
Abstract
Uebelmannia is a cactus genus represented by three microendemic species with patchy distributions in campo rupestre landscapes in the Espinhaço Range in eastern Brazil. It is one of the ten genera of Cactaceae listed as threatened with extinction due to habitat loss and illegal overcollection. Assessment of the genetic diversity and population structure of this threatened genus is crucial to provide guidelines for both in situ and ex situ conservation and management efforts. Here, we genotyped 12 microsatellite loci from samples covering the entire distribution of this genus (485 individuals from 20 localities) to investigate the genetic diversity, spatial population structure, and demography of Uebelmannia species. The results identified moderate-to-high levels of genetic diversity in Uebelmannia, comparable to the wide-range cacti from Cerrado biome. The results confirmed an extremely high population structure even at small geographic scales, with population clusters exhibiting high inbreeding and genetic signatures of a recent bottleneck. Based on this study, we suggest some conservation strategies, including in situ management for populations at the borders of protected areas and ex situ seed collection, for further management of this genus. Furthermore, the results suggest the use of a precautionary approach for translocation plans and highlight that effective conservation management of Uebelmannia should target genetically clustered populations instead of species or subspecies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The extraordinary biodiversity of campo rupestre (CR) landscapes in eastern Brazil has long drawn the attention of conservation biologists (e.g., Giulietti et al. 1997; BFG 2018). Recently, researchers interpreted CR as a bonafide representation of the old stable landscapes (Silveira et al. 2016; Mucina 2018) and were alarmed by the unprecedented impacts of human disturbances to these areas. This has led to increasing interest in research in various areas to investigate the plant life in this megadiverse and highly endemic vegetation complex (Morellato and Silveira 2018).
Campo rupestre is montane, grassy-shrubby mosaic vegetation occurring on rocky outcrops of quartzite sandstone or ironstone along with sandy, stony, and waterlogged grasslands (Silveira et al. 2016). The core distribution of CR occurs primarily on high plateaus and isolated mountain tops along the Espinhaço Range in eastern Brazil, with smaller disjunct areas found in the central Brazilian highlands. Although CR covers approximately 0.78% (66,447 km2, Silveira et al. 2016) of the land in Brazil, it harbors 14.7% of the Brazilian vascular flora, with c. 2000 endemic species (BFG 2018), of which 255 are at risk of extinction (Monteiro et al. 2018). Several environmental, ecological, and evolutionary patterns are observed in CR, such as strong environmental filters (nutrient-poor soils, seasonal droughts, and high irradiance; Fernandes 2016), high biodiversity and narrow endemism (Conceição et al. 2016), phylogenetic conservatism (Zappi et al. 2017), predominance of old lineages (Silveira et al. 2016), and species dispersal limitations (Morellato and Silveira 2018). These features have supported the existence of CR as an ancient and stable landscape (Silveira et al. 2016; Mucina 2018) based on OCBIL (old climatically buffered infertile landscape) theory (Hopper 2009). Considering the ancient and stable heterogeneous topography of CR, the diversification patterns found among CR taxa suggest that these landscapes contain climate (Bonatelli et al. 2014) and/or fire microrefugia (Conceição et al. 2016; Mucina 2018), museums of ancient lineages (Zappi et al. 2017) and cradles of continuing diversification of endemic lineages (Bitencourt and Rapini 2013). Taken together, these features establish CR as a priority area for the conservation of Brazilian flora (Loyola et al. 2014; Monteiro et al. 2018). An important challenge in the implementation of conservation efforts in CR is the high beta diversity due to the abundance of microendemic taxa, increasing the number of protect areas that need effective conservation.
