
Abstract
Fragmentation of natural populations can have negative effects at the genetic level, thus threatening their evolutionary potential. Many of the negative genetic impacts of population fragmentation can be ameliorated by gene flow and it has been suggested that in wind-pollinated tree species, high or even increased levels of gene flow are a feature of fragmented populations, although several studies have disputed this. We have used a combination of nuclear microsatellites and allele-specific PCR (AS-PCR) analysis of chloroplast single nucleotide polymorphisms (SNPs) to examine the levels and patterns of genetic diversity and population differentiation in fragmented populations of juniper (Juniperus communis) in Ireland and inform conservation programs for the species. Significant population differentiation was found for both chloroplast and nuclear markers, indicating restricted gene flow, particularly over larger geographic scales. For conservation purposes, the existence of genetically distinct clusters and geographically localised chloroplast haplotypes suggests that the concept of provenance should be taken into account when formulating augmentation or reintroduction strategies. Furthermore, the potential lack of seed dispersal and seedling establishment means that ex-situ approaches to seed and seedling management may have to be considered.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat loss and fragmentation is one of the greatest threats to global biodiversity (Wilcox and Murphy 1985; Saunders et al. 1991). The potentially deleterious ecological effects of fragmentation on species and communities include changes in resource availability, reduction in population numbers and loss of connectivity leading to population isolation. Within species, fragmentation of natural populations can have negative effects at the genetic level, thus threatening their evolutionary potential (Young et al. 1996). Theoretical and empirical population genetic studies have predicted that fragmentation will lead to a loss of genetic diversity due to inbreeding (Keller and Waller 2002), population isolation and restricted gene flow (Schaal and Leverich 1996; Couvet 2002) and small effective population sizes (Ellestrand and Elam 1993) and that these may lead to a decline in fitness or even, ultimately, extinction (Newman and Pilson 1997; Frankham and Ralls 1998; Keller and Waller 2002).
Many of the negative genetic impacts of population fragmentation can be ameliorated by gene flow (Allendorf 1983). In tree species, which are generally believed to harbour high levels of within-population genetic diversity, studies on impacted populations have yielded conflicting results regarding the effects of fragmentation. It has been suggested using both direct and indirect estimates of gene flow that the removal of potential physical barriers to pollen movement allows for high or even increased levels of gene flow in wind-pollinated tree species (Foré et al. 1992; White et al. 2002; Dick et al. 2003; Bacles et al. 2005) but other studies have suggested that this is not always the case (Sork et al. 2002; Koenig and Ashley 2003; Jump and Peñuelas 2006).
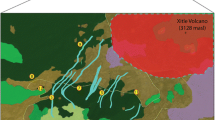
In this study, we have examined the genetic diversity in fragmented populations of juniper (Juniperus communis) in Ireland. Coniferous trees are generally highly heterozygous, outbreeding and wind-pollinated, and thus should exhibit high levels of intrapopulation genetic diversity but low levels of genetic differentiation between populations. Information on the genetic diversity of extant juniper populations and how this diversity is partitioned is important for conservation purposes, since many extant populations exhibit a highly fragmented distribution. This is exemplified by the distribution of juniper in Ireland, where the majority of populations are restricted to the extreme western regions of the island (Fig. 1). The species is one of only three native conifers in Britain, the others being yew (Taxus baccata) and Scots pine (Pinus sylvestris), and one of only two in Ireland since natural populations of Scots pine became extinct on the island several hundred years ago (Bradshaw and Browne 1987). Juniper exhibits a variety of morphological forms ranging from prostrate and creeping to erect, tree-like shrubs and two subspecies, ssp. communis and ssp. nana, are currently believed to exist in the UK, although previous molecular and biochemical analyses failed to discriminate between the two (Vines 1998; Filipowicz et al. 2006). Plants are dioecious, with wind-pollinated female cones, or “berries”, producing seeds that are primarily dispersed by birds. Despite the potential for high levels of dispersal of both pollen and seeds, the species has shown a serious reduction in distribution across the UK and Ireland and populations are believed to have declined by up to 60% since 1960 (Ward 1973; Preston et al. 2002; Thomas et al. 2007). This decline can be attributed to a wide range of factors including climate change, intensification of agriculture, especially grazing, and urbanisation (Clifton et al. 1997; Sanz-Elorza et al. 2003; Verheyen et al. 2005). Recruitment levels appear to be low, with a recent survey of juniper in Northern Ireland finding an age structure highly skewed towards mature and old trees with very little evidence of berries (Preston et al. 2007). As a consequence of population decline, juniper is protected under Section 8 of the Wildlife and Countryside Act in Britain and corresponding legislation in Northern Ireland.

