had similar results using the full-genomic HCV.
On the other hand, there is evidence that HCV induces hepcidin downregulation. Clinical studies with infected HCV patients (Drakesmith and Prentice 2008; Fujita et al. 2007) and in vitro analysis of plasma from acute phase HCV-infected donors (Armitage et al. 2014; H. Liu et al. 2012) report decrease in hepcidin levels. Differences between virus genotypes and the nature of the host are the most obvious causes for such discrepant results: patients infected with HCV-1b genotype presented higher hepatic iron concentration compared to those infected with HCV-2a and HCV-2b (Georgopoulou et al. 2014). Nonetheless, genotypic heterogeneity is not the only reason for these distinct patterns. There is evidence that HCV Core protein and NS5A control hepcidin encoding gene (Hamp) expression in distinct stages of virus life cycle. Recently, Dimitriadis et al. (2021) showed that NS5A protein inhibits hepcidin production, probably hijacking zinc ions from the host. They also confirmed Foka et al. (2014) results indicating that the Core protein enhanced hepcidin production through activation of the proteins STAT3 and SMAD4, which are members of protein families involved in signaling pathways, respectively, JAK/STAT and Transforming Growth Factor-β (TGF-β)/SMAD.
The JAK/STAT signaling pathway is central for the expression of a number of biomolecules. Janus Kinase (JAK) interact with cytokine receptors in a noncovalently way, mediating phosphorylation of tyrosine amino acids to recruit one or more proteins from the Signal Transducer and Activator of Transcription (STAT) family, which are then transported to the cell nucleus and regulate specific genes (Hu et al. 2021). It is known that IL-6 promotes hepcidin transcription through JAK/STAT3 activation in response to tumors (Johnston and Grandis 2011) and infections (Xu et al. 2021). What Foka’s work (2014) suggests is that Core protein stimulates IL-6 production as an Acute Phase Protein regulator, which activates the JAK/STAT3 pathway and induces Hamp gene expression. Hepcidin is an APP type II.
Hepcidin transcription can be regulated by TGF-β, a superfamily of proteins in control of several biological functions and which signals are transduced to the cell nucleus by SMAD protein family (Itoh et al. 2000; Lifshitz and Frenkel 2013). Each SMAD participates in distinct signaling pathways, but SMAD4 is a central mediator. When iron levels are high, endothelial cells from the liver activate transcription factors, which in turn activate production and secretion of Bone Morphogenetic Protein 6 (BMP6), one member of TGF-β superfamily. BMP6 binds to a complex of BMP receptors and hemojuvelin (HJV) coreceptors, stimulating phosphorylation of SMAD1, SMAD5 and SMAD8, which then form an heteropolymeric complex with SMAD4, promoting Hamp gene expression (Xu et al. 2021). Hemojuvelin protein can compete with BMP6 for the HJV coreceptors, inhibiting hepcidin transcription, unless the zinc-dependent Matriptase-2 (MT2) catalyzes HJV degradation (Agarwal and Yee 2019; Knez et al. 2017; Kondaiah et al. 2019). That being said, it is possible that the Core protein modulates hepcidin production affecting TGF-β/BMP/SMAD mechanism, as suggested by Dimitriadis et al. (2021). Nevertheless, their hypothesis of zinc hijacking by NS5A protein as the cause for HPN downregulation still needs further investigation, although the authors attributed MT2 malfunction to oxidative stress.
HCV Core protein can interact with lipids and dysregulates host lipid metabolism, taking the Very Low-Density Proteins (VLDP) engineering system to construct its own lipoproteins (Lavie and Dubuisson 2017). Apparently, HCV inhibits the secretion of VLDL, resulting in fatty accumulation in the liver, the organ with highest VLDL levels (Sidorkiewicz and Brown 2021). Liver is also the major iron storage organ (Kohgo et al. 2008). About 30% of people with non-alcoholic fatty liver disease (NAFLD) have disturbed iron homeostasis (Datz et al. 2017; Dongiovanni et al. 2011; Fernandez et al. 2022) and HCV is among the major risk factors to the development of liver diseases such as chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) (Kwon et al. 2014; Llovet et al. 2021; Nash et al. 2010). Nishina et al. (2010) observed steatosis in the liver of HCV-infected mice under iron overload conditions. López-Prieto et al. (2013) noticed FTN and IL-6 levels proportional to the amount of liver fat in HCV-infected patients. Despite the strong correlations mentioned, it is unclear how iron homeostasis dysregulation influences HCV-induced liver damage at the molecular level.
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
The cohort of viruses exerting strong influence on iron homeostasis was joined by a new member recently. Work reporting a direct correlation between ferritin levels and severity of COVID-19 symptoms (Colafrancesco et al. 2020; Perricone et al. 2020) have received attention and motivated a number of studies searching for evidences of SARS-Cov-2 influence on iron homeostasis. Whether ferritin upregulation is controlled by the virus or driven by the common molecular mechanisms inducing acute phase proteins is still controversial. However, hyperferritinemia is a hallmark symptom of COVID-19 (Szabo et al. 2022), and the abnormally high FTN levels are not explained solely by the standard host response to infection.
In a cross-sectional study with 164 patients with pneumonia complications, considerably higher levels of ferritin and hepcidin were found in COVID-19 patients compared to influenza and bacterial infection (Hegelund et al. 2022). Blood sample analysis of 34 COVID-19 patients confirmed this proportional relation and found that hephaestin (HEPH) and HIF-1α levels are inversely proportional to ferritin amounts and symptom severity (Aslan et al. 2023). HEPH is a ferroxidase enzyme required for iron transport from intestinal enterocytes into the circulation, and it couples to the iron exporter FPT to oxidize Fe(II) to Fe(III) (Doguer et al. 2017; Petrak and Vyoral 2005; Vulpe et al. 1999). Also, HIF is a heterodimeric transcription factor whose 1α subunit is an oxygen sensor. HIF-1α suffers degradation in presence of normal O2 levels but, under hypoxia conditions, is free to activate EPO secretion from kidney and stimulate erythroferrone production (Hirota 2019; Shah and Xie 2014). As mentioned above, erythroferrone is the hepcidin suppressor. If HPN is suppressed, FPT is not degraded and iron can be exported from the cell and transported into circulation to be used in erythropoiesis. Hypoxia is one of the most common symptoms of COVID-19 (Cavezzi et al. 2020), so an increase in HIF-1α and HEPH to attenuate oxygen deficiency should be expected.