Cactaceae is a common component of the xeromorphic phytophysiognomy of CR, usually growing on bare rock or white sandy soils. Of the 42 cactus species endemic to CR (Zappi and Taylor 2008), 28 from 10 genera are listed in the threatened categories of both the IUCN (IUCN 2018) and the Brazilian Red List floras (Martinelli and Moraes 2013), highlighting CR as a hotspot of threatened cacti (Goettsch et al. 2015). Among these threatened taxa, the genus Uebelmannia contains three microendemic species whose populations extend over an area of c. 8000 km2 in the southern Espinhaço Range. All three species occur in small and patchy populations and are categorized as critically endangered or endangered (IUCN 2018) and listed in Appendix I of the Convention on International Trade and Endangered Species as a direct consequence of their rarity and illegal overcollection (Zappi and Taylor 2008). In addition, Uebelmannia is the only remaining taxon representing the early-divergent lineage within Cactaceae, sister to the clade giving rise to most of the Brazilian cactus diversity (Hernández-Hernández et al. 2011). This phylogenetic distinctiveness increases the importance of its conservation even further. Concerning the level of threats and the particularities of the different components of Brazilian cactus diversity (e.g., high taxonomic richness, endemicity, ecological singularity, rarity, and conservation status), since 2011, the Chico Mendes Institute for Biodiversity Conservation (ICMBio, Brazilian government) has implemented the National Action Plan for the Conservation of Cacti (PAN Cactaceae; Ribeiro-Silva et al. 2011) together with Brazilian researchers. Members of the genus Uebelmannia inhabiting CR landscapes in eastern Brazil represent one of the threatened taxa included in the PAN Cactaceae.
Among the three species of this genus, Uebelmannia pectinifera is the one with the broadest distribution, occurring on the western side of the southern Espinhaço Range. This species is subdivided into three subspecies, the nominate form, U. pectinifera subsp. flavispina, and U. pectinifera subsp. horrida, a taxon formerly known from only a single locality on the northernmost limit of the species distribution. Three new localities of U. pectinifera subsp. horrida were recently disclosed by G. Olsthoorn (pers. comm.) and explored in this work, showing that the distribution range of this taxon extends to the south into the Sempre Vivas National Park. Uebelmannia gummifera occurs on the eastern side of the southern Espinhaço Range, with two subspecies, the nominate form and U. gummifera subsp. meninensis. Uebelmannia buiningii has the narrowest range in the genus, occurring in a few populations adjacent to the U. gummifera range. All three species are characterized by having solitary, globose or cylindrical stems bearing yellow flowers apically, and diurnal flowers that attract hymenopterans as pollinators (Schulz and Machado 2000). Recently, Teixeira et al. (2018) reported that U. buiningii is a self-incompatible species and does not form fruits or seeds without pollination, with two bee species (Dialictus opacus and Plebeia sp.) acting as effective pollinators. Similarly, one of the authors of the current study (L.Y.S.A., pers. comm., unpublished results) has investigated the reproductive biology of U. pectinifera and also observed that it is a self-incompatible species.
To provide guidelines for the conservation of the Uebelmannia genus, we used nuclear microsatellite markers to assess the level and distribution of genetic diversity, level of inbreeding, and recent bottlenecks across its entire range of distribution. The main objectives of the study were to address the following questions: (i) Are Uebelmannia populations experiencing genetic erosion due to their narrow and patchy distribution? (ii) What is the level of spatial genetic structure, and does it agree with the taxonomic divisions within this genus? Finally, based on the results of this study, we proposed management guidelines for this microendemic, phylogenetically important and endangered cactus genus.
Materials and methods
Sampling
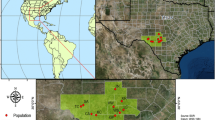
We collected root tips of 485 reproductively mature plants from 20 different localities of U. buiningii (n = 67), U. gummifera (n = 164), and U. pectinifera (n = 254). The sampling strategy was to cover the entire taxonomic diversity and distributional range of the whole Uebelmannia genus (Table 1; Fig. 1). Sampling in the protected areas of Serra Negra State Park and Sempre Vivas National Park was carried out in accordance with Brazilian law through special permits provided to one of the authors of the current study (E.M.M.) by the Minas Gerais State Forestry Institute (permit number COL-073/11) and the Chico Mendes Biodiversity Conservation Institute (permit number 28464), respectively. Genomic DNA was extracted using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) and quantified using 1% agarose gel.

