
Abstract
The Caatinga biome is one of the largest areas of the South American seasonally dry tropical forest that has been severely affected by unsustainable natural resource use. Furthermore, the biome has been identified as an ecologically sensitive region that is particularly susceptible to climate changes. One of the most economically important native palm tree for traditional communities from the semi-arid Caatinga is the carnauba palm, Copernicia prunifera, which offers diverse natural resources, yet its natural populations suffer intense exploitation. To inform conservation and population management strategies, we sought to determine if remaining natural populations of this species in an intensively exploited area in Northeast Brazil displayed evidence of negative genetic impacts because of exploitation and how this might interact with expected environmental changes. Mantel’s test revealed a positive and significant correlation between geographic and genetic distances, suggesting natural populations are structured by isolation by distance, while also experiencing genetic barriers as identified through Monmonier's algorithm. The studied populations showed evidence of genetic bottlenecks, while future climate scenarios suggest that potentially suitable habitats for C. prunifera within its native range will be reduced. Significant genetic differentiation among populations resulted in three distinct genetic groups which are consistent with ecological niche modelling. In addition to the need for in situ conservation of C. prunifera populations to minimize the loss of important alleles, the creation of germplasm banks for ex situ conservation and strategies for developing planted productive forests are urgently required to maintain natural populations and ensure sustainability resources for traditional communities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Indiscriminate exploitation of natural forest resources has significantly decreased the size of many natural populations, resulting in fragmented habitats and population isolation (DeFries et al. 2005). Studies have debated the impact of habitat fragmentation and population reduction on genetic diversity in natural populations (Aguilar et al. 2008; Jump and Penuelas 2006; Honnay and Jacquemyn 2007). The fragmentation can significantly affect the movement of animals, pollen, and seeds (Tewksbury et al. 2002), which can alter populations’ genetic structure (Bacles et al. 2004; Sebbenn et al. 2011). The reduced size of natural areas and fragmentation may also lead to a loss of the genetic diversity contained within and among populations (Young et al. 1996; Newman and Pilson 1997).
Anthropogenic disturbances can have a significant impact on population genetic diversity and structure (Santos et al. 2015; Omondi et al. 2016). Consequently, studies of genetic structure and diversity in populations of key biological resources are needed to understand how diversity is distributed within and between populations and factors that affect this distribution (Schwartz et al. 2007). The influence of these factors vary with life-history traits and include effective population size, mode of reproduction, and breeding systems (Degen and Roubik 2004), as well as the geographical range of the species (Rouger and Jump 2014). Furthermore, gene flow also has an impact on genetic structure within and among populations (Provan et al. 2008; Araújo et al. 2017), which is influenced not only by the ability of dispersers and pollinators to reach other populations, but also by geographical barriers that may exist between populations (Dias et al. 2016).
The Caatinga biome represents one of the largest areas of the South American seasonally dry tropical forest. It has been severely deforested as a result of wood consumption, livestock grazing, and fire, and more than half of all ‘poor’ Brazilians in the country live within the biome (Silveira-Neto 2014). Furthermore, most areas of Caatinga are ecologically sensitive, with particularly amplified responses to climate variability (Seddon 2016), and are currently experiencing a trajectory of drying (da Silva 2004). Native to the Caatinga, Copernicia prunifera, known as carnauba palm, is economically significant because of the commercially important wax (carnauba wax) that covers its leaves (IBGE 2018), especially younger leaves. However, extensive and unsustainable harvesting practices, agricultural expansion, and an absence of sustainable management programmes represent major threats to the long-term continuation of C. prunifera populations. Continued unsustainable harvesting of non-timber forest products (NTFPs) is expected to have cascading ecological impacts, from individual and population to community and ecosystem function (Ticktin 2004). Over-exploitation of carnauba populations has had a negative impact on associated wild fauna, for example forcing wild triatomines to seek other habitats (Lima and Sarquis 2008).
Copernicia prunifera populations have rapidly declined because of anthropogenic disturbance over the last century primarily due to deforestation and agricultural expansion (D’alva 2004). The use of carnauba wax dates back to the 18th Century for the production of candles. From the second half of the 19th Century, the discovery of new uses for the wax intensified its exportation and allowed the development of economically important extractive, agroindustrial and commercial activities. From the 1960s, the modernization of agriculture led to the deforestation of extensive areas of the Caatinga, significantly reducing the C. prunifera habitat (D’alva 2004), while exploitation of carnauba has increased. An additional, and substantial, contemporary threat relates to a changing climate given that the whole of the species’ distribution is located in semi-arid regions subject to desertification (MMA 2005). Ecological niche modeling (ENM) allows correlating a set of environmental variables with the geographical occurrence of a species. The ENM become a useful method to address ecology issues such as conservation practices, indicating regions with habitat suitability under ongoing climate change (Zacarías-Correa et al. 2020).
Assessments of genetic diversity for key species can provide important contributions when defining conservation strategies and developing management programs (Duarte et al. 2015) and should be taken into consideration in development of public policies aimed at conserving biodiversity (Laikre et al. 2010). Molecular markers based on amplification of DNA provide valuable tools to study genetic structure and diversity between individuals and within and between populations (Nybom 2004). The use of inter-simple sequence repeat (ISSR) markers provides a quick and simple method to effectively analyse the genetic diversity of natural populations across a large number of polymorphic bands. This method is low cost and does not require prior information of the genome, which is particularly important for genera such as Copernicia as there is no previous knowledge of microsatellite regions of the genome (Reddy et al. 2002). While ISSR markers cannot differentiate heterozygous from homozygous individuals since they are dominant markers, they do permit the analysis of multiple loci in a single reaction (Wolfe 2005) and can be an alternative in cases where a high number of null alleles exist in microsatellite markers (Rosa et al. 2017).