In any case, it is possible that the virus seriously disrupts iron homeostasis in critical patients who were unable to induce iron circulation due to hyperferritinemia. Recent meta-analysis confirmed higher levels of ferritin and hepcidin associated with severity and mortality of COVID-19 (Peng et al. 2022). Additionally, Zhou et al (2022) found that serum iron, Total Iron Binding Capacity (TIBC) and hemoglobin levels are inversely proportional to disease severity. Dysfunction of hemoglobin may be another contributing factor for hypoxia in COVID-19 (Cavezzi et al. 2020), and hemoglobin denaturation can be caused by the interaction of its beta-chain with virus surface proteins (Lippi and Mattiuzzi 2020), since the virus needs porphyrin to replicate (Chaudhry et al. 2021). Zeinivand et al. (2023) found decreased levels of hemoglobin and neutrophiles in COVID-19 patients. They also mention abnormal levels of proteins associated with iron metabolism in long COVID patients, namely, ceruloplasmin and transferrin.
A major symptom of COVID-19 is the cytokine storm, including IL-6, the hepcidin inducer (Coomes and Haghbayan 2020). Both IL-6 and IL-1 are linked to severe COVID-19 (Fara et al. 2020) and are involved in ferritin synthesis (Mahroum et al. 2022; Taneri et al. 2020). Given IL-6 role on hepcidin transcription via JAK/STAT3 signaling, further evaluation of SARS-CoV-2 effects on this pathway would be interesting. It could help to shed light on the mechanism resulting in iron dysregulation and the consequent hyperferritinemia. This condition can induce ROS production and lipid peroxidation, culminating in ferroptosis. Pasini et al. (2021) compared ferroptosis biomarkers with clinical conditions of patients suffering severe COVID-19; decreased glutathione peroxidase 4 (GPX4) expression with concomitant increases in fatty acid peroxide levels and oxidative stress are present in both cases. Possibly, GPX4 inhibition in SARS-CoV-2 infection is due to selenium deficiency, since Vero-E6 cells infected with this virus had significant reduction in mRNA expression of endoplasmic reticulum-resident selenoproteins (Li et al. 2023). GPX4 is a selenium protein that protects cells from ferroptosis (Yao et al. 2021). Despite that, so far there is no molecular level research showing a cause-effect relationship between these phenomena.
As well as in the HCV case, different stages of SARS-CoV-2 infection and COVID-19 development can affect iron homeostasis in distinct ways. Thermal analysis of proteins extracted from SARS-CoV-2 infected cells showed low levels of Tf and Tf saturation concurrently with increased ferritin 24 h after infection, yet infected cells and controls showed no differences in Tf concentration after 48 h (Telek et al. 2022). Study with mild to critical condition patients revealed hyperferritinemia in 35% of them until after 60 days post-COVID-19 infection, as well as IDA associated with IA until 360 days after infection (Sonnweber et al. 2022). There are works showing association between IDA and thrombocytopenia (Franchini et al. 2008; Jimenez et al. 2021), a common complication of COVID-19 (Alharbi et al. 2022; Bhattacharjee and Banerjee 2020). Another in vitro research observed that high levels of IL-6 and HPN promotes expression of angiotensin-converting enzyme 2 (ACE-2) lung tissue cells (Toe et al. 2022). ACE-2 binds to the virus spike glycoprotein, allowing coronavirus to enter the cell (Gupta et al. 2022). It is probable that SARS-CoV-2 S protein has a cysteine-rich sequence similar to cysteine-rich domain of human hepcidin, which could contribute to block cell iron export by ferroportin (Di Paola et al. 2020; Ehsani 2020). However, except for Di Paola et al. (2020) and Ehsani (2020) in silico analysis, there is no direct evidence of this. According to Ebrahimi et al. (2022) SARS-CoV-2 RdRp is an iron-sulfur cluster enzyme. Additionally, Wang et al. (2021) demonstrated that Sars-CoV-2 inhibits thioredoxin reductase, which is responsible for RNR recycling. It should be noted that coronaviruses are ssRNA positive, which are made from NTP. Therefore, it is advantageous for the virus to inhibit the only enzyme that converts NTP to dNTP, leaving more NTP available for its replication.
Iron chelators in viral infections
It is possible to either defend or oppose repositioning of known iron chelators as complement antiviral adjuvants. Therefore, it is important to understand how viral infections affect iron homeostasis in order to determine whether iron chelation can be effective in inhibiting viral replication without causing more damage to the host.
Inflammation related to viral infections can induce an increase in free intracellular iron, opening way for opportunistic microorganisms’ infection. Attempts to treat this condition with deferoxamine (DFO) increased the rate of mortality (Makarov et al. 2020), probably because DFO is a bacterial siderophore itself (Benite et al. 2002). On the other hand, according to Drakesmith and Prentice (2008) (Drakesmith and Prentice 2008), iron chelation with DFO prevents the generation of ROS, with consequent inhibition of NF-κB activation, which mediates transcription of HIV genes. van Asbeck et al. (2001) question this hypothesis since in their studies, DFO inhibited RNR in mononuclear cells and no evidence of NF-κB inhibition was found. Attempts to use deferiprone (DFP) to treat hepatic fibrosis caused by HCV infection were not successful (Chen et al. 2006), but deferasirox (DFX) inhibited the replication of West Nile virus in both cells and mosquitoes, its natural vector (Duchemin and Paradkar 2017). However, DFP restored T lymphocytes resistance against SARS-CoV-2 infection by increasing expression of the beta subunit of the interferon alpha beta receptor in the surface of this cells (Li et al. 2023).
Drug repositioning with favipiravir (FAV), an RNA antiviral used to treat influenza, was proposed to treat COVID-19 because it may interact with SARS-CoV-2 RdRp (Chedid et al. 2021; Naydenova et al. 2021). Pari and Yousef (2021) used Density Functional Theory (DFT) to show FAV also acts as an iron chelator. Unfortunately, there are no further investigations to confirm it, such as a direct spectrophotometric experiment. Maybe access to the drug is not so simple, since it is teratogenic and a risk factor for individuals with renal or hepatic impairment (Almutairi et al. 2023), which are common conditions in systemic disease patients, such as COVID-19.
Off-label use of tocilizumab (TCZ, an antibody against IL-6 receptor) associated with DFX as adjuvant had good outcomes in COVID-19 patients severely ill (Birlutiu et al. 2021), but it is not clear how much of this effect results from DFX, since TCZ can also affect iron homeostasis through regulation of HPN through IL-6 receptor inhibition. In in vitro assays, Fe(III)-thiosemicarbazones complexes (100 μM) inhibited 30% of SARS-CoV-2 activity, and computational modeling suggests these compounds interact with one of the virus proteases (Atasever Arslan et al. 2021). As mentioned before, micromolar doses of LCF have antiviral activity against some viruses and can prevent SARS-CoV-2 infection in Vero-E6 cells. DFO conjugated with 6-aminoquinoline (DFOQUN) in order to increase the chelator lipophilicity (Carvalho et al. 2021) presented similar results in preventing SARS-CoV-2 infection in Vero-E6 cells (Pereira et al. 2022): 10 μM of DFOQUN was enough to have a 50% antiviral activity with excellent safety to the host cell (survival rate = 128.9%). Moreover, DFOQUN affinity constant for iron (~ 1030) is superior to LCF’s (~ 1020), and the rate of viral particles per cell in DFOQUN in vitro assays (MOI = 0.05) was higher than the used by Campione et al (2021) in LCF experiments (MOI = 0.01). It is worth mentioning that docking molecular studies have shown that quinoline iron complexes exhibit higher average bond enthalpy with SARS-CoV-2 RdRp compared to the drugs extensively tested during the COVID-19 pandemic, namely chloroquine, remdesevir, galidesivir, tenofovir, sofosbuvir, ribavirin, among others (Aggarwal and Maji 2022).