Map of natural occurrence and sampling localities of the genus Uebelmannia. Sampling localities are coded according to Table 1 and taxa are labeled by symbols according to the inset. Black dots represent the natural occurrence of the genus according to the Global Biodiversity Information Facility (GBIF) records. Expected heterozygosity predicted by empirical Bayesian kriging across the Uebelmannia distribution range is depicted according to the color scale in the inset. The limits of the conservation units in the studied area are shown by the green lines
Microsatellite analysis
We used a total of 12 perfect (non-composed or interrupted) dinucleotide nuclear microsatellite loci characterized by Moraes et al. (2014) for Uebelmannia species. The PCR conditions and thermocycling parameters followed Moraes et al. (2014) with minor adjustments (Table A1 in the Online Resource 1). Amplicons were visualized on 3% agarose gel and subsequently run on a Fragment Analyzer Automated CE System (Advanced Analytical Technologies, Ames, IA, USA) using the 35–500 bp dsDNA Reagent Kit (Advanced Analytical Technologies). Finally, the alleles were scored using PROSIZE v2.0 (Advanced Analytical Technologies).
The genetic diversity at each sampled locality was assessed according to the number of alleles (A), effective number of alleles (ne), number of private alleles (Pa), and expected (HE) and observed heterozygosities (HO) using GENALEX v6.501 (Peakall and Smouse 2012). The allelic richness (AR) and inbreeding coefficient (FIS) were computed in FSTAT v2.9.3.2 (Goudet 2001). Deviations from Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were assessed using ARLEQUIN v3.5.2.2 (Excoffier and Lischer 2010), and the significance levels of these tests were adjusted according to the sequential Bonferroni correction for multiple comparisons (Rice 1989). All loci were checked for the presence of null alleles as implemented in MICRO-CHECKER v2.2.3 (Van Oosterhout et al. 2004). To avoid the possible bias introduced by null alleles, we estimated the global FST based on the corrected data for null alleles in FREENA (Chapuis and Estoup 2007). We used a probabilistic interpolation method and the empirical Bayesian kriging (EBK) approach as implemented in ArcGIS 10.6 (Krivoruchko 2012) to generate a geographic map of the expected heterozygosity across the Uebelmannia range.
Spatial population structure
Genetic differentiation among all Uebelmannia samples (global differentiation) and within species were assessed using the standard fixation indices based on the infinite alleles model (FST and \({G}_{{\textrm{ }\textrm{ST}}}^{\prime\prime}\) ) and the stepwise mutation model (RST) using ARLEQUIN and GENALEX. We also used AMOVA in ARLEQUIN to explore the level of genetic variance in the hierarchical models assuming taxonomic and geographic groupings inferred by clustering analyses.
We explored the population genetic structure using different approaches. Since each approach involves its own assumptions and issues (Excoffier and Heckel 2006; Putman and Carbone 2014; Janes et al. 2017), we inferred the population genetic structure according to congruent clustering results. First, we used the Bayesian clustering approach in STRUCTURE v2.3.4 (Pritchard et al. 2000), assuming no admixture model and correlated allele frequencies (Appendix A1 in the Online Resource 1). To explore the hierarchical structuring, in the first run, we used all 20 sampled localities. In a second round, we used the subsets of data corresponding to the identified genetic clusters in the first round. To supplement the results of STRUCTURE, we performed discriminant analysis of principal components (DAPC, Jombart et al. 2010) as implemented in ‘adegenet’ (Jombart 2008).
Among the clustering methods considering spatial distribution, we used GENELAND v4.0.5 (Guillot et al. 2005), TESS v2.3 (Chen et al. 2007), and BAPS v6.0 (Corander et al. 2008) (Appendix A2 in the Online Resource 1). For these analyses, we selected the uncorrelated or correlated frequency models according to the presence or absence of isolation by distance, respectively. Finally, we employed BAPS under the model of spatial clustering of groups to estimate the most likely number of genetic clusters in the populations. We also used STRUCTURE to assess the evidence of migration between the sampled localities, incorporating geographic information into the analysis. The occurrence of migration is inferred from the establishment of a minimum probability (0.5) that the genotype of a particular individual belongs to a specific population. Individuals with values below this cut-off were considered migrants or descendants of migrants.