Map showing locations of populations sampled. Inset map shows distribution of juniper in Ireland
The aims of the present study were to use biparentally inherited nuclear markers and paternally inherited chloroplast markers (Neale and Sederoff 1989; Neale et al. 1991; Wagner 1992) to elucidate the levels and patterns of genetic diversity in juniper in Ireland to inform conservation and management strategies. We utilised a combination of nuclear microsatellites and a cheap, high-throughput method of analysing single nucleotide polymorphisms (SNPs) in the chloroplast genome to assess the effects of gene flow patterns in shaping the present-day genetic architecture of extant juniper populations throughout its known Irish range.
Materials and methods
Sampling and DNA extraction
Samples were obtained from 19 populations in 12 regions representing the majority of the distribution of juniper in Ireland (Table 1). Where sample numbers within populations are small, these reflect small numbers of accessible plants. Samples were stored at −20°C and DNA was extracted from needle tissue using the Qiagen DNeasy Plant Mini Kit, after an initial 8 min grinding at 30 Hz using a Retsch MM300 mixer mill. DNA was quantified visually on 1% agarose gels stained with ethidium bromide and diluted to a concentration of 50 ng μl−1 for subsequent PCR.
Nuclear microsatellite analysis
All samples were genotyped for nuclear microsatellite loci JC16, JC32 and JC35. Primer sequences and PCR protocols are given in Michalczyk et al. (2006). The other two primers described in the same paper, JC31 and JC37, could not be reliably amplified and thus were not used in the present study. PCR was carried out on an MWG thermal cycler in a total volume of 10 μl containing 100 ng genomic DNA, 10 pmol of 32P-end labelled forward primer, 10 pmol of reverse primer, 1× PCR reaction buffer (5 mM Tris–HCl (pH 9.1), 1.6 mM [NH4]2SO4, 15 μg/μl BSA), 2.5 mM MgCl2 and 0.5 U Taq polymerase (Genetix). Products were resolved on 6% denaturing polyacrylamide gels containing 1× TBE and 8 M urea after addition of 10 μl of 95% formamide loading buffer. Gels were run at 70 W constant power for 2 h, transferred to 3MM Whatman blotting paper and exposed to X-ray film overnight at −20°C. In all cases, previously analysed samples were included as controls to compare product sizes across gels.
Chloroplast single nucleotide polymorphism allele-specific PCR (SNP AS-PCR) analysis
An initial screen for chloroplast variation was carried out using a single individual from each of the populations studied (Table 2). The following eight regions were analysed: trnT-trnF (Taberlet et al. 1991); trnD-trnT, psbC-trnS (Demesure et al. 1995); atpH-atpI, atpI-rpoC2, petB-petD (Grivet et al. 2001); trnV intron (Wang et al. 2003); trnG-trnS (Zhang et al. 2005). PCR was carried out on a MWG Primus thermal cycler using the following parameters: initial denaturation at 94°C for 3 min followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min (48°C for petB-petD), extension at 72°C for 2 min and a final extension at 72°C for 5 min. PCR was carried out in a total volume of 20 μl containing 200 ng genomic DNA, 20 pmol of forward primer, 20 pmol of reverse primer, 1× PCR reaction buffer (7.5 mM Tris–HCl (pH9.0), 2.0 mM [NH4]2SO4, 5.0 mM KCl, 2.0 mM MgCl2) and 2.0 U BIOTOOLS DNA polymerase. Five micro litre PCR product was resolved on 1.5% agarose gels and visualised by ethidium bromide staining and the remaining 15 μl sequenced commercially (Macrogen, Korea). Sequences were aligned using the clustalw program in the BioEdit software package.
To facilitate inexpensive, large-scale genotyping of SNPs, mutations detected in the chloroplast sequences were converted into allele-specific PCR (AS-PCR) primer sets. These mainly used the nested competitive primer approach of Soleimani et al. (2003) but a pair of specific PCR primers was also used to screen for length variation in the trnT-trnD region using standard PCR. For nested competitive primer design, the selective primer was designed so that the 3′ nucleotide of the primer was the SNP position and had an annealing temperature of 58°C. Compatible flanking primers, also with annealing temperatures of 58°C, were designed approximately 100 bp upstream and downstream of the SNP. In total, five SNPs were assayed in all samples using these approaches (Table 2). The AS-PCR protocol was as follows: initial denaturation at 94°C for 3 min followed by 11 touchdown cycles of denaturation at 94°C for 60 s, annealing at 65°C for 60 s (−0.7°C per cycle), extension at 72°C for 60 s followed by 24 cycles of denaturation at 94°C for 60 s, annealing at 58°C for 60 s, extension at 72°C for 60 s and a final extension at 72°C for 5 min. PCR was carried out in a total volume of 10 μl containing 100 ng genomic DNA, 10 pmol of forward primer, 10 pmol of reverse primer, 10 pmol SNP-selective primer, 1× PCR reaction buffer (5 mM Tris–HCl (pH9.1), 1.6 mM [NH4]2SO4, 15 μg/μl BSA), 200 μM each dNTP, 2.5 mM MgCl2 and 0.5 U Taq polymerase (Genetix). The trnT-trnD PCR protocol was as follows: initial denaturation at 94°C for 3 min followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s, extension at 72°C for 30 s and a final extension at 72°C for 5 min. For both assays, PCR products were resolved on a 2% agarose gels and visualised by ethidium bromide staining.