Despite being ineffective against coronaviruses in vivo, chloroquine (CQ) and their analogues affect proliferation of SARS-CoV-2 in vitro by inhibiting glycosylation of the ACE2 receptor, rendering the endocytic pathway impossible (Ayipo et al. 2021). Inhibition of the endocytic pathway occurs due to the alkalinization of endosomes. Roldan et al. (2020) argue that the pH increase caused by chloroquine and hydroxychloroquine (HCQ) may disturb iron homeostasis, inhibiting endocytosis of the Tf-TfR complex and, consequently, iron absorption. The 6-aminoquinoline moiety of DFOQUN is a CQ analog. Thus, it is possible that there is a synergistic effect of iron chelation and inhibition of the endocytic pathway. Further studies are necessary to confirm this hypothesis.
Concluding remarks
Iron chelators have been tested as broad-spectrum antivirals because they inhibit iron sequestration by antigens. Despite the lack of consensus on the effects of viral infections on iron homeostasis, the specificities of HIV, HCV, and SARS-CoV-2 discussed in this review are very intriguing. Mechanisms involved in the processes of entry, replication, and/or assembly of these particles exert a strong influence on pathways that directly or indirectly control iron modulation. Viral polymorphism is an obstacle to the effective application of narrow-spectrum antivirals. In this sense, the concomitant use of these antivirals with iron chelators might be an alternative treatment for diseases caused by such organisms. The chelating agents may also prevent the disease from reaching critical conditions and, consequently, hospitalization.
Data availability
Data can be available upon request from the authors.
References
Agarwal AK, Yee J (2019) Hepcidin. Adv Chronic Kidney Dis 26(4):298–305. https://doi.org/10.1053/j.ackd.2019.04.005
Aggarwal N, Maji S (2022) Potential applicability of Schiff bases and their metal complexes during COVID-19 pandemic: a review. Rev Inorg Chem 42(4):363–383. https://doi.org/10.1515/revic-2021-0027
Agoro R, Taleb M, Quesniaux VFJ, Mura C (2018) Cell iron status influences macrophage polarization. PLoS ONE. https://doi.org/10.1371/journal.pone.0196921
Alharbi MG, Alanazi N, Yousef A, Alanazi N, Alotaibi B, Aljurf M, El Fakih R (2022) COVID-19 associated with immune thrombocytopenia: a systematic review and meta-analysis. Expert Rev Hematol 15(2):157–166. https://doi.org/10.1080/17474086.2022.2029699
Almutairi AO, El-Readi MZ, Althubiti M, Alhindi YZ, Ayoub N, Alzahrani AR, Al-Ghamdi SS, Eid SY (2023) Liver injury in favipiravir-treated COVID-19 patients: retrospective single-center cohort study. Trop Med Infect Dis 8(2):129. https://doi.org/10.3390/tropicalmed8020129
Andrés CMC, Pérez de la Lastra JM, Juan CA, Plou FJ, Pérez-Lebeña E (2022) The Role of Reactive Species on Innate Immunity. Vaccines 10(10):1735. https://doi.org/10.3390/vaccines10101735
Andrews NC (2000) Iron homeostasis: insights from genetics and animal models. Nat Rev Genet 1(3):208–217. https://doi.org/10.1038/35042073
Antunes SA, Canziani MEF (2016) Hepcidin: an important iron metabolism regulator in chronic kidney disease. J Bras Nefrol 38(3):351–355. https://doi.org/10.5935/0101-2800.20160053
Armitage AE, Stacey AR, Giannoulatou E, Marshall E, Sturges P, Chatha K, Smith NMG, Huang XJ, Xu XN, Pasricha SR, Lie N, Wu H, Webster C, Prentice AM, Pellegrino P, Williams I, Norris PJ, Drakesmith H, Borrow P (2014) Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proc Natl Acad Sci USA 111(33):12187–12192. https://doi.org/10.1073/pnas.1402351111
Aslan ES, Aydın H, Tekin YK, Keleş S, White KN, Hekim N (2023) Association between iron metabolism and SARS-COV-2 infection, determined by ferritin, hephaestin and hypoxia-induced factor-1 alpha levels in COVID-19 patients. Mol Biol Rep. https://doi.org/10.1007/s11033-022-08221-3
Atasever Arslan B, Kaya B, Şahin O, Baday S, Saylan CC, Ülküseven B (2021) The iron(III) and nickel(II) complexes with tetradentate thiosemicarbazones. Synthesis, experimental, theoretical characterization, and antiviral effect against SARS-CoV-2. J Mol Struct 1246:131166. https://doi.org/10.1016/j.molstruc.2021.131166
Ayipo YO, Yahaya SN, Alananzeh WA, Babamale HF, Mordi MN (2021) Pathomechanisms, therapeutic targets and potent inhibitors of some beta-coronaviruses from bench-to-bedside. Infect Genet Evol 93:104944. https://doi.org/10.1016/j.meegid.2021.104944
Baker HM, Baker EN (2004) Lactoferrin and Iron: structural and dynamic aspects of binding and release. Biometals 17(3):209–216
Barupala DP, Dzul SP, Riggs-Gelasco PJ, Stemmler TL (2016) Synthesis, delivery and regulation of eukaryotic heme and Fe–S cluster cofactors. Archiv Biochem Biophys 592:60–75. https://doi.org/10.1016/j.abb.2016.01.010
Benite AMC, Machado SP (2002) Sideróforos: uma resposta dos microorganismos. Quim Nova 25(6b):1155–1164. https://doi.org/10.1590/s0100-40422002000700016
Bhattacharjee S, Banerjee M (2020) Immune thrombocytopenia secondary to COVID-19: a systematic review. SN Compr Clin Med 2(11):2048–2058. https://doi.org/10.1007/s42399-020-00521-8
Birlutiu V, Birlutiu RM, Chicea L (2021) Off-label tocilizumab and adjuvant iron chelator effectiveness in a group of severe COVID-19 pneumonia patients. Medicine 100(18):e25832. https://doi.org/10.1097/MD.0000000000025832
Biron CA, Dalod M, Salazar-Mather TP (2014) Innate Immunity and Viral Infections. Immunol Infect Dis. https://doi.org/10.1128/9781555817978.ch11
Bruzzese A, Martino EA, Mendicino F, Lucia E, Olivito V, Bova C, Filippelli G, Capodanno I, Neri A, Morabito F, Gentile M, Vigna E (2023) Iron chelation therapy. Eur J Haematol 110(5):490–497. https://doi.org/10.1111/ejh.13935