We tested the presence of isolation by distance (IBD) through redundancy analysis (RDA) following the approach described by Meirmans (2015; Appendix A3 in the Online Resource 1). Conditioned by the presence of a significant correlation, we then used the pairwise genetic differentiation matrix to construct a map of local differentiation in LOCALDIFF (Duforet-Frebourg and Blum 2014) considering two simulated neighbors at a distance of 0.1 and 100 posterior replicates.
We investigated the recent reduction in the effective population size by assessing whether the populations deviated from mutation-drift equilibrium with BOTTLENECK v1.2.02 (Cornuet and Luikart 1996), as detailed in Appendix A4 (Online Resource 1). We also used the modified Garza-Williamson index in ARLEQUIN v3.5.2.2 (Excoffier and Lischer 2010), assuming a reduction in population size of M ≤ 0.68 (Garza and Williamson 2001). In addition, estimates of contemporary effective population size (Ne) were based on a single-sample approach using the LD method (Waples and Do 2008), as implemented in NEESTIMATOR v2.01 (see details in Appendix A5 in the Online Resource 1; Do et al. 2014).
Results
Genetic diversity
The mean genetic diversity at the species level was generally higher for U. gummifera and U. buiningii than for U. pectinifera (Table 2). Within the populations, we found a low number of effective alleles (1.03–3.03) and allelic richness (1.09–3.95) for U. pectinifera at most sampled localities. In contrast, we found moderate genetic diversity in U. buiningii (AR = 3.89–4.83; ne = 2.46–2.84) and U. gummifera (AR = 3.99–5.59; ne = 2.98–4.48). Furthermore, the highest number of sampled localities (six) harboring private alleles was found for U. gummifera (Table 2). The observed heterozygosity was moderate for U. buiningii (HO = 0.404–0.477) and U. gummifera (HO = 0.318–0.427), while a wide range of values among localities (HO = 0.000–0.443) was observed for U. pectinifera. According to the EBK approach, the highest levels of HE were predicted in the U. buiningii and U. gummifera ranges (Fig. 1). Interestingly, U. pectinifera showed moderate to low HE predictions for the populations in the Sempre Vivas National Park and outside the protected areas, respectively.
A significant amount of inbreeding was suggested by the significant positive values of FIS for almost all localities of the genus (FIS = 0.222–1.000; Table 2) except for the cases of Ub-1 and Ub-2 (U. buiningii). At the species level, U. buiningii showed the lowest mean value of inbreeding (0.147), followed by U. pectinifera (0.336) and then U. gummifera (0.433). All species had at least one population with a locus deviating from HWE. In addition, null alleles were found at all sampled localities (Table 2). A high number of locus pairs showing LD was found in only two populations: U. buiningii (Ub-2) and U. gummifera (Ugm-2; Table 2). Lastly, FREENA estimates for all loci were almost the same for the original (FST = 0.359) and null allele-corrected (FST = 0.364) datasets. Therefore, we carried out all the subsequent analyses without the exclusion of any loci except when the missing data from locus Upec214 (not transferred, Table A1 in the Online Resource 1) caused the performance of the analysis to be problematic.
Genetic and spatial structure
The global genetic differentiation was significantly high for all loci in Uebelmannia (FST = 0.36, RST = 0.57, and \({G}_{{\textrm{ }\textrm{ST}}}^{\prime\prime} = 0.73\); Table A2 in the Online Resource 1). Within species, U. pectinifera showed the highest estimates of genetic divergence among populations (FST = 0.45, RST = 0.63, and \({G}_{{\textrm{ }\textrm{ST}}}^{\prime\prime} = 0.76\)), while U. gummifera showed low to moderate estimates of the fixation indices (FST = 0.10, RST = 0.28, and \({G}_{{\textrm{ }\textrm{ST}}}^{\prime\prime} = 0.30\)). Estimates of genetic differentiation in U. buiningii were low and significant for most fixation indices (FST = 0.05, \({G}_{{\textrm{ }\textrm{ST}}}^{\prime\prime} = 0.13\)). There was no IBD when considering the genetic variation in U. buiningii, U. gummifera, and U. pectinifera separately or for the combination of U. buiningii and U. gummifera. However, we found very low IBD (RDA × FST = 16%) when all three species were combined. Nonstationary patterns of IBD showed the highest local genetic differentiation in a small area of the U. buiningii and U. gummifera ranges located in the northwestern border of Serra Negra State Park (Fig. 2).