Given the importance of C. prunifera to local communities and the potential impacts of its overexploitation on resource sustainability and biodiversity, we sought to determine if recent rapid increases in the exploitation of C. prunifera populations are associated with negative impacts on the genetics of the species. We hypothesised that genetic bottlenecks would accompany high levels of genetic differentiation among populations due to unsustainable management practices over the years in a harvest-intense area. Furthermore, we sought to determine the extent to which landscape boundaries result in current genetic discontinuities within the species and potential interactions of exploitation and habitat suitability predicted by ENM.
Material and methods
Target species
Copernicia prunifera individuals can be found in river valleys and in seasonally flooded areas in the semi-arid region of northeastern Brazil, where they generally form monodominant populations known as carnaubais. The species is highly resistant to the prolonged absence of water and permanent floods (Arruda and Calbo 2004). The wax produced from its leaves is used in cosmetics, pharmaceutical capsules, electronics, food products, polishing waxes, and coatings (Sousa et al. 2015), and the stems are commonly used in house construction (Fig. S1). The production value of its wax and fibers brings in more than $55 million per year, according to the official government data (IBGE 2018). The species presents multiple inflorescences, which are made up of yellowish and hermaphroditic flowers (Silva et al. 2017). Flowering is subannual, with greater intensity between November and February and ripe fruits between January and March (Rocha et al. 2015). The flowers are visited by insects like the irapuá bee (Trigona spinipes) and the maribondo-caboclo wasp (Polistes canadensis), and the species has a mixed mating system that is preferentially allogamous (Silva et al. 2017). Fruits are likely dispersed by the palm tanager (sanhaçu-do-coqueiro; Tangara palmarum) (Silva et al. 2017) and bats (Sousa et al. 2015), demonstrating the relevant interactions between species (animal-plant) that need to be preserved.
Sampling
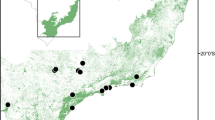
This study was conducted in eleven natural populations located in Rio Grande do Norte and Ceará States, Brazil, which represents one of the areas in which the species is most intensely harvested in Northeast Brazil (D’alva 2004; IBGE 2018). One-hundred and eighty individuals were sampled (Table 1 and Fig. 1), and sampling ranged from 11 to 24 individuals per population, which is consistent with other studies using ISSR markers (Duarte et al. 2015; Rosa et al. 2017). Pairwise distance between populations ranged from 4.6 km between SER and LGP to 310.4 km between LGP and AR1 (Fig. 1). Small pieces of leaves were cut using a tree trimmer, placed in plastic tubes containing 2 mL CTAB 2X (cationic hexadecyltrimethylammonium bromide), labelled, and stored in a freezer at – 20 °C until DNA extraction.

Geographic location of the sampled Copernicia prunifera populations in northeast Brazil (a), and altitudinal gradients (b). Populations are identified according to genetic groups established by Structure (see Fig. 3 and Fig. 4). Group distribution is shown in comparison with ecological niche modelling for the species at present day (c) and the future scenario (d). Red corresponds to regions with the highest probability of C. prunifera occurrence, blue corresponds to the least suitable regions, green lines correspond to rivers. Both figures (c and d) show the main genetic boundaries indicating three barriers among populations (dotted lines a–a, b–b, c–c) obtained with Monmonier’s maximum difference algorithm (see Fig. S3). The coordinates of each population are shown in Table 1
Historical anthropogenic disturbances
Although change in population size was not measured directly, a previous ethnoecological and ethnobotanical survey indicates substantial population decrease over recent decades (Sousa et al. 2015) that has accelerated since the 1960s (D’alva 2004). All sampled populations have been subjected to recent disturbance, showing signs of fire, intensive leaf extraction, timber harvesting, and trampling by cattle resulting in damage to regeneration (Fig. S1). Government data showing powder and wax production derived from C. prunifera are given in Table 1 and are based on the Brazilian Institute of Geography and Statistics Automatic Recovery System—SIDRA (IBGE 2018).
DNA extraction, PCR, and electrophoresis
DNA extraction was performed using the CTAB method, as described by Doyle (1990). We tested 29 ISSR primers and selected seven that best amplified C. prunifera DNA. For polymerase chain reaction (PCR), the Veriti automatic thermocycler was used with a volume of 12 μL containing genomic DNA. The PCR mix was composed of buffer (10x), BSA (1.0 mg mL−1), MgCl2 (50 mM), dNTP (2.5 mM), primer (2 μM), Taq polymerase (5.0 U μL), DNA (diluted 1:50), ISSR primer (2 µM), and ultrapure water. The reaction sequence consisted of denaturation at 94 °C for 2 min followed by 37 cycles of 94 °C for 15 s, 47 °C for 30 s, and 72 °C for 1 min. The process was completed with a final step at 72 °C for 7 min and then cooled to 4 °C. Amplification products were subjected to 1.5% horizontal agarose gel electrophoresis, stained with GelRed™ in 1 X TAE (Tris–Acetate-EDTA) buffer at a voltage of 100 V for two and a half hours against a 1 kb molecular weight size marker. Subsequently, the gels were visualised and photographed in ultraviolet light using the E-Box VX2 (Vilber Lourmat, Marne la Valle, France).
Genetic diversity
Polymorphic information content (PIC) was calculated to test the ability of the ISSR primers to distinguish polymorphism between individuals, with the absence or presence of bands as indicators. For the calculation, we used the formula proposed by Anderson et al. (1993): \({\text{PIC}}_{{{\text{i }}}} = 1 - \sum\nolimits_{{{\text{j}} = 1}}^{{\text{n}}} {{\text{P}}_{{{\text{ij}}}}^{2} }\), where \({\text{P}}_{{{\text{ij}}}}\) is the frequency of allele “j” in marker “I”. To estimate the genetic diversity parameters, we used the software PopGene v.1.32 (Yeh et al. 1997) to assess the total number of observed alleles (na), number of effective alleles (ne), Nei's (1973) genetic diversity (h), and Shannon index (I) for each population. The Bayesian approach to determine genetic diversity (hs, Holsinger 1999) was also estimated using the program Hickory v.1.1 (Holsinger and Lewis 2007).