Campione E, Lanna C, Cosio T et al (2021) Lactoferrin against SARS-CoV-2. In vitro and in silico evidences. Front Pharmacol 12:4–9. https://doi.org/10.3389/fphar.2021.666600
Candelaria PV, Leoh LS, Penichet ML, Daniels-Wells TR (2021) Antibodies targeting the transferrin receptor 1 (TfR1) as direct anti-cancer agents. Front Immunol 12:607692. https://doi.org/10.3389/fimmu.2021.607692
Carvalho RRV, Peres TV, Liria CW, Machini MT, Aschner M, Espósito BP (2021) Conjugates of desferrioxamine and aromatic amines improve markers of iron-dependent neurotoxicity. Biometals 34(2):259–275. https://doi.org/10.1007/s10534-020-00277-7
Castro L, Tórtora V, Mansilla S, Radi R (2019) Aconitases: non-redox iron-sulfur proteins sensitive to reactive species. Acc Chem Res 52(9):2609–2619. https://doi.org/10.1021/acs.accounts.9b00150
Cavezzi A, Troiani E, Corrao S (2020) COVID-19: hemoglobin, iron, and hypoxia beyond inflammation: a Narrative Review. Clin Practice 10(2):1271. https://doi.org/10.4081/cp.2020.1271
Chaudhry ZR, Rasheed S, Shakir S, Rashid E, Ansari M, Rasheed F (2021) Corona virus lowers hemoglobin more in severe infection than mild COVID-19 infection. Prof Med J 28(8):1211–1214. https://doi.org/10.29309/TPMJ/2021.28.08.6523
Chedid NGB, Ferraz LG, Carestiato T (2021) FAVIPIRAVIR: Tratamento da COVID-19. https://www.gov.br/inpi/pt-br/servicos/patentes/tecnologias-para-covid-19/Arquivos%20Teste%20deb/copy_of_ESTUDO5.pdf. Accessed on 21 Nov 2023.
Chen A-C, Peng C-T, Wu S-F, Wu K-H, Chiang I-P, Tsai C-H (2006) Effect of deferiprone on liver iron overload and fibrosis in hepatitis C Virus-infected thalassemia. Hemoglobin 30(2):209–214. https://doi.org/10.1080/03630260600642518
Chen TT, Li L, Chung D-H, Allen CDC, Torti SV, Torti FM, Cyster JG, Chen C-Y, Brodsky FM, Niemi EC, Nakamura MC, Seaman WE, Daws MR (2005) TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med 202(7):955–965. https://doi.org/10.1084/jem.20042433
Chhabra R, Saha A, Chamani A, Schneider N, Nanjundan M, Shah R (2020) Iron pathways and iron chelation approaches in viral, microbial, and fungal infections. Pharmaceuticals 13(10):1–23. https://doi.org/10.3390/ph13100275
Coffey R, Ganz T (2017) Iron homeostasis: An anthropocentric perspective. J Biol Chem 292(31):12727–12734. https://doi.org/10.1074/jbc.R117.781823
Colafrancesco S, Alessandri C, Conti F, Priori R (2020) COVID-19 gone bad: a new character in the spectrum of the hyperferritinemic syndrome? Autoimmun Rev 19(7):102573. https://doi.org/10.1016/j.autrev.2020.102573
Coomes EA, Haghbayan H (2020) Interleukin-6 in Covid-19: a systematic review and meta-analysis. Rev Med Virol 30(6):1–9. https://doi.org/10.1002/rmv.2141
Cruvinel WM, Mesquita Júnior D, Araújo JAP, Catelan TTT, Souza AWS, Silva NP, Andrade LEC (2010) Immune system – Part I. Fundamentals of innate immunity with emphasis on molecular and cellular mechanisms of inflammatory response. Rev Bras Reumatol 50(4):434–447. https://doi.org/10.1590/S0482-50042010000400008
Dalamaga M, Karampela I, Mantzoros CS (2020) Commentary: could iron chelators prove to be useful as an adjunct to COVID-19 treatment regimens? Metabolism 108:154260. https://doi.org/10.1016/j.metabol.2020.154260
Datz C, Müller E, Aigner E (2017) Iron overload and non-alcoholic fatty liver disease. Minerva Endocrinol 42(2):173–183. https://doi.org/10.23736/S0391-1977.16.02565-7
Di Paola L, Hadi-Alijanvand H, Song X, Hu G, Giuliani A (2020) The discovery of a putative allosteric site in the SARS-CoV-2 Spike protein using an integrated structural/dynamic approach. J Proteome Res 19(11):4576–4586. https://doi.org/10.1021/acs.jproteome.0c00273
Dickson KB, Zhou J (2020) Role of reactive oxygen species and iron in host defense against infection. Front Biosci (Landmark Ed) 25(8):1600–1616. https://doi.org/10.2741/4869
Dimitriadis A, Foka P, Kyratzopoulou E, Karamichali E, Petroulia S, Tsitoura P, Kakkanas A, Eliadis P, Georgopoulou U, Mamalaki A (2021) The Hepatitis C virus NS5A and core proteins exert antagonistic effects on HAMP gene expression: the hidden interplay with the MTF-1/MRE pathway. FEBS Open Bio 11(1):237–250. https://doi.org/10.1002/2211-5463.13048
Doguer C, Ha JH, Gulec S, Vulpe CD, Anderson GJ, Collins JF (2017) Intestinal hephaestin potentiates iron absorption in weanling, adult, and pregnant mice under physiological conditions. Blood Adv 1(17):1335–1345. https://doi.org/10.1182/bloodadvances.2017008359
Dongiovanni P, Fracanzani AL, Fargion S, Valenti L (2011) Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J Hepatol 55(4):920–932. https://doi.org/10.1016/j.jhep.2011.05.008
Drakesmith H, Prentice A (2008) Viral infection and iron metabolism. Nat Rev Microbiol 6(7):541–552. https://doi.org/10.1038/nrmicro1930
Duchemin J-B, Paradkar PN (2017) Iron availability affects West Nile virus infection in its mosquito vector. Virol J 14(1):103. https://doi.org/10.1186/s12985-017-0770-0
Ebrahimi KH, Ciofi-Baffoni S, Hagedoorn PL, Nicolet Y, Le Brun NE, Hagen WR, Armstrong FA (2022) Iron–sulfur clusters as inhibitors and catalysts of viral replication. Nat Chem 14(3):253–266. https://doi.org/10.1038/s41557-021-00882-0
Ehsani S (2020) COVID-19 and iron dysregulation: distant sequence similarity between hepcidin and the novel coronavirus spike glycoprotein. Biol Direct 15(1):19. https://doi.org/10.1186/s13062-020-00275-2