Local genetic differentiation within the genus Uebelmannia based on LOCALDIFF
The clustering methods revealed much subdivision at the highest hierarchical level of population structure in Uebelmannia, resulting in 15 (GENELAND and DAPC) or more genetic clusters: 17 in BAPS and 21 in TESS (Fig. 3; Figs. A1 and A2 in the Online Resource 1). We could not find the most likely K groups at the genus level using STRUCTURE because we obtained multiple peaks of ΔK and the inferred clusters for the same K were instable across different runs (Fig. A3 in the Online Resource 1). The highest number of genetic clusters was observed in U. pectinifera, with 10 clusters according to GENELAND and 11 for TESS, BAPS, and DAPC, where individuals from each sampled locality were assigned to a distinct genetic cluster. Uebelmannia buiningii individuals were consistently assigned to a unique genetic cluster according to all clustering methods. In contrast, the clustering results for U. gummifera were inconsistent, showing three clusters according to DAPC, four for TESS and GENELAND, and five for BAPS, which partially corresponded to the geographic distribution and subspecies status of the populations.

Population genetic clustering in Uebelmannia with color plots from a TESS results using the no admixture model and K = 21 and b GENELAND results for the uncorrelated model and K = 15. Black bars show U. pectinifera, U. gummifera, and U. buiningii, with population code in Table 1. Each color corresponds to a distinct genetic cluster, and each bar corresponds to the proportion of an individual’s genotype in the genetic clusters
Additional clustering analyses taking into account each species separately (Fig. 4; Figs. A4 and A5 in the Online Resource 1) supported U. pectinifera as a highly structured species subdivided into 10 (DAPC), 11 (GENELAND and BAPS), or 12 (TESS) clusters, each one generally corresponding to a single sampled locality. On the other hand, STRUCTURE assigned individual genotypes into only two genetic clusters, with most of the U. pectinifera individuals showing admixed membership proportions (Fig. 4; Figs. A4 and A5 in the Online Resource 1). For U. gummifera, the clustering methods showed three (STRUCTURE and DAPC), six (GENELAND and BAPS), and seven (TESS) genetic clusters. For the K = 3 model, both STRUCTURE and DAPC assigned individuals of the subspecies meninensis into a single cluster, while individuals of the subspecies gummifera were subdivided into two additional clusters corresponding to the eastern and western portions of the range of the subspecies. The analyses resulting in high K values (six or seven clusters) identified most U. gummifera individuals as having admixed genotypes from several clusters (GENELAND and TESS) or according to each sampled locality (BAPS). The clustering analyses generated contrasting results for U. buiningii (Fig. 4; Figs. A4 and A5 in the Online Resource 1). STRUCTURE and GENELAND identified three and four clusters, respectively, with most individuals showing admixed genotypes from all genetic clusters. BAPS identified two clusters (Fig. A5 in the Online Resource 1), assigning individuals from the neighboring localities Ub-2 and Ub-3 into the same genetic cluster, while DAPC assigned individuals of each sampled locality into one distinct cluster. Although TESS identified four genetic clusters for U. buiningii, the largest membership proportion of each individual was assigned to the same cluster.