Genetic structure and discontinuity
Genetic differentiation among populations was calculated using both Nei’s (1978) standard genetic distance (Ds) and a Bayesian approach (theta), in which we assessed the theta-II statistic (Holsinger and Lewis 2007) that corresponds to theta-B of Holsinger and Wallace (2004). This provides the best estimate of the proportion of genetic diversity due to differences among contemporaneous populations in the program Hickory v1.1 (Holsinger and Lewis 2007). Mantel’s test was performed using GenAlex v.6.503 (Peakall and Smouse 2012), resampled using the Monte Carlo method (999 permutations), to test for the existence of a correlation between geographic distance and both Nei’s genetic distance (Ds, 1978) and theta-II (Holsinger and Lewis 2007).
The program Ntsys (Rohlf 1993) was used to produce a dendrogram based on the unweighted pair-group method using arithmetic averages (UPGMA) to simplify interpretation of genetic identity based on Nei’s (1978) distance obtained with PopGene. The stability of the clusters was verified with bootstrap analysis using 1000 permutations implemented in the program Bood-P, version 1.2 (Coelho 2001). Bayesian analysis was performed using the program Structure v.2.3.4 (Pritchard 2000) to infer the number of genetic groups (K) that represent the sampled populations. Ten independent runs for each K (ranging from 1 to 13) were conducted, with the estimates of K based on the model of mixed ancestry (admixture) and the frequency of correlated alleles. Each run was comprised of 250,000 simulations via Markov Chain Monte Carlo (MCMC) and a burn-in of 500,000 iterations. The number of K populations was identified according to the method ∆K (Evanno et al. 2005), as implemented in the Structure Harvester program (Earl and Vonholdt 2012). We used the program Arlequin 3.5 (Excoffier and Lischer 2010) for the analysis of molecular variance (AMOVA) to understand how genetic variation is partitioned within and among clusters (according to Bayesian analysis), using 10,000 permutations to test for significance.
Subsequently, a fully Bayesian clustering approach was implemented in the program Barrier 2.2 (Manni et al. 2004) to identify any potential discontinuity of genetic data across the geographical area. The sampled populations were connected by Delaunay's triangulation according to their geographical coordinates. Monmonier's algorithm was implemented to identify zones with the greatest genetic differences (Ds).
Environmental variables
BIOCLIM variables (Booth et al. 2014) included in the model to predict the availability of suitable environments for the species were obtained from the WorldClim database, version 2.0 (worldclim.org/; Fick and Hijmans 2017). Climate projections (average for 2061-2080) were downloaded from WorldClim version 1.4 (Hijmans et al. 2005). Projections were based on the representative concentration pathway 8.5 or ‘business as usual’ scenario (Riahi et al. 2011) from the Earth system configuration of the 2nd Hadley Centre Global Environmental Model (HadGEM2-ES, Collins et al. 2011). Climate distributions were projected at a spatial resolution of 30 arc-s (~ 1 km2). To derive a model with a reduced set of variables, we used Pearson's correlation coefficient for each pairwise comparison to eliminate highly correlated, redundant variables (r ≥ 0.85 or r ≤ − 0.85, Table S1), with the program ENMTools 1.4.3 (Warren et al. 2010). Then, a reduced final set of six current bioclimatic variables that maximized training gain (Quipildor et al. 2018) and the area under the curve (AUC) were utilized, based on the preliminary MaxEnt model (Table S1).
Niche modeling
We obtained C. prunifera occurrence records (n = 35) using self-collected data and from Brazil’s speciesLink network (splink.cria.org.br; Canhos et al. 2015), an e-infrastructure that provides free and open access to primary biodiversity data and associated tools. Errors, duplicates, and records of cultivated plants were identified and eliminated inside a geographic area of approximately 260,500 km2, in order to avoid bias caused by uneven sampling. The distribution model to predict the availability of suitable environments for the species was obtained using the machine-learning maximum entropy model, Maxent version 3.4.1 (Phillips and Dudík 2008). Ten replicates of multiple runs of cross-validation were used, in which the occurrence data are randomly divided into a number of equal-sized groups (Phillips and Dudík 2008). As a threshold, we chose the 10th percentile training presence to optimize the correct discrimination between presence and pseudo-absences in the test data, using the raw output of Maxent (Merow et al. 2013). We explored a range of regularization coefficient values (1.0 to 5.0) to compare competing models (Merow et al. 2013). The Bayesian (BIC) and sample size corrected Akaike information criteria (AICc) were employed for model selection (Warren and Seifert 2011), showing that 2.0 was the most appropriate level of regularization (Table S2).
Detection of genetic bottlenecks
Recent reductions in effective population size were assessed using the Bottleneck program, version 1.2 (Cornuet and Luikart 1996). The Infinite Allele Model (IAM) and Stepwise Mutation Model (SMM), based on Kimura and Crow (1964) and Kimura and Otha (1978), respectively, were used to infer the presence of genetic bottlenecks. The mutation model of the ISSR loci is an intermediary between IAM and SMM (Luikart et al. 1998), thus we used both models. The sign test was applied (α = 0.05) based on the frequency of alleles to determine the existence of recent, significant genetic bottlenecks (Cornuet and Luikart 1996).
Results
Genetic polymorphism
The seven selected primers amplified 101 loci. The number of loci per primer ranged from 13 to 18 with an average of 14.4 (Table 2). The PIC of each primer used varied from 0.339 to 0.446, with an average of 0.418.