Esan MO, van Hensbroek MB, Nkhoma E, Musicha C, White SA, ter Kuile FO, Phiri KS (2013) Iron supplementation in HIV-infected Malawian children with anemia: a double-blind, randomized. Controll Trial Clin Infect Dis 57(11):1626–1634. https://doi.org/10.1093/cid/cit528
Fara A, Mitrev Z, Rosalia RA, Assas BM (2020) Cytokine storm and COVID-19: a chronicle of pro-inflammatory cytokines. Open Biol. https://doi.org/10.1098/rsob.200160
Fernandez M, Lokan J, Leung C, Grigg A (2022) A critical evaluation of the role of iron overload in fatty liver disease. J Gastroenterol Hepatol 37(10):1873–1883. https://doi.org/10.1111/jgh.15971
Fillebeen C, Muckenthaler M, Andriopoulos B, Bisaillon M, Mounir Z, Hentze MW, Koromilas AE, Pantopoulos K (2007) Expression of the subgenomic hepatitis C virus replicon alters iron homeostasis in Huh7 cells. J Hepatol 47(1):12–22. https://doi.org/10.1016/j.jhep.2007.01.035
Foka P, Dimitriadis A, Kyratzopoulou E, Giannimaras DA, Sarno S, Simos G, Georgopoulou U, Mamalaki A (2014) A complex signaling network involving protein kinase CK2 is required for hepatitis C virus core protein-mediated modulation of the iron-regulatory hepcidin gene expression. Cell Mol Life Sci 71(21):4243–4258. https://doi.org/10.1007/s00018-014-1621-4
Franchini M, Targher G, Montagnana M, Lippi G (2008) Iron and thrombosis. Ann Hematol 87(3):167–173. https://doi.org/10.1007/s00277-007-0416-1
Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, Tanaka H, Kobayashi Y, Iwasa M, Watanabe S, Adachi Y, Kaito M (2007) Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol Med 13(1–2):97–104. https://doi.org/10.2119/2006-00057.Fujita
Ganz T (2019) Erythropoietic regulators of iron metabolism. Free Radical Biol Med 133:69–74. https://doi.org/10.1016/j.freeradbiomed.2018.07.003
Garrido-Rodríguez V, Álvarez-Ríos AI, Olivas-Martínez I, Pozo-Balado M, Bulnes-Ramos Á, Leal M, Pacheco YM (2022) Dysregulation of iron metabolism modulators in virologically suppressed HIV-infected patients. Front Immunol 13:5. https://doi.org/10.3389/fimmu.2022.977316
Gawlik K, Gallay PA (2014) HCV core protein and virus assembly: what we know without structures. Immunol Res 60(1):1–10. https://doi.org/10.1007/s12026-014-8494-3
Georgopoulou U, Dimitriadis A, Foka P, Karamichali E, Mamalaki A (2014) Hepcidin and the iron enigma in HCV infection. Virulence 5(4):465–476. https://doi.org/10.4161/viru.28508
González-Candelas F, López-Labrador FX (2010) Clinical relevance of genetic heterogeneity in HCV. Future Virol 5(1):33–49. https://doi.org/10.2217/fvl.09.63
Grotto HZW (2008) Iron metabolism: An overview on the main mechanisms involved in its homeostasis. Rev Bras Hematol Hemoter 30(5):390–397
Gupta Y, Maciorowski D, Medernach B, Becker DP, Durvasula R, Libertin CR, Kempaiah P (2022) Iron dysregulation in COVID-19 and reciprocal evolution of SARS-CoV-2: natura nihil frustra facit. J Cell Biochem 123(3):601–619. https://doi.org/10.1002/jcb.30207
Halliwell B, Gutteridge JMC (2007) Free radicals in biology and medicine, 4th edn. Oxford University Press, Oxford
Harigae H (2018) Iron metabolism and related diseases: an overview. Int J Hematol 107(1):5–6. https://doi.org/10.1007/s12185-017-2384-0
Hegelund MH, Glenthøj A, Ryrsø CK, Ritz C, Dungu AM, Sejdic A, List KCK, Krogh-Madsen R, Lindegaard B, Kurtzhals JAL, Faurholt-Jepsen D (2022) Biomarkers for iron metabolism among patients hospitalized with community-acquired pneumonia caused by infection with SARS-CoV-2, bacteria, and influenza. APMIS 130(9):590–596. https://doi.org/10.1111/apm.13259
Hirota K (2019) An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radical Biol Med 133:118–129. https://doi.org/10.1016/j.freeradbiomed.2018.07.018
Hu X, Li J, Fu M, Zhao X, Wang W (2021) The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Therapy 6(1):402. https://doi.org/10.1038/s41392-021-00791-1
Itoh S, Itoh F, Goumans MJ, Dijke PT (2000) Signaling of transforming growth factor-β family members through Smad proteins. Eur J Biochem 267(24):6954–6967. https://doi.org/10.1046/j.1432-1327.2000.01828.x
Janeway C, Travers P, Walport M, Shlomchik M (2001) Immunobiology, 5th edn. Garland Science, New York
Jimenez K, Leitner F, Leitner A, Scharbert G, Schwabl P, Kramer A-M, Krnjic A, Friske J, Helbich T, Evstatiev R, Khare V, Gasche C (2021) Iron deficiency-induced thrombocytosis increases thrombotic tendency in rats. Haematologica 106(3):782–794. https://doi.org/10.3324/haematol.2019.245092
Johnston PA, Grandis JR (2011) STAT3 signalling: anticancer strategies and challenges. Mol Interventions 11(1):18–26. https://doi.org/10.1124/mi.11.1.4
Jones JE, Le Sage V, Lakdawala SS (2021) Viral and host heterogeneity and their effects on the viral life cycle. Nat Rev Microbiol 19(4):272–282. https://doi.org/10.1038/s41579-020-00449-9
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:401. https://doi.org/10.3389/fimmu.2014.00461
Kell DB, Heyden EL, Pretorius E (2020) The biology of lactoferrin, an iron-binding protein that can help defend against viruses and bacteria. Front Immunol 11:7. https://doi.org/10.3389/fimmu.2020.01221
Kerkhoff AD, Meintjes G, Opie J, Vogt M, Jhilmeet N, Wood R, Lawn SD (2016) Anaemia in patients with HIV-associated TB: relative contributions of anaemia of chronic disease and iron deficiency. Int J Tuberc Lung Dis 20(2):193–201. https://doi.org/10.5588/ijtld.15.0558
Kiihn LC, Hentze MW (1992) Coordination of cellular iron metabolism by post-transcriptional gene regulation. J Inorg Biochem 47(3-4):183–195. https://doi.org/10.1016/0162-0134(92)84064-t