Data analysis
Nuclear microsatellite allele sizes were scored using a 10 bp ladder and were checked by comparison with previously sized control samples. Levels of polymorphism measured as allelic richness (A R) and expected heterozygosity (H E) were calculated using the fstat software package (V2.9.3.2; Goudet 2001) and the popgene software package (V1.32; Yeh et al. 1997) respectively. Polymorphisms at the five chloroplast SNPs were combined to give multi-locus haplotypes. For both nuclear and chloroplast markers, interpopulation differentiation and differentiation between regions (see Table 1) were estimated from allele and haplotype frequencies using Φ-statistics, which give an analogue of F-statistics (Weir and Cockerham 1984) calculated within the analysis of molecular variance (AMOVA) framework (Excoffier et al. 1992), using the arlequin software package (V3.01; Excoffier et al. 2005). To facilitate comparisons with future studies, we also calculated a standardized value of population differentiation, F′ST(N), from the nuclear microsatellite data set, as this statistic is independent of the levels of variation detected within populations (Hedrick 2005). Population pairwise estimates of gene flow based on nuclear microsatellites were calculated using the private alleles method (Slatkin 1985; Barton and Slatkin 1986) as implemented in the genepop software package (V3.4; Raymond and Rousset 1995). Population pairwise F ST values were also calculated using genepop and significance of population differentiation was estimated using the genic differentiation option in genepop after sequential Bonferroni correction for multiple tests. To further identify possible spatial patterns of gene flow, the software package baps (V3.2; Corander et al. 2003) was used to identify clusters of genetically similar populations using a Bayesian approach. Ten replicates were run for all possible values of the maximum number of clusters (K) up to K = 19, the number of populations sampled in the study, with a burn-in period of 10,000 iterations followed by 50,000 iterations. Multiple independent runs always gave the same outcome. Finally, a test for isolation by distance (IBD; Rousset 1997) was carried out using a Mantel test to assess the relationship between genetic distance, measured as F ST/(1−F ST), and geographical distance in genepop.
Results
Levels of within-population genetic variation
The three microsatellite loci used in this study were moderately to highly polymorphic, with numbers of alleles ranging from 8 (JC16) to 35 (JC32). Within-population levels of expected heterozygosity averaged across loci ranged from 0.460 in the ROS population to 0.765 in the PNO population (Table 3). Levels of allelic richness averaged across loci ranged from 2.733 in the MOY population to 4.265 in the PNO population.
Analysis of a total of 4,735 bp of sequence from eight regions of the chloroplast genome in one individual per population revealed only four substitutions and two indel mutations (Table 2). AS-PCR analysis of these mutations in the complete sample gave rise to six haplotypes (Fig. 2). All populations were variable and within-population chloroplast diversity values ranged from 0.200 in the BAR population to 0.600 in the MOY population (Table 4).

Distribution of chloroplast AS-PCR haplotypes
Population structuring and levels of gene flow
The analysis of molecular variance (AMOVA) revealed significant differences between populations for both nuclear (Φ ST(N) = 0.0957; P < 0.001) and chloroplast (Φ ST(C) = 0.2491; P < 0.001) markers (Table 5). The standardised estimate of population differentiation based on nuclear microsatellite markers, F′ST(N), was 0.429. The three-level AMOVA suggested that the majority of between-population variation for nuclear markers was due to differences between regions (Φ CT(N) = 0.0755; P < 0.001) but that between-region differentiation was not a significant factor for chloroplast markers (Φ CT(C) = 0.0526; NS). Despite this, there was evidence of some geographical substructuring of chloroplast haplotypes: Haplotype 3 was found only in the three populations from the Mournes area in the northeast (CAS, GUL and ANN), Haplotype 4 was restricted to the far northwest populations (FAN and PNO) and Haplotype 6 was only found in one population (CAP) from the far southwest.
Levels of gene flow between pairs of populations calculated from private alleles at nuclear microsatellite loci ranged from 0.38 (GUL/BAR) to 3.95 (FAN/MON) with a mean value of 1.15 (Table 6) and a global value (i.e. across all populations) of 1.09. Although it has been suggested that calculation of Nm values gives an indirect estimate of historical, rather than contemporary, levels of gene flow, the approach has been widely used and comparison with other studies in outcrossing coniferous tree species may be informative (see Discussion). Over half (87 of 171) of the values were less than 1.00, which represents the theoretical threshold for population differentiation due to genetic drift (Wright 1951). 162 of 171 population-pairwise F ST values were significantly different from zero, with values ranging from 0.002 (CAP/CAR) to 0.453 (ROS/CUR) and a mean of 0.103. Six of the nine non-significant F ST values were between populations from the same region. No evidence for isolation by distance was detected.