Genetic diversity
The percentage of polymorphic loci of the populations ranged from 16.83% in SER to 79.21% in SMG. The mean Nei’s genetic diversity (h) was 0.213, the mean Bayesian genetic approach (hs) was 0.236, and the Shannon index (I) was 0.312 (Table 3). The estimates of hs based on Bayesian approach were less variable (Coefficient of Variation = 19.89%) than Nei's genetic diversity h (CV = 36.30%) and Shannon index I (CV = 36.11%) (Fig. S2).
We found a positive and significant correlation between estimates of h and hs (rPearson = 0.986; P < 0.0001), between estimates of h and I (rPearson = 0.999; P < 0.0001), and between hs and I (rPearson = 0.986; P < 0.0001). The populations SMG, MOS, ICA, and RUS presented higher values of Nei’s genetic diversity (h ≥ 0.280 Table 3). The Shannon index (I) showed that the SMG, MOS, ICA, AR1, and RUS populations have higher values (I ≥ 0.400).
The greatest genetic distance was between SMG and SER (0.581) according to Nei’s Ds (Table S3), and between APD and SER (0.657) according to theta-II genetic distance (Table S4). The smallest genetic distance was between AR1 and AR2 for both methods (Ds = 0.017; theta-II = 0.005). The mean Ds was 0.213 and the mean theta-II was 0.375.
Population genetic structure and ENM
According to Bayesian inference, the full statistical model had the smallest DIC (Table S5; Spiegelhalter et al. 2002). Thus, the analyses of genetic diversity (hs) and pairwise genetic differentiation among populations (theta-II) were run using the full statistical model.
The Mantel test revealed the existence of a positive and significant correlation between geographic and genetic distances using both Nei’s (r = 0.423; P = 0.006) and theta-II genetic distance (r = 0.449; P = 0.003) (Fig. 2). C. prunifera populations are geographically structured and the results obtained from Bayesian analysis suggest the existence of three genetic groups (ΔK = 3; Fig. 3); this structure is congruent with the UPGMA dendrogram and Bayesian subdivisions (Fig. 4).

Relationship between geographic distances and Nei’s genetic distance (A) and theta-II genetic distance (B) for Copernicia prunifera populations

Plot of the mean values of LnP (D) of the Bayesian analysis (solid line) and ΔK analysis (dotted line). The bars indicate standard deviations of LnP (D) values

UPGMA dendrogram based on Nei’s genetic identity (left). Bootstrap values, when ≥ 50%, are given at each of the forks in the dendrogram. Bayesian analysis with the proportion of genotypes in the sampled populations (right), whereas the dark horizontal lines delimit populations. SE – Southeast (red); NC—North Coast (blue); NW—Northwest groups (green)
The AMOVA indicated the existence of significant population structure, with 14.61% variation among the Northwest, North Coast, and Southeast groups (\(\Phi_{{{\text{CT}}}}\), P = 0.005), 25.84% among populations within groups (\(\Phi_{{{\text{SC}}}}\), P < 0.0001), and 59.56% within populations (\(\Phi_{{{\text{ST}}}}\), P < 0.0001) (Table 4). The Southeast group had a smaller total h (0.151), hs (0.206), and I (0.221) than the Northwest group (h = 0.221; hs = 0.235; I = 0.324) and North Coast group (h = 0.281; hs = 0.285; I = 0.414).
The mapping of Ds using Delaunay's triangulation showed three genetic discontinuities (barriers) that separated even geographically proximal populations, as follows: (1) SER and LGP; (2) MAC; (3) ICA, SMG, AR1, AR2, RUS, MOS, APD and JUC, as shown in Fig. 1 and Fig. S3. The identified genetic discontinuities correspond to the most unfavourable geographical range for the species according to niche modelling (barrier a-a, Fig. 1c and d) and to altitudinal gradients (barriers b–b and c–c, Fig. 1b). According to the ENM analyses, the most favourable region for the occurrence of C. prunifera is in the Northwest of the sample area (Fig. 1c). The species does not grow well at high altitude, where the current range was identified as unsuitable for the species (Fig. 1b and c). The environmental variables that most influenced the current range were minimum temperature of coldest month (bio06) and mean temperature of warmest quarter (bio10) (Table S6). For the future scenario, the most influential variables were bio06, and the annual temperature range (bio07). In the future scenario, the extent of potentially suitable habitat for C. prunifera within its native range is reduced (Fig. 1d).
Genetic bottlenecks
Populations SER, MAC, JUC, APD, and RUS revealed a highly significant deficit in heterozygosity under both IAM and SMM models, thus demonstrating the occurrence of population bottlenecks (Table 5). MOS, ICA, and AR1 populations showed a significant bottleneck based on the IAM model and only the LGP population showed a significant genetic bottleneck based on the SMM model. Populations AR2 and SMG demonstrate equilibrium between mutation and drift.
Discussion
The markers used in the present study were moderately informative (Botstein et al. 1980), with PIC values ranging from 0.339 to 0.446. We found a high percentage of polymorphic loci for the whole population (99.09%), which demonstrates that the ISSR molecular markers used in this study are effective for estimating genetic diversity. ISSR markers have been used successfully in recent studies of genetic diversity (Pádua et al. 2021; Torres-Silva et al. 2021). Based on AMOVA, greater genetic variation occurred within than among populations. However, the genetic differentiation among populations was relatively high (\(\Phi_{{{\text{ST}}}} = 0.371;\;37.1\%\)) according to the expectations for species with similar life-history traits (Nybom 2004), and likely related to the large geographical distances between populations as discussed below.