Knez M, Graham RD, Welch RM, Stangoulis JCR (2017) New perspectives on the regulation of iron absorption via cellular zinc concentrations in humans. Crit Rev Food Sci Nut 57(10):2128–2143. https://doi.org/10.1080/10408398.2015.1050483
Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J (2008) Body iron metabolism and pathophysiology of iron overload. Int J Hematol 88(1):7–15. https://doi.org/10.1007/s12185-008-0120-5
Kondaiah P, Yaduvanshi PS, Sharp PA, Pullakhandam R (2019) Iron and zinc homeostasis and interactions: Does enteric zinc excretion cross-talk with intestinal iron absorption? Nutrients 11(8):78. https://doi.org/10.3390/nu11081885
Kumari N, Ammosova T, Diaz S, Lin X, Niu X, Ivanov A, Jerebtsova M, Dhawan S, Oneal P, Nekhai S (2016) Increased iron export by ferroportin induces restriction of HIV-1 infection in sickle cell disease. Blood Adv 1(3):170–183. https://doi.org/10.1182/bloodadvances.2016000745
Kung Y-A, Chiang H-J, Li M-L, Gong Y-N, Chiu H-P, Hung C-T, Huang P-N, Huang S-Y, Wang P-Y, Hsu T-A, Brewer G, Shih S-R (2022) Acyl-coenzyme a synthetase long-chain family member 4 is involved in viral replication organelle formation and facilitates virus replication via ferroptosis. mBio 13(1):e02717-12. https://doi.org/10.1128/mbio.02717-21
Kwon Y-C, Ray RB, Ray R (2014) Hepatitis C virus infection: establishment of chronicity and liver disease progression. EXCLI J 13:977–996
Lavie M, Dubuisson J (2017) Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimie 141:62–69. https://doi.org/10.1016/j.biochi.2017.06.009
Li Q, Chen Z, Zhou X, Li G, Zhang C, Yang Y (2023) Ferroptosis and multi-organ complications in COVID-19: mechanisms and potential therapies. Front Genetics 14:1187985. https://doi.org/10.3389/fgene.2023.1187985
Li X, Wang M, Cheng A et al (2020) Enterovirus Replication Organelles and Inhibitors of Their Formation. Front Microbiol 11:1817. https://doi.org/10.3389/fmicb.2020.01817
Lifshitz V, Frenkel D (2013) TGF-β. Handbook of biologically active peptides. https://doi.org/10.1016/B978-0-12-385095-9.00225-6
Lippi G, Mattiuzzi C (2020) Hemoglobin value may be decreased in patients with severe coronavirus disease 2019. Hematol Transf Cell Therapy 42(2):116–117. https://doi.org/10.1016/j.htct.2020.03.001
Liu C-C, Ahearn JM (2005) Acute-phase proteins and inflammation: immunological and clinical implications. Measuring Immun 48:131–143. https://doi.org/10.1016/B978-012455900-4/50272-5
Liu H, Trinh TL, Dong H, Keith R, Nelson D, Liu C (2012) Iron regulator hepcidin exhibits antiviral activity against hepatitis C Virus. PLoS ONE. https://doi.org/10.1371/journal.pone.0046631
Liu W, Zhang S, Nekhai S, Liu S (2020) Depriving iron supply to the virus represents a promising adjuvant therapeutic against viral survival. Curr Clin Microbiol Rep 7(2):13–19. https://doi.org/10.1007/s40588-020-00140-w
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS (2021) Hepatocellular Carcinoma. Nat Rev Dis Primers 7(1):6. https://doi.org/10.1038/s41572-020-00240-3
López-Prieto J, González-Reimers E, Alemán-Valls MR, de la Vega-Prieto MJ, Abreu-González P, Pelazas-González R, Hernández-Luis R, Jorge-Ripper C, Santolaria-Fernández F (2013) Iron and proinflammatory cytokines in chronic hepatitis C virus infection. Biol Trace Elem Res 155(1):5–10. https://doi.org/10.1007/s12011-013-9760-2
Machado PRL, Araújo MIAS, Carvalho L, Carvalho EM (2004) Mecanismos de resposta imune às infecções. An Bras Dermatol 79(6):647–664. https://doi.org/10.1590/S0365-05962004000600002
Mahroum N, Alghory A, Kiyak Z, Alwani A, Seida R, Alrais M, Shoenfeld Y (2022) Ferritin – from iron, through inflammation and autoimmunity, to COVID-19. J Autoimmun 126:102778. https://doi.org/10.1016/j.jaut.2021.102778
Makarov V, Riabova O, Ekins S, Pluzhnikov N, Chepur S (2020) The past, present and future of RNA respiratory viruses: Influenza and coronaviruses. Pathogens and Disease 78(7):046. https://doi.org/10.1093/femspd/ftaa046
Mancinelli R, Rosa L, Cutone A, Lepanto MS, Franchitto A, Onori P, Gaudio E, Valenti P (2020) Viral hepatitis and iron dysregulation: molecular pathways and the role of lactoferrin. Molecules 25(8):1997. https://doi.org/10.3390/molecules25081997
Merino KM, Allers C, Didier ES, Kuroda MJ (2017) Role of monocyte/macrophages during HIV/SIV infection in adult and pediatric acquired immune deficiency syndrome. Front Immunol 8:1693. https://doi.org/10.3389/fimmu.2017.01693
Miyachi H, Kobayashi Y, Relja B, Fujita N, Iwasa M, Gabazza EC, Takei Y (2011) Effect of suppressor of cytokine signaling on hepcidin production in hepatitis C virus replicon cells. Hepatol Res 41(4):364–374. https://doi.org/10.1111/j.1872-034X.2011.00777.x
Naif HM (2013) Pathogenesis of HIV infection. Infectious Dis Rep 5(S1):26–30. https://doi.org/10.4081/idr.2013.s1.e6
Nash KL, Woodall T, Brown AS, Davies SE, Alexander GJ (2010) Hepatocellular carcinoma in patients with chronic hepatitis C virus infection without cirrhosis. World J Gastroenterol 16(32):4061. https://doi.org/10.3748/wjg.v16.i32.4061
Naydenova K, Muir KW, Wu L-F, Zhang Z, Coscia F, Peet MJ, Castro-Hartmann P, Qian P, Sader K, Dent K, Kimanius D, Sutherland JD, Löwe J, Barford D, Russo CJ (2021) Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc Nat Acad Sci 118(7):78. https://doi.org/10.1073/pnas.2021946118
Ni S, Yuan Y, Kuang Y, Li X (2022) Iron metabolism and immune regulation. Front Immunol 13:816282. https://doi.org/10.3389/fimmu.2022.816282
Nishina S, Korenaga M, Hidaka I, Shinozaki A, Sakai A, Gondo T, Tabuchi M, Kishi F, Hino K (2010) Hepatitis C virus protein and iron overload induce hepatic steatosis through the unfolded protein response in mice. Liver Int 30(5):683–692. https://doi.org/10.1111/j.1478-3231.2010.02210.x