The BAPS analysis identified nine genetic clusters (Fig. 3a). In general, populations from the same region were assigned to the same cluster with the exception of the Lough Derg populations, where the PMN population was assigned to a cluster of its own whereas the CAR population was grouped with the FAN and MON populations. The Voronoi tessellation (Fig. 3b) further highlights the spatial organisation of the genetic clusters, with clusters containing multiple populations usually comprising geographically proximal populations. The only exceptions to this are the grouping of the PNO and CUR populations, and the grouping of the CAR, FAN and MON populations as described above.

(a) Colour-coded assignment of populations to nine clusters using the BAPS software package. (b) Voronoi tessellation showing spatial organisation of populations in nine clusters delineated by BAPS. Colours as in (a)
Discussion
Allele-specific PCR as a tool for population and conservation genetics
To our knowledge, this represents the first population genetics study to utilise AS-PCR for high-throughput screening of SNP variation. SNP genotyping techniques range from simple, PCR-based assays that can be resolved on standard agarose gels such as PCR-RFLP, to more complicated methods requiring the use of fluorescently labelled primers and/or dideoxynucleotides and polyacrylamide gel or capillary electrophoresis such as single base extension (SBE) or allele-specific primer extension (ASPE; Morin et al. 2004). Although PCR-RFLP approaches are cheap and technically simple, only a small fraction of SNPs give rise to restriction site changes. Whilst not as amenable to multiplexing as other SNP assays and not as straightforward when applied to diploid nuclear genes, the three-primer AS-PCR technique allows reliable and cost-effective genotyping of organellar SNP variation for large-scale population genetic analyses, particularly where the SNP does not result in a restriction site gain or loss.
Population differentiation and restricted gene flow
Although levels of gene flow in outcrossing, wind-pollinated tree species such as juniper are expected to be high, the findings of the present study are contrary to this. Our value for population differentiation based on nuclear loci (Φ ST(N) = 0.0957) is slightly higher than the average value for outcrossing gymnosperm species (0.073) quoted by Hamrick and Godt (1996) but slightly lower than the mean value quoted for biparentally inherited markers in conifers (0.116) by Petit et al. (2005). The standardised value of population differentiation, F′ST(N), was much higher (0.429), reflecting the high levels of within-population variation detected by microsatellites. Population differentiation based on chloroplast markers was also high (Φ ST(C) = 0.2491). Previous studies using chloroplast markers in conifers have tended to find around 10% or less of the total genetic variation partitioned between populations (e.g. Provan et al. 1998 (Φ ST(C) = 0.032 in Pinus sylvestris); Vendramin et al. 2000 (R ST(C) ≈ 0.1 in Picea abies); Richardson et al. 2002 (Φ ST(C) = 0.046 in Pinus albicaulis); Robledo Arnuncio et al. 2005 (Φ ST(C) = 0.031 in Pinus sylvestris); Naydenov et al. 2005 (Φ ST(C) = 0.110 in Pinus banksiana), 2006 (Φ ST(C) = 0.061 in Pinus nigra)) and in cases where high levels of population differentiation have been reported, these inflated values tend to be the result of long-term isolation of populations (e.g. Vendramin et al. 1998 (Φ ST(C) = 0.254 in Pinus pinaster); Jaramillo-Correa et al. 2006 (Φ ST(C) = 0.295 in Picea chihuahuana)). Where data are available for both the nuclear and chloroplast genomes in gymnosperms, as is the case in this study, differentiation between populations is expected to be more marked for chloroplast markers than for nuclear markers (Ennos 1994; Hu and Ennos 1997, 1999). Empirical studies, however, have generally found comparable levels of differentiation in both classes of markers which have been attributed to the high dispersal capabilities of pollen in conifers (Dong and Wagner 1994; Latta and Mitton 1997; Viard et al. 2001; Ribeiro et al. 2002). In the present study, differentiation based on chloroplast markers (Φ ST(C) = 0.2491) was much higher than that calculated for nuclear markers (Φ ST(N) = 0.0957), which is consistent with the action of genetic drift on the smaller effective population size of the uniparentally transmitted, haploid chloroplast genome. The limited dispersal suggested by both the nuclear and chloroplast Φ ST values is also reflected in the baps analysis, which delineated nine genetic clusters that are largely congruent with the spatial organisation of populations studied.