Historical range and recent changes to the size and distribution of populations can influence the diversity within and genetic differentiation between populations (da Silva et al. 2015). According to Monmonier’s algorithm, our analysis indicates that populations from the Southeast group (LGP, SER, and MAC) are more isolated than the other population groups, with less genetic diversity (Table 3) and were clustered by Structure as sharing genotypes (Fig. 1 and Fig. 3). The Bayesian analysis revealed that C. prunifera populations occurring in the most favourable region of the species’ geographical range showed the highest levels of genetic diversity (Northwest and North Coast groups). The likely absence of genetic discontinuities in the Northwest region and the indication that this is the most favourable area of the species’ range may have enabled the maintenance of high levels of genetic diversity in these populations. This finding is of particular interest for the understanding of the local adaptation of C. prunifera populations and to make conservation decisions, since the genetically informed ecological niche models (gENMs) improve the predictions of species distributions under ongoing climate change (Ikeda et al. 2017).
The high suitability in the Northwest and the average suitability in the Southeast can be explained by the native range. C. prunifera populations generally occur at river valleys (Fig. 1c; green lines) and seasonally flooded areas in the semi-arid. Furthermore, the Northwest populations belong to the Caatinga biome, a seasonally dry tropical forest. On the other hand, the populations in the Southeast are influenced by the Atlantic Forest biome, a rainforest. The humidity coming from the ocean currents of the Atlantic Ocean (Xie and Carton 2004) added to the presence of the Atlantic Forest (da Silva and Tabarelli 2000) probably are not enough to provide high suitability for the wide distribution of the species in the Southeast of the sample area. However, in the future scenario, the extent of potentially suitable habitat for C. prunifera within its native range is reduced, mainly in the coastal region of the Northwest and Southeast occurrence area (Fig. 1d), which is also subject to the greatest anthropogenic pressure (e.g. urban and agricultural expansion, wind power plants) from human populations (Scarano and Ceotto 2015).
The Mantel test confirms that the most geographically remote sampled populations were also less genetically similar. Nei’s (1978) standard genetic distance between populations had an average of 0.21, which is high for species with animal-ingested seed dispersal mechanisms (\({\text{G}}_{{{\text{ST}}}} = 0.16\) ; Nybom 2004). Although bats are potential dispersers (Sousa et al. 2015), C. prunifera individuals present an aggregated spatial pattern and spatial genetic structure up to 12.3 m which may be related to restricted seed dispersal (Pinheiro et al. 2017a). The greatest genetic similarity was found between populations AR1 and AR2, and between RUS and MOS, which are geographically proximal to each other and belong to the Northwest group. Despite the considerable geographic distance between the ICA and SMG populations, they are nearest the coast and grouped by both the dendrogram and Bayesian analysis. However, phylogeographic data are necessary to better understand the colonization history of the species in different habitats (e.g. Zhang et al. 2020).
Alongside potential future reductions in habitat suitability, as well as overexploitation and anthropogenic disturbances, it is essential to identify populations that have undergone reductions in effective population size to understand the risks of possible local extinction due to reduced population size (Cobo-Simón et al. 2020). A reduction in effective population size may lead to a reduction in genetic diversity within populations, likely as a result of genetic drift after demographic bottlenecks (Jacquemyn et al. 2009), especially given the predicted reduction in suitable habitat for C. prunifera under ongoing climate change. Most of the populations showed a genetic bottleneck (Table 5), which is likely due to the significant anthropogenic pressure related to intense exploitation of carnauba wax in these areas since the 18th Century, as well as deforestation for the expansion of agriculture (D’alva 2004; Sousa et al. 2015). Although the SMG population showed no evidence of a recent bottleneck, it is currently affected by extensive anthropogenic impacts due to the expansion of wind power generation and the occurrence of fires in the neighbouring vicinity (personal observations), which may result in future genetic bottlenecks.
Although C. prunifera is not currently listed as an endangered species (Martinelli and Moraes 2013), it has been substantially affected by the expansion of agricultural activities over time, contributing to reductions in its natural populations (D’alva 2004; Sousa et al. 2015). In addition to recent reductions in population size and loss of diversity, we can infer that the studied populations have high genetic divergence, indicating current genetic isolation. Consequently, conservation measures for natural C. prunifera populations are needed to minimize further loss of alleles and to ensure sustainability resources for traditional communities. While herein we assessed neutral diversity, parallel losses in functional diversity might have consequences for the future of the species as its environment continues to change. Climate change will have profound effects on the semi-arid region (Marengo et al. 2017; Pinheiro et al. 2017b), and alterations in the potentially suitable habitats showed in our study should be considered (Fig. 1). In addition to in situ conservation of natural populations, and given the substantial economic importance of this species, one strategy would be the creation of germplasm banks for ex situ conservation, with seeds coming from the most diverse populations. Since the seeds are recalcitrant (Araújo et al. 2013), we recommend in vivo germplasm banks. Another approach could include the preservation of several populations across the geographic distribution of the species, considering the divergent genetic groups identified herein.