Pari AA, Yousefi M (2021) DFT examination of electronic and structural features of favipiravir for iron chelation. Biointerface Res Appl Chem 12(4):5081–5088. https://doi.org/10.33263/BRIAC124.50815088
Pasini AMF, Stranieri C, Girelli D, Busti F, Cominacini L (2021) Is ferroptosis a key component of the process leading to multiorgan damage in COVID-19? Antioxidants 10(11):1677. https://doi.org/10.3390/antiox10111677
Peace CG, O’Neill LAJ (2021) Ironing out vaccine efficacy. Med 2(2):113–114. https://doi.org/10.1016/j.medj.2021.01.003
Peng D, Gao Y, Zhang L, Liu Z, Wang H, Liu Y (2022) The relationship between hepcidin-mediated iron dysmetabolism and COVID-19 severity: a meta-analysis. Front Public Health 10:881412. https://doi.org/10.3389/fpubh.2022.881412
Pereira TA, Moraes CB, Barbosa CG, Peres BM, Freitas Jr LHG, Espósito BP (2022) Lipophilic iron chelators on tumor cells and SARS-CoV-2. Communication in the XX Brazilian Meeting on Inorganic Chemistry
Perricone C, Bartoloni E, Bursi R, Cafaro G, Guidelli GM, Shoenfeld Y, Gerli R (2020) COVID-19 as part of the hyperferritinemic syndromes: the role of iron depletion therapy. Immunol Res 68(4):213–224. https://doi.org/10.1007/s12026-020-09145-5
Petrak J, Vyoral D (2005) Hephaestin : a ferroxidase of cellular iron export. Int J Biochem Cell Biol 37(6):1173–1178. https://doi.org/10.1016/j.biocel.2004.12.007
Pierce BS, Elgren TE, Hendrich MP (2003) Mechanistic implications for the formation of the diiron cluster in ribonucleotide reductase provided by quantitative EPR spectroscopy. J Am Chem Soc 125(29):8748–8759. https://doi.org/10.1021/ja021290h
Posey JE, Gherardini FC (2000) Lack of a role for iron in the lyme disease pathogen. Science 288(5471):1651–1653. https://doi.org/10.1126/science.288.5471.1651
Preston AE, Drakesmith H, Frost JN (2021) Adaptive immunity and vaccination – iron in the spotlight. Immunotherapy Advances 1(1):007. https://doi.org/10.1093/immadv/ltab007
Roldan EQ, Biasiotto G, Magro P, Zanella I (2020) The possible mechanisms of action of 4-aminoquinolines (chloroquine/hydroxychloroquine) against Sars-Cov-2 infection (COVID-19): a role for iron homeostasis? Pharmacol Res 158:104904. https://doi.org/10.1016/j.phrs.2020.104904
Ruddell RG, Hoang-Le D, Barwood JM, Rutherford PS, Piva TJ, Watters DJ, Santambrogio P, Arosio P, Ramm GA (2009) Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 49(3):887–900. https://doi.org/10.1002/hep.22716
Salter-Cid L, Brunmark A, Li Y, Leturcq D, Peterson PA, Jackson MR, Yang Y (1999) Transferrin receptor is negatively modulated by the hemochromatosis protein HFE: implications for cellular iron homeostasis. Proc Natl Acad Sci USA 96:5434–5439. https://doi.org/10.1073/pnas.96.10.5434
Schalinske KL, Blemings KP, Steffen DW, Chen OS, Eisenstein RS (1997) Iron regulatory protein 1 is not required for the modulation of ferritin and transferrin receptor expression by iron in a murine pro-B lymphocyte cell line. Proc Natl Acad Sci USA 94(20):10681–10686. https://doi.org/10.1073/pnas.94.20.10681
Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC (2008) The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 7(3):205–214. https://doi.org/10.1016/j.cmet.2007.11.016
Shah YM, Xie L (2014) Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology 146(3):630–642. https://doi.org/10.1053/j.gastro.2013.12.031
Sidorkiewicz M, Brown RJ (2021) Hepatitis C Virus uses host lipids to its own advantage. Metabolites 11(5):273. https://doi.org/10.3390/metabo11050273
Singh AK (2018) Erythropoiesis. In Textbook of Nephro-Endocrinology, 2nd edn. Academic Press. https://doi.org/10.1016/B978-0-12-803247-3.00012-X
Sjöberg B-M (1997) Ribonucleotide reductases :a group of enzymes with different metallosites and a similar reaction mechanism. In Metal Sites in Proteins Models - Iron Centers. Springer. https://doi.org/10.1007/3-540-62870-3_5
Sonnweber T, Grubwieser P, Sahanic S, Böhm AK, Pizzini A, Luger A, Schwabl C, Koppelstätter S, Kurz K, Puchner B, Sperner-Unterweger B, Hüfner K, Wöll E, Nairz M, Widmann G, Tancevski I, Löffler-Ragg J, Weiss G (2022) The impact of iron Dyshomeostasis and Anaemia on long-term pulmonary recovery and persisting symptom burden after COVID-19: a prospective observational cohort study. Metabolites 12(6):158. https://doi.org/10.3390/metabo12060546
Stoffel NU, Uyoga MA, Mutuku FM, Frost JN, Mwasi E, Paganini D, van der Klis FRM, Malhotra IJ, LaBeaud AD, Ricci C, Karanja S, Drakesmith H, King CH, Zimmermann MB (2020) Iron deficiency anemia at time of vaccination predicts decreased vaccine response and iron supplementation at time of vaccination increases humoral vaccine response: a birth cohort study and a randomized trial follow-up study in Kenyan infants. Front Immunol 11:58. https://doi.org/10.3389/fimmu.2020.01313
Szabo R, Petrisor C, Bodolea C, Simon R, Maries I, Tranca S, Mocan T (2022) Hyperferritinemia, Low circulating iron and elevated hepcidin may negatively impact outcome in COVID-19 patients: a pilot study. Antioxidants 11(7):89. https://doi.org/10.3390/antiox11071364
Szymczak A, Zalewska M, Rymer W, Jankowska EA (2022) Asymptomatic Human Immunodeficiency Virus-1 infection with high CD4+ T cell count does not alter iron metabolism or hepcidin levels: the pilot study. Infect Dis Therapy 11(1):265–275. https://doi.org/10.1007/s40121-021-00560-1