To date, there have only been two published population-level genetic studies on Juniperus communis. Oostermeijer and de Knegt (2004) used allozymes to assess the levels and distribution of genetic diversity in twelve populations from fragmented heathlands in the Netherlands and found low (F ST(N) = 0.026) levels of population differentiation. In contrast, a study using AFLPs on eight populations from England and Wales suggested a high level of genetic structuring, although summary statistics for population differentiation (e.g. F ST/G ST/Φ ST) were not calculated (van der Merwe et al. 2000). Of particular note, though, is a study on the congeneric J. przewalskii, which revealed very high levels of differentiation using chloroplast markers (G ST = 0.772; Zhang et al. 2005). Although no evidence of isolation by distance was evident at the global scale (i.e. across all populations) in the present study, suggesting the predominance of genetic drift over gene flow, individual values for inter-population differentiation and gene flow suggest that there may be adequate gene flow at local scales to prevent population divergence. Six of the nine non-significant pairwise F ST values were between populations from the same geographical region and the average value of Nm between populations from the same region (1.94) was almost double that of the average figure between populations from different regions (1.09). Values of Nm in conifers tend to be much higher, with values of Nm > 3 being the norm (Ledig 1998). Although Nm values give an indication of historical gene flow, the decline in juniper populations over the last few hundred years means that these values probably overestimate contemporary levels of gene flow and thus the degree of connectivity between extant populations is even lower. Zhang et al. (2005) reported that field studies on J. tibetica revealed no wind-mediated pollen dispersal beyond 2 km and in all six cases in the present study where populations were separated by less than this distance (BRO versus TRI, GOR versus TRI, BRO versus GOR, GUL versus CAS, ANN versus CAS and GUL versus ANN), population-pairwise F ST values were non-significant. Seeds in juniper are primarily dispersed by thrushes of the genus Turdus (Livingston 1972; Snow and Snow 1988) but a study on thrush communities in fragmented Juniperus thurifera populations has suggested that a decrease in abundance of frugivorous birds from smaller patches of woodland has had a negative impact on dispersal and seedling recruitment (Santos and Telleria 1994; Santos et al. 1999). Taken together, the potentially limited capacity for dispersal within and between fragmented populations via both pollen and seeds may explain the high levels of genetic differentiation found in Irish juniper populations. Ennos (1994) described a method to calculate the relative rates of interpopulation seed and pollen flow using a combination of maternal and biparentally inherited markers. In conifers, this generally uses data from the mitochondrial genome since the chloroplast genome is almost always transmitted paternally, unlike in angiosperms where maternal inheritance of the chloroplast genome occurs in the vast majority of taxa. Such calculations are unlikely to be feasible for the present study, however, since previous evidence suggests that the mitochondrial genome may be paternally inherited in the Cupressaceae, which includes juniper. Neale et al. (1989) described paternal inheritance of mitochondrial DNA in the coast redwood Sequoia sempervirens and cytological studies have shown the cytoplasmic inheritance of paternal mitochondria in other members of the Cupressaceae (Camefort 1970; Chesnoy 1973).
Conservation implications
Juniper populations in both the UK and Ireland have been in decline for many years now and one of the goals of the Species Action Plan is to maintain and re-establish natural populations. Information from population genetic studies is now considered an integral part of conservation programmes (Haig 1998) and the findings of this study are particularly relevant to the conservation of juniper in Ireland, where populations tend to be highly fragmented. The relatively high levels of genetic differentiation between populations and the apparent geographical structuring of this variation coupled with the occurrence of geographically localized haplotypes suggest that the concept of provenance should be taken into account when formulating conservation strategies for Irish populations of juniper. One obvious starting point for the designation of distinct management units would be the genetic clusters identified by the BAPS analysis which tend to reflect the limited levels of gene flow at larger geographic scales as described above. Of particular note for conservation purposes are the populations from the Mournes area: the region is geographically distinct and isolated from the remainder of the populations in Ireland, which have a predominantly western distribution, and almost a third (18 of 59) plants studied from this area exhibited an endemic chloroplast haplotype.
One of the main perceived threats to juniper populations is the lack of recruitment from seed (reviewed in Thomas et al. 2007) and establishment of seedlings has been shown to be negatively affected by both grazing (Ward 1973; Gilbert 1980) and climatic factors (Rosen 1988, 1995; Garcia et al. 1999). Overgrazing may present a particular problem to many of the populations examined in this study, particularly those occurring in montane and rough pasture hillsides where effective fencing is problematic. Coastal populations tend to be out of the reach of many grazing animals but their persistence on cliff faces exposes them to windthrow and, consequently, many of these populations comprise limited numbers of stunted trees. These threats to seedling establishment are further exacerbated by low levels of seed viability coupled with limited dispersal. Verheyen et al. (2005) showed that only 3% of seeds collected from a managed nature reserve in Belgium were viable and seed viability may be even more limited for Irish populations, which have an age structure skewed towards mature and old plants, since older stands tend to have lower reproductive capacity (Dearnley and Duckett 1999; Preston et al. 2007). The limited dispersal suggested by the findings of the present study is reflected by field observations which suggest that thrushes responsible for seed dispersal in juniper tend to favour larger, berry-rich populations over smaller, isolated populations even where individual plants within smaller populations produce large numbers of berries (Garcia et al. 2001). Consequently, the small, isolated populations comprising mainly senescent plants examined in this study may be at particular risk of ongoing loss of diversity and extinction. Conservation efforts aimed at the maintenance and reintroduction of these populations may be most effective when ex-situ management of seed and seedlings is implemented.