In order to avoid or minimize the deleterious effects of bottlenecks observed in most populations, one approach to mitigation would be to enhance gene flow between populations (Luikart et al. 1998). However, given the likely interaction between genetic and demographic decline, we suggest that in situ conservation to induce natural regeneration is a priority. Nevertheless, most of the populations are likely to be subjected to limitations in terms of palm establishment, for example due to NTFP extraction and soil compaction and trampling through animal husbandry. Consequently, management strategies should also focus on practical measures to improve regeneration success, such as pausing extractive activity during reproductive periods and introducing rotation cycles for leaf harvesting to recover over-exploited areas. Also, there is a need to consider the current social and economic conditions of harvesters to reach successful ‘social’ forests (Pritchard and Brockington 2019). This means that harvesters in poorer areas need additional support, including longer-term investments, to keep the equilibrium between the socioeconomic demand and forest conservation (Poudyal et al. 2018; Oldekop et al. 2019). These strategies can occur alongside the development of productive C. prunifera forests to support a more sustainable resource supply by reducing pressure from wild harvesting. The sustainable management of non-timber C. prunifera products is urgently needed to limit the negative impacts resulting from the deforestation of these populations which can contribute to developing a sustainable supply that can provide financial income for rural communities into the future.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 14:1811–1820. https://doi.org/10.1111/j.1365-294X.2008.03971.x
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36:181–186. https://doi.org/10.1139/g93-024
Araújo MRG, Melo Júnior AF, Menezes EV, Brandão MM, Cota LG, Oliveira DA, Royo VA, Vieira FA (2017) Fine-scale spatial genetic structure and gene flow in Acrocomia aculeata (Arecaceae): analysis in an overlapping generation. Biochem Syst Ecol 71:147–154. https://doi.org/10.1016/j.bse.2017.02.005
Araújo LHB, Silva RAR, Dantas EX, Sousa RF, Vieira FA (2013) Germinação de sementes da Copernicia prunifera: biometria, pré-embebição e estabelecimento de mudas. Enciclopédia Biosfera 9(17):1517–1528
Arruda GMT, Calbo MER (2004) Effects of flooding on carnaúba growth, gas exchange and root porosity (Copernicia prunifera (Mill.) H.E. Moore). Acta Bot Bras 18(2):219–224. https://doi.org/10.1590/S0102-33062004000200002
Bacles CFE, Lowe AJ, Ennos RA (2004) Genetic effects of chronic habitat fragmentation on tree species: the case of Sorbus aucuparia in a deforested Scottish landscape. Mol Ecol 13(3):573–584. https://doi.org/10.1046/j.1365-294X.2004.02093.x
Booth TH, Nix HA, Busby JR, Hutchinson MF (2014) BIOCLIM: the first species distribution modelling package, its early applications and relevance to most current MAXENT studies. Divers Distrib 20(1):1–9. https://doi.org/10.1111/ddi.12144
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Canhos DAL, Sousa-Baena MS, de Souza S, Maia LC, Stehmann JR, Canhos VP, De Giovanni R, Bonacelli MBM, Los W, Peterson AT (2015) The importance of biodiversity e-infrastructures for megadiverse countries. Plos Biol 13:1–7. https://doi.org/10.1371/journal.pbio.1002204
Cobo-Simón I, Méndez-Cea B, Jump AS, Seco J, Gallego FJ, Linares JC (2020) Understanding genetic diversity of relict forests. Linking long-term isolation legacies and current habitat fragmentation in Abies pinsapo Boiss. For Ecol Manag 461:117947. https://doi.org/10.1016/j.foreco.2020.117947
Coelho ASG (2001) Bood-P, dendrogram assessment based on distances/genetic similarities estimates through the bootstrap procedure. Universidade Federal de Goiás, Goiânia
Collins WJ, Bellouin N, Doutriaux-Boucher M, Gedney N, Halloran P, Hinton T et al (2011) Development and evaluation of an Earth-SYSTEM model–HadGEM2. Geosci Model Dev 4(4):1051–1075. https://doi.org/10.5194/gmd-4-1051-2011
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
D’alva OA (2004) O extrativismo da carnaúba no Ceará. Dissertation, Universidade Federal do Ceará
da Silva VDPR (2004) On climate variability in Northeast of Brazil. J Arid Environ 58(4):575–596. https://doi.org/10.1016/j.jaridenv.2003.12.002
da Silva JMC, Tabarelli M (2000) Tree species impoverishment and the future flora of the Atlantic forest of Northeast Brazil. Nature 404(6773):72–74. https://doi.org/10.1038/35003563
da Silva CC, Ribeiro MC, Cortes MC, Galetti M, Collevatti RG (2015) Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm. Heredity 115:216–224. https://doi.org/10.1038/hdy.2015.30
DeFries R, Hansen A, Newton AC, Hansen MC (2005) Increasing isolation of protected areas in tropical forests over the past twenty years. Ecol Appl 15:19–26. https://doi.org/10.1890/03-5258
Degen B, Roubik D (2004) Effects of animal pollination on pollen dispersal, selfing, and effective population size of tropical trees: a simulation study. Biotropica 36(2):165–179. https://doi.org/10.1111/j.1744-7429.2004.tb00309.x
Dias EF, Moura M, Schaefer H, Silva L (2016) Geographical distance and barriers explain population genetic patterns in an endangered island perennial. AoB Plants. https://doi.org/10.1093/aobpla/plw072
Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus 12(1):13–15
Duarte JF, Carvalho D, Vieira FA (2015) Genetic conservation of Ficus bonijesulapensis R.M. Castro in a dry forest on limestone outcrops. Biochem Syst Ecol 59:54–62. https://doi.org/10.1016/j.bse.2015.01.008
Earl DA, Vonholdt BM (2012) Structure Harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climatol 37:4302–4315. https://doi.org/10.1002/joc.5086