Taneri PE, Gómez-Ochoa SA, Llanaj E, Raguindin PF, Rojas LZ, Roa-Díaz ZM, Salvador D, Groothof D, Minder B, Kopp-Heim D, Hautz WE, Eisenga MF, Franco OH, Glisic M, Muka T (2020) Anemia and iron metabolism in COVID-19: a systematic review and meta-analysis. Eur J Epidemiol 35(8):763–773. https://doi.org/10.1007/s10654-020-00678-5
Telek E, Ujfalusi Z, Kemenesi G, Zana B, Jakab F, Hild G, Lukács A, Hild G (2022) A possible way to relate the effects of SARS-CoV-2-induced changes in transferrin to severe COVID-19-associated diseases. Int J Mol Sci 23(11):78. https://doi.org/10.3390/ijms23116189
Toe QK, Issitt T, Mahomed A, Almaghlougth F, Bahree I, Sturge C, Hu X, Panselinas I, Burke-Gaffney A, Wort SJ, Quinlan GJ (2022) Human pulmonary artery endothelial cells upregulate ACE2 expression in response to iron-regulatory elements: Potential implications for SARS-CoV-2 infection. Pulmonary Circ 12:e12068. https://doi.org/10.1002/pul2.12068
Tong WH, Rouault TA (2007) Metabolic regulation of citrate and iron by aconitases: role of iron-sulfur cluster biogenesis. Biometals 20(3–4):549–564. https://doi.org/10.1007/s10534-006-9047-6
Torrents E (2014) Ribonucleotide reductases: essential enzymes for bacterial life. Front Cell Infect Microbiol 4:52. https://doi.org/10.3389/fcimb.2014.00052
Troxell B, Xu H, Yang XF (2012) Borrelia burgdorferi, a pathogen that lacks iron, encodes manganese-dependent superoxide dismutase essential for resistance to streptonigrin. J Biol Chem 287(23):19284–19293. https://doi.org/10.1074/jbc.M112.344903
Vahey MD, Fletcher DA (2019) Low-fidelity assembly of influenza a virus promotes escape from host cells. Cell 176(1–2):281-294.e19. https://doi.org/10.1016/j.cell.2018.10.056
van Asbeck BS, Georgiou NA, van der Bruggen T, Oudshoorn M, Nottet HSLM, Marx JJM (2001) Anti-HIV effect of iron chelators: different mechanisms involved. J Clin Virol 20(3):141–147. https://doi.org/10.1016/S1386-6532(00)00122-0
Vulpe CD, Kuo Y-M, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21(2):195–199. https://doi.org/10.1038/5979
Waheed A, Grubb JH, Zhou XY, Tomatsu S, Fleming RE, Costaldi ME, Britton RS, Bacon BR, Sly WS, Doisy EA (2001) Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc Nat Acad Sci 99(5):3117–3122. https://doi.org/10.1073/pnas.0427014
Wang MP, Joshua B, Jin NY, Du SW, Li C (2022) Ferroptosis in viral infection: the unexplored possibility. Acta Pharmacol Sin 43(8):1905–1915. https://doi.org/10.1038/s41401-021-00814-1
Wang Y, Huang J, Sun Y, Stubbs D, He J, Li W, Wang F, Liu Z, Ruzicka JA, Taylor EW, Rayman MP, Wan X, Zhang J (2021) SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem Toxicol 153:112286. https://doi.org/10.1016/j.fct.2021.112286
Ward RJ, Crichton RR, Taylor DL, Della Corte L, Srai SK, Dexter DT (2011) Iron and the immune system. J Neural Transm 118(3):315–328. https://doi.org/10.1007/s00702-010-0479-3
Wei M, Cheng A, Wang M (2012) The small subunit of ribonucleotide reductase gene and protein of herpes viruses. Rev Med Microbiol 23(4):82–85. https://doi.org/10.1097/MRM.0b013e3283573668
Wisaksana R, De Mast Q, Alisjahbana B, Jusuf H, Sudjana P, Indrati AR, Sumantri R, Swinkels D, Van Crevel R, Van Der Ven A (2013) Inverse relationship of serum hepcidin levels with CD4 cell counts in HIV-infected patients selected from an Indonesian prospective cohort study. PLoS ONE 8(11): e79904. https://doi.org/10.1371/journal.pone.0079904
World Health Organization (WHO) (2023) Anaemia. https://www.who.int/news-room/fact-sheets/detail/anaemia. Accessed on 4 Oct 2023.
Xu Y, Alfaro-Magallanes VM, Babitt JL (2021) Physiological and pathophysiological mechanisms of hepcidin regulation: clinical implications for iron disorders. Br J Haematol 193(5):882–893. https://doi.org/10.1111/bjh.17252
Yang Y, Bazhin AV, Werner J, Karakhanova S (2013) Reactive oxygen species in the immune system. Int Rev Immunol 32(3):249–270. https://doi.org/10.3109/08830185.2012.755176
Yao Y, Chen Z, Zhang H et al (2021) Selenium–GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol 22(9):1127–1139. https://doi.org/10.1038/s41590-021-00996-0
Zeinivand M, Sharifi M, Hassanshahi G, Nedaei SE (2023) Deferoxamine has the potential to improve the COVID-19-related inflammatory response in diabetic patients. Int J Pept Res Ther 29(4):63. https://doi.org/10.1007/s10989-023-10516-3
Zhou S, Li H, Li S (2022) The associations of iron related biomarkers with risk, clinical severity and mortality in SARS-CoV-2 patients: a meta-analysis. Nutrients 14(16):3406. https://doi.org/10.3390/nu14163406
Zou D-M, Sun W-L (2017) Relationship between Hepatitis C Virus infection and iron overload. Chin Med J 130(7):866–871. https://doi.org/10.4103/0366-6999.202737
Acknowledgements
The authors wish to thank the financial support from the CNPq, under Contract Number 150539/2022-3 and from FAPESP, under Contracts Number 2018/19684-0 and 2021/10894-5.
Funding
This research was funded by the National Council for Scientific and Technological Development (CNPq) under contract number 150539/2022-3 and by The São Paulo Research Foundation (FAPESP), in Brazil, contracts number 2018/19684-0 and 2021/10894-5.
Author information
Authors and Affiliations
Contributions
Conceptualization, TAP and BPE; methodology, TAP and BPE; resources, TAP and BPE; data curation, BPE; writing—original draft preparation, TAP; writing—review and editing, TAP and BPE; supervision, BPE; project administration, TAP; funding acquisition, TAP and BPE All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Institutional review board
Not applicable.
Informed consent
Not applicable.
Sample availability
Samples of the compounds are not available from the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