References
Allendorf FW (1983) Isolation, gene flow and genetic differentiation among populations. In: Schonewalk-Cox CM, Chambers SM, MacBryde B, Thomas L (eds) Genetics and conservation. Benjamin-Cummings, London, pp 51–65
Bacles CFE, Burczyk J, Lowe AJ, Ennos RA (2005) Historical and contemporary mating patterns in remnant populations of the forest tree Fraxinus excelsior L. Evolution 59:979–990
Barton NH, Slatkin M (1986) A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 56:409–415
Bradshaw RHW, Browne P (1987) Changing patterns in the post-glacial distribution of Pinus sylvestris in Ireland. J Biogeogr 14:237–248
Camefort H (1970) Structural peculiarities of female gamete of Cryptomeria japonica - formation of plastes-reticulum complexes during maturation period of gamete. C R Hebd Seances Acad Sci D 271:49–52
Chesnoy L (1973) Paternal origin of organelles in Chamaecyparis lawsoniana A. Murr. proembryo (Cupressaceae). Caryologia 25(S):221–232
Clifton SJ, Ward LK, Ranner DS (1997) The status of juniper Juniperus communis L. in north-east England. Biol Conserv 79:67–77
Corander J, Waldmann P, Sillanpää MJ (2003) Bayesian analysis of genetic differentiation between populations. Genetics 163:367–374
Couvet D (2002) Deleterious effects of restricted gene flow in fragmented populations. Conserv Biol 16:369–376
Dearnley TC, Duckett JG (1999) Juniper in the Lake District National Park: a review of condition and regeneration. Watsonia 22:261–267
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–131
Dick CW, Etchelecu G, Austerlitz F (2003) Pollen dispersal of tropical trees (Dinizia excelsa: Fabaceae) by native insects and African honeybees in pristine and fragmented Amazonian rain forest. Mol Ecol 12:753–764
Dong JS, Wagner DB (1994) Paternally inherited chloroplast polymorphism in Pinus: estimation of diversity and population subdivision and tests of disequilibrium for a maternally inherited mitochondrial polymorphism. Genetics 136:1187–1194
Ellstrand NC, Elam DC (1993) Population genetic consequences of small population size—implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Ennos RA (1994) Estimating the relative rates of pollen and seed migration among plant populations. Heredity 72:250–259
Excoffier L, Laval LG, Schneider S (2005) ARLEQUIN, Version 3.0: an integrated software package for population genetic data analysis. Evol Bioinform Online 1:47–50
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial DNA restriction data. Genetics 131:479–491
Filipowicz N, Piotrowski A, Ochocka JR, Asztemborska M (2006) The phytochemical and genetic survey of common and dwarf juniper (Juniperus communis and Juniperus nana) identifies chemical races and close taxonomic identity of the species. Planta Med 72:850–853
Foré SA, Hickey RJ, Vankat JL, Guttman SI, Schaeffer RL (1992) Genetic structure after forest fragmentation: a landscape ecology perspective on Acer saccharum. Can J Bot 70:1659–1668
Frankham R, Ralls K (1998) Inbreeding leads to extinction. Nature 392:441–442
Garcia D, Zamora R, Gómez JM, Hódar JA (1999) Bird rejection of unhealthy fruits reinforces the mutualism between juniper and its avian dispersers. Oikos 85:536–544
Garcia D, Zamora R, Gómez JM, Hódar JA (2001) Frugivory at Juniperus communis depends more on population characteristics than on individual attributes. J Ecol 89:639–647
Gilbert OL (1980) Juniper in Upper Teesdale. J Ecol 68:1013–1024
Goudet J (2001) FSTAT, version 2.9.3, A program to estimate and test gene diversities and fixation indices. http://www2.unil.ch/popgen/softwares/fstat.htm
Grivet D, Heinze B, Vendramin GG, Petit RJ (2001) Genome walking with consensus primers: application to the large single copy region of chloroplast DNA. Mol Ecol Notes 1:345–349
Haig SM (1998) Molecular contributions to conservation. Ecology 79:413–425
Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond B 351:1291–1298
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Hu X-S, Ennos RA (1997) On estimation of the ratio of pollen to seed flow among plant populations. Heredity 79:541–552
Hu X-S, Ennos RA (1999) Impacts of seed and pollen flow on population genetic structure for plant genomes with three contrasting modes of inheritance. Genetics 152:441–450
Jaramillo-Correa JP, Beaulieu J, Ledig FT, Bousquet J (2006) Decoupled mitochondrial and chloroplast DNA population structure reveals Holocene collapse and population isolation in a threatened Mexican-endemic conifer. Mol Ecol 15:2787–2800
Jump AS, Peñuelas J (2006) Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci USA 103:8096–8100