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Holsinger KE (1999) Analysis of genetic diversity in geographically structured populations: a Bayesian perspective. Hereditas 130:245–255. https://doi.org/10.1111/j.1601-5223.1999.00245.x
Holsinger KE, Lewis PO (2007) Hickory: a package for analysis of population genetic data v1.1. University of Connecticut, Mansfield
Holsinger KE, Wallace LE (2004) Bayesian approaches for the analysis of population genetic structure: an example from Platanthera leucophaea. Mol Ecol 13:887–894. https://doi.org/10.1111/j.1365-294X.2004.02052.x
Honnay O, Jacquemyn H (2007) Susceptibility of common and rare plant species to the genetic consequences of habitat fragmentation. Conserv Biol 21:823–831. https://doi.org/10.1111/j.1523-1739.2006.00646.x
IBGE —Instituto Brasileiro de Geografia Estatística. SIDRA—Banco de tabelas estatísticas. 2018. Avaible https://sidra.ibge.gov.br/pesquisa/pevs/quadros/brasil/2018. Accessed date: 31 May 2020
Ikeda DH, Max TL, Allan GJ, Lau MK, Shuster SM, Whitham TG (2017) Genetically informed ecological niche models improve climate change predictions. Glob Change Biol 23(1):164–176. https://doi.org/10.1111/gcb.13470
Jacquemyn H, Brys R, Adriaens D, Honnay O, Roldán-Ruiz I (2009) Effects of population size and forest management on genetic diversity and structure of the tuberous orchid Orchis mascula. Conserv Genet 10:161–168. https://doi.org/10.1007/s10592-008-9543-z
Jump AS, Penuelas J (2006) Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci 103(21):8096–8100. https://doi.org/10.1073/pnas.0510127103
Kimura M, Crow J (1964) The number of alleles that can be maintained in a finite population. Genetics 49(4):725–738
Kimura M, Otha T (1978) Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci 75(6):2868–2872
Laikre L, Allendorf FW, Aroner LC et al (2010) Neglect of genetic diversity in implementation of the convention on biological diversity. Conserv Biol 24:86–88. https://doi.org/10.1111/j.1523-1739.2009.01425.x
Lima MM, Sarquis O (2008) Is Rhodnius nasutus (Hemiptera; Reduviidae) changing its habitat as a consequence of human activity? Parasit Res 102:797–800. https://doi.org/10.1007/s00436-007-0823-1
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89(3):238–247. https://doi.org/10.1093/jhered/89.3.238
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol 76:173–190. https://doi.org/10.1353/hub.2004.0034
Marengo JA, Torres RR, Alves LM (2017) Drought in Northeast Brazil—past, present, and future. Theor Appl Climatol 129(3):1189–1200. https://doi.org/10.1007/s00704-016-1840-8
Martinelli G, Moraes MA (2013) Livro vermelho da flora do Brasil. Instituto de Pesquisas Jardim Botânico do Rio de Janeiro, Rio de Janeiro
Merow C, Smith MJ, Silander JA Jr (2013) A practical guide to MaxEnt for modeling species’ distributions: what it does, and why inputs and settings matter. Ecography 36(10):1058–1069. https://doi.org/10.1111/j.1600-0587.2013.07872.x
MMA (2005) Secretaria de Recursos Hídricos, Programa de ação nacional de combate à desertificação e mitigação dos efeitos da seca PAN-Brasil, Brasilia
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evolution 51:354–362. https://doi.org/10.1111/j.1558-5646.1997.tb02422.x
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155. https://doi.org/10.1111/j.1365-294X.2004.02141.x
Oldekop JA, Sims KR, Karna BK, Whittingham MJ, Agrawal A (2019) Reductions in deforestation and poverty from decentralized forest management in Nepal. Nat Sustain 2(5):421–428. https://doi.org/10.1038/s41893-019-0277-3
Omondi SF, Odee DW, Ongamo GO, Kanya JI, Khasa DP (2016) Genetic consequences of anthropogenic disturbances and population fragmentation in Acacia senegal. Conserv Genet 17(6):1235–1244. https://doi.org/10.1007/s10592-016-0854-1
Pádua JAR, Rocha LF, Brandão MM, Vieira FA, Carvalho D (2021) Priority areas for genetic conservation of Eremanthus erythropappus (DC.) MacLeish in Brazil. Genet Resour Crop Evol. https://doi.org/10.1007/s10722-021-01144-1
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Phillips SJ, Dudík M (2008) Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31:161–175. https://doi.org/10.1111/j.0906-7590.2008.5203.x
Pinheiro LG, Chagas KPT, Freire ASM, Ferreira MC, Fajardo CG, Vieira FA (2017a) Anthropization as a determinant factor in the genetic structure of Copernicia prunifera (Arecaceae). Genet Mol Res. https://doi.org/10.4238/gmr16039768
Pinheiro EAR, Van Lier QDJ, Bezerra AHF (2017b) Hydrology of a water-limited forest under climate change scenarios: the case of the Caatinga Biome. Braz for 8(3):62. https://doi.org/10.3390/f8030062
Poudyal M, Jones JPG, Rakotonarivo OS, Hockley N, Gibbons JM, Mandimbiniaina R, Rasoamanana A, Andrianantenaina NS, Ramamonjisoa BS (2018) Who bears the cost of forest conservation? PeerJ 6:e5106. https://doi.org/10.7717/peerj.5106
Pritchard R, Brockington D (2019) Regrow forests with locals’ participation. Nature 569(7758):630–631. https://doi.org/10.1038/d41586-019-01664-y
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Provan J, Beatty GE, Hunter AM, McDonald RA, McLaughlin E, Preston SJ, Wilson S (2008) Restricted gene flow in fragmented populations of a wind-pollinated tree. Conserv Genet 9:1521–1532
Quipildor VB, Kitzberger T, Ortega-Baes P, Quiroga MP, Premoli AC (2018) Regional climate oscillations and local topography shape genetic polymorphisms and distribution of the giant columnar cactus Echinopsis terscheckii in drylands of the tropical Andes. J Biogeogr 45(1):116–126. https://doi.org/10.1111/jbi.13106
Reddy MP, Sarla N, Siddiq EA (2002) Inter simple sequence repeat (ISSR) polymorphism and its application plant breeding. Euphytica 128:9–17. https://doi.org/10.1023/A:1020691618797
Riahi K, Rao S, Krey V, Cho C, Chirkov V, Fischer G et al (2011) RCP 8.5—A scenario of comparatively high greenhouse gas emissions. Clim Change 109(1–2):33–57. https://doi.org/10.1007/s10584-011-0149-y