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Koenig WD, Ashley MV (2003) Is pollen limited? The answer is blowin’ in the wind. Trends Ecol Evol 18:157–159
Latta RG, Mitton JB (1997) A comparison of population differentiation across four classes of gene marker in limber pine (Pinus flexilis James). Genetics 146:1153–1163
Ledig FT (1998) Genetic variation in Pinus. In: Richardson DM (ed) Ecology and biology of Pinus. Cambridge University Press, Cambridge, pp 251–280
Livingston RB (1972) Influence of birds, stones and soil on the establishment of pasture juniper Juniperus communis and red cedar J. virginiana in New England pasture communities. Ecology 53:1141–1147
Michalczyk IM, Sebastiani F, Buonamici A, Cremer E, Mengel C, Ziegenhagen B, Vendramin GG (2006) Characterization of highly polymorphic nuclear microsatellite loci in Juniperus communis L. Mol Ecol Notes 6:346–348
Morin PA, Luikart G, Wayne RK, the SNP Workshop Group (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216
Naydenov K, Tremblay MF, Fenton N (2005) Chloroplast microsatellite differentiation in jack pine (Pinus banksiana) populations in Quebec. Belg J Bot 138:181–191
Naydenov K, Tremblay MF, Fenton NJ, Alexandrov A (2006) Structure of Pinus nigra Arn. populations in Bulgaria revealed by chloroplast microsatellites and terpenes analysis: provenance tests. Biochem Syst Ecol 34:562–574
Neale DB, Sederoff RR (1989) Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theor Appl Genet 77:212–216
Neale DB, Marshall KA, Sederoff RR (1989) Chloroplast and mitochondrial DNA are paternally inherited in Sequoia sempervirens D. Don Endl. Proc Natl Acad Sci USA 86:9347–9349
Neale DB, Marshall KA, Harry DE (1991) Inheritance of chloroplast and mitochondrial DNA in incense cedar (Calocedrus decurrens). Can J For Res 21:717–720
Newman D, Pilson D (1997) Increased probability of extinction due to decreased effective population size: experimental populations of Clarkia pulchella. Evolution 51:354–362
Oostermeijer JGB, de Knegt B (2004) Genetic population structure of the wind-pollinated, dioecious shrub Juniperus communis in fragmented Dutch heathlands. Plant Species Biol 19:175–184
Petit RJ, Dumimil J, Fineschi S, Hampe A, Salvini D, Vendramin GG (2005) Comparatie organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701
Preston CD, Pearman DA, Dines TD (2002) New atlas of the British and Irish flora. Oxford University Press, UK
Preston SJ, Wilson C, Jennings S, Provan J, McDonald RA (2007) The status of juniper (Juniperus communis) in Northern Ireland in 2005. Ir Nat J 28:372–378
Provan J, Soranzo N, Wilson NJ, McNicol JW, Forrest GI, Cottrell J, Powell W (1998) Gene-pool variation in Caledonian and European Scots pine (Pinus sylvestris L.) revealed by chloroplast simple sequence repeats. Proc R Soc Lond B 265:1697–1705
Raymond M, Rousset F (1995) Genepop (version 1.2): population genetic software for exact tests and ecumenicism. J Hered 86:248–249
Ribeiro MM, Mariette S, Vendramin GG, Szmidt AE, Plomion C, Kremer A (2002) Comparison of genetic diversity estimates within and among populations of maritime pine using chloroplast simple-sequence repeat and amplified fragment length polymorphism data. Mol Ecol 11:869–877
Richardson BA, Brunsfield J, Klopfenstein NB (2002) DNA from bird-dispersed seed and wind-disseminated pollen provides insights into postglacial colonization and population genetic structure of whitebark pine (Pinus albicaulis). Mol Ecol 11:215–227
Robledo Arnuncio JJ, Collada C, Alia R, Gil L (2005) Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area. J Biogeogr 32:595–605
Rosen E (1988) Development and seedling establishment within a Juniperus communis stand on Oland, Sweden. Acta Bot Neerl 37:193–201
Rosen E (1995) Periodic droughts and long-term dynamics of Alvar grassland vegetation on Oland, Sweden. Folia Geobot Phytotaxon 30:131–140
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Santos T, Telleria JL (1994) Influence of forest fragmentation on seed consumption and dispersal of Spanish juniper Juniperus thurifera. Biol Conserv 70:129–134
Santos T, Telleria JL, Virgos E (1999) Dispersal of Spanish juniper Juniperus thurifera by birds and mammals in a fragmented landscape. Ecography 22:193–204
Sanz-Elorza M, Dana ED, Gonzalez A, Sobrino E (2003) Changes in the high-mountain vegetation of the central Iberian peninsula as a probable sign of global warming. Ann Bot 92:273–280
Saunders DA, Hobbs RJ, Margules CR (1991) Biological consequences of ecosystem fragmentation: a review. Conserv Biol 5:18–32
Schaal BA, Leverich WJ (1996) Molecular variation in isolated plant populations. Plant Species Biol 11:33–40
Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39:53–65
Snow BK, Snow D (1988) Birds and berries: a study of an ecological interaction. Poyser, Carton, Staffs, UK
Soleimani VD, Baum BR, Johnson DA (2003) Efficient validation of single nucleotide polymorphisms in plants by allele-specific PCR, with an example from barley. Plant Mol Biol Rep 21:281–288
Sork VL, Davis FW, Smouse PE, Apsit VJ, Dyer RJ, Fernandez JF, Kuhn B (2002) Pollen movement in declining populations of California valley oak, Quercus lobata: where have all the fathers gone? Mol Ecol 11:1657–1668
Taberlet P, Gielly L, Patou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Thomas PA, El-Barghathi M, Polwart A (2007) Biological flora of the British Isles: Juniperus communis L. J Ecol 95:1404–1440
van der Merwe M, Winfield MO, Arnold GM, Parker JS (2000) Spatial and temporal aspects of the genetic structure of Juniperus communis populations. Mol Ecol 9:379–386
Vendramin GG, Anzidei M, Madaghiele A, Bucci G (1998) Distribution of genetic diversity in Pinus pinaster Ait. as revealed by chloroplast microsatellites. Theor Appl Genet 97:456–463
Vendramin GG, Anzidei M, Madaghiele A, Sperisen C, Bucci G (2000) Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 43:68–78
Verheyen K, Schreurs K, Vanholen B, Hermy M (2005) Intensive management fails to promote recruitment in the last large population of Juniperus communis (L.) in Flanders (Belgium). Biol Conserv 124:113–121
Viard F, El-Kassaby YA, Ritland K (2001) Diversity and genetic structure in populations of Pseudotsuga menziesii (Pinaceae) at chloroplast microsatellite loci. Genome 44:336–344
Vines T (1998) The relationship between Juniperus communis ssp. communis and Juniperus communis ssp. nana as revealed by morphometrics and RAPD marker data. B.Sc. thesis, University of Edinburgh
Wagner DB (1992) Nuclear, chloroplast and mitochondrial DNA polymorphisms as biochemical markers in population genetic analyses of forest trees. New Forest 6:373–390
Wang WP, Hwang CY, Lin TP, Hwang SY (2003) Historical biogeography and phylogenetic relationships of the genus Chamaecyparis (Cupressaceae) inferred from chloroplast DNA polymorphism. Plant Syst Evol 241:13–28
Ward LK (1973) The conservation of juniper: present status of juniper in Southern England. J Appl Ecol 14:163–178
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
White GM, Boshier DH, Powell W (2002) Increased pollen flow counteracts fragmentation in a tropical dry forest: an example from Swietenia humilis Zuccarini. Proc Natl Acad Sci USA 99:2038–2042
Wilcox BA, Murphy DD (1985) Conservation strategy—the effects of fragmentation on extinction. Am Nat 125:879–887
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Yeh FC, Yang R-C, Boyle T, Ye Z-H, Mao JX (1997) POPGENE, the user-friendly shareware for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Canada. http://www.ualberta.ca/~fyeh/
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zhang Q, Chiang TY, George M, Liu JQ, Abbott RJ (2005) Phylogeography of the Qinghai-Tibetan Plateau endemic Juniperus przewalskii (Cupressaceae) inferred from chloroplast DNA sequence variation. Mol Ecol 14:3513–3524
Acknowledgements
The authors are extremely grateful to everybody who provided samples for this study, including Robin Govier, Clare Heardman, Stuart Jennings, Christine Maggs, Ian Montgomery, Declan O’Donnell, Neil Reid and Conor Wilson. We are particularly indebted to Mike Wyse-Jackson who continually supplied us with material from a variety of locations throughout the duration of the study. We are grateful to Valerie Hall, Tim Hipkiss, Peter McEvoy, Christine Maggs, Ian Montgomery, Staffan Roos and Mark Wright for helpful discussions and to Ian Montgomery, Rob Paxton and two anonymous referees and the Subject Editor for comments on an earlier version of the manuscript. This research was supported through the Quercus partnership between Queen’s University Belfast and the Environment and Heritage Service, Northern Ireland and by an Environmental Sciences Association of Ireland (ESAI) student bursary to Conor Wilson.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Provan, J., Beatty, G.E., Hunter, A.M. et al. Restricted gene flow in fragmented populations of a wind-pollinated tree. Conserv Genet 9, 1521–1532 (2008). https://doi.org/10.1007/s10592-007-9484-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-007-9484-y