Rocha TGF, Silva RAR, Dantas EX, Vieira FA (2015) Phenology of Copernicia prunifera (Arecaceae) in a caatinga area of Rio Grande do Norte. Cerne 21:673–681. https://doi.org/10.1590/01047760201521041758
Rohlf FJ (1993) Numerical taxonomy and multivariate analysis system. Exeter Software, New York
Rosa Jd, Weber GG, Cardoso R, Górski F, Da-Silva PR (2017) Variability and population genetic structure in Achyrocline flaccida (Weinm.) DC., a species with high value in folk medicine in South America. PLoS ONE 12(8):e0183533. https://doi.org/10.1371/journal.pone.0183533
Rouger R, Jump AS (2014) A seascape genetic analysis reveals strong biogeographical structuring driven by contrasting processes in the polyploid saltmarsh species Puccinellia maritima and Triglochin maritima. Mol Ecol 23:3158–3170. https://doi.org/10.1111/mec.12802
Santos AS, Cazetta E, Morante Filho JC, Baumgarten J, Faria D, Gaiotto FA (2015) Lessons from a palm: genetic diversity and structure in anthropogenic landscapes from Atlantic forest Brazil. Conserv Genet 16(6):1295–1302
Scarano FR, Ceotto P (2015) Brazilian Atlantic forest: impact, vulnerability, and adaptation to climate change. Biodivers Conserv 24(9):2319–2331. https://doi.org/10.1007/s10531-015-0972-y
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33. https://doi.org/10.1016/j.tree.2006.08.009
Sebbenn AM, Carvalho ACM, Freitas MLM, Moraes SMB, Gaino APSC, Silva JM, Jolivet C, Moraes ML (2011) Low levels of realized seed and pollen gene flow and strong spatial genetic structure in a small, isolated and fragmented population of the tropical tree Copaifera langsdorffii Desf. Heredity 106:134–145. https://doi.org/10.1038/hdy.2010.33
Seddon AW, Macias-Fauria M, Long PR, Benz D, Willis KJ (2016) Sensitivity of global terrestrial ecosystems to climate variability. Nature 531(7593):229–232. https://doi.org/10.1038/nature16986
Silva RAR, Fajardo CG, Vieira FA (2017) Mating system and intrapopulational genetic diversity of Copernicia prunifera (Arecaceae): a native palm from Brazilian semiarid. Genet Mol Res. https://doi.org/10.4238/gmr16039764
Silveira-Neto RM (2014) Crescimento pró-pobre no nordeste do Brasil: uma análise dos períodos (1991–2000 e 2000–2010). Estudos Econ 44(3):497–526. https://doi.org/10.1590/S0101-41612014000300003
Sousa RF, Silva RAR, Rocha TGF, Santana JAS, Vieira FA (2015) Ethnoecology and ethnobotany of the palm carnauba wax in Brazilian semi-arid. Cerne 21(4):587–594. https://doi.org/10.1590/01047760201521041764
Spiegelhalter DJ, Best NG, Carlin BP, van der Linde A (2002) Bayesian measures of model complexity and fit. J R Stat Soc Series B 64:583–639. https://doi.org/10.1111/1467-9868.00353
Tewksbury JJ, Levey DJ, Haddad NM, Sargent S, Orrock JL, Weldon A, Danielson BJ, Brinkerhoff J, Damschen EI, Townsend P (2002) Corridors affect plants, animals, and their interactions in fragmented landscapes. Proc Natl Acad Sci 99:12923–12926. https://doi.org/10.1073/pnas.202242699
Ticktin T (2004) The ecological implications of harvesting non-timber forest products. J Appl Ecol 41(1):11–21. https://doi.org/10.1111/j.1365-2664.2004.00859.x
Torres-Silva G, Schnadelbach AS, Bezerra HB, Lima-Brito A, Resende SV (2021) In vitro conservation and genetic diversity of threatened species of Melocactus (Cactaceae). Biodivers Conserv 30(4):1067–1080. https://doi.org/10.1007/s10531-021-02132-8
Warren DL, Glor RE, Turelli M (2010) ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33(3):607–611. https://doi.org/10.1111/j.1600-0587.2009.06142.x
Warren DL, Seifert SN (2011) Ecological niche modeling in Maxent: the importance of model complexity and the performance of model selection criteria. Ecol Appl 21(2):335–342. https://doi.org/10.1890/10-1171.1
Wolfe AD (2005) ISSR techniques for evolutionary biology. Molecular evolution: producing the biochemical data. Elsevier, Amsterdam, pp 134–144
Xie SP, Carton JA (2004) Tropical Atlantic variability: patterns, mechanisms, and impacts. Earth’s Climate: the Ocean-Atmosphere Interaction. Geophys Monogr 147:121–142. https://doi.org/10.1029/147GM07
Yeh FC, Yang RC, Boyle TBJ, Ye ZH, Mao JX (1997) PopGene, the user-friendly shareware for population genetic analysis. University of Alberta, Edmonton
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11(10):413–418. https://doi.org/10.1016/0169-5347(96)10045-8
Zacarías-Correa AG, Lira-Noriega A, Pérez-Calix E, Samain MS, Wolfe AD (2020) Back to the future of a rare plant species of the Chihuahuan desert: tracing distribution patterns across time and genetic diversity as a basis for conservation actions. Biodivers Conserv 29:1821–1840. https://doi.org/10.1007/s10531-020-01962-2
Zhang HX, Wang Q, Jia SW (2020) Genomic phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): insights into habitat fragmentation in Arid Northwestern China. Diversity 12(9):335. https://doi.org/10.3390/d12090335
Acknowledgements
The authors acknowledge the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for their financial support [Grant Number 471099/2012-0]. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. FAV was supported by a scholarship from CAPES, Programa de Pós-Doutorado no Exterior, [Grant Number 88881.118979/2016-01]. This research is registered in the Sistema Nacional de Gestão do Patrimônio Genético e do Conhecimento Tradicional Associado (SisGen) under code A0FBC35.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Communicated by Daniel Sanchez Mata.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
dos Santos, J.R.M., de Almeida Vieira, F., Fajardo, C.G. et al. Overexploitation and anthropogenic disturbances threaten the genetic diversity of an economically important neotropical palm. Biodivers Conserv 30, 2395–2413 (2021). https://doi.org/10.1007/s10531-021-02200-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-021-02200-z