Population genetic clustering in U. buiningii, U. gummifera, and U. pectinifera with color plots from STRUCTURE, GENELAND (correlated model), and TESS (admixture model). Each color corresponds to a distinct genetic cluster, and each bar corresponds to the proportion of an individual’s genotype in the genetic clusters, with the population codes in Table 1. STRUCTURE results were inconsistent in U. buiningii and U. pectinifera due to no resolution of ΔK or multiple peaks and no stability in clustering groups. GENELAND also showed artificial clusters for all populations in U. gummifera likely due to low MCMC convergence
The hierarchical AMOVA results were in line with clustering analyses and showed higher genetic variance among populations within species or subspecies than among these taxonomic groups (Table A3 in the Online Resource 1). In general, the genetic variance was higher among than within groups (i.e., FCT > FSC) when each species was subdivided into several clusters. Specifically, the AMOVA results supported a population structure composed of at least 15 clusters for Uebelmannia, 10 clusters for U. pectinifera, four clusters for U. gummifera, and a single cluster for U. buiningii..
Demographic analyses
The ‘USEPOPINFO’ model in STRUCTURE identified eight individuals as migrants among the 485 sampled individuals (Table A4 in the Online Resource 1). Three individuals sampled as U. buiningii were identified as migrants from a U. gummifera population (Ugg-2), and one was identified as being from another locality of U. buiningii (Ub-2). The remaining identified migrants included one individual sampled as U. gummifera and identified as originating from a U. pectinifera population and three U. pectinifera individuals originating from U. gummifera or from other localities where U. pectinifera occurred.
Based on the BOTTLENECK analysis, only the western populations of U. gummifera subsp. gummifera (Ugg-1 and Ugg-2) and one population of U. pectinifera (Uph-3) exhibited excess heterozygosity in relation to the expectation under the TPM model. In contrast, the M-ratio tests supported a recent population size reduction in all tested groups and populations, as indicated by the consistently low values of the Garza-Williamson index (Table 3). These contrasting results were expected due to the lower statistical power of the heterozygosity-excess approach in comparison to M-ratio tests in detecting bottlenecks (Peery et al. 2012).
The effective population size estimate of the cohesive genetic cluster of U. buiningii resulted in Ne = 18.30 (95% CIjackknife 9.00–38.30). In the U. pectinifera and U. gummifera tests, all the genetic clusters except Uph-2 (Ne = 19.20; 95% CIjackknife 7.20–322.90) showed infinite estimates at the upper limit of the CI (Table 4), suggesting that the effect of sampling error is larger than any signal of LD or genetic drift (Waples and Do 2010). On the other hand, similar values at the lower limit of the CI were found in U. gummifera, ranging from 61.60 to 64.90, while they were lower and ranged from 2.90 to 32.70 in U. pectinifera.
Discussion
Genetic diversity and assessment of population structure
The CR landscapes of eastern Brazil, with their astonishing biodiversity, endemism and high number of threatened plant species, have been considered to be a top priority for conservation investment and research regarding plant life in Brazil (Monteiro et al. 2018). Here, we investigated the level and distribution of genetic diversity in the Uebelmannia genus, a highly prioritized, microendemic, and threatened CR taxon. Our findings provide guidelines for conservation of the Uebelmannia species and improve the understanding of the conservation genetics of endemic habitat specialists among CR plants. We used 12 nuclear microsatellite loci coupled with broad-scale sampling, covering the entire distribution of Uebelmannia. The results showed moderate-to-high levels of genetic diversity, extremely high population structure, and genetic signatures of recent bottlenecks in this genus. The results further confirmed that U. buiningii is the only species in the genus deviating from this scenario; however, it has the narrowest range among the species of the genus.
Although small populations are particularly likely to have low levels of genetic variability (Frankham 1996), Uebelmannia maintains a unique pattern of moderate-to-high levels of HE and AR. The mean values of HE (0.394) and AR (2.988) for U. pectinifera are the lowest among the studied plant species in CR using microsatellite markers (Khan et al. 2018b and references therein). However, the other two species, U. buiningii and U. gummifera, showed higher genetic diversity, suggesting that these species are not suffering from genetic erosion. Such unexpected patterns of genetic diversity have also been observed in other microendemic cacti in CR (Moraes et al. 2012; Bonatelli et al. 2014; Khan et al. 2018b), a characteristic in line with the predictions of OCBIL theory (Hopper 2009). The indication of higher genetic diversity within U. buiningii and U. gummifera than in U. pectinifera suggests that habitat fragmentation either as a result of anthropogenic degradation (Goettsch et al. 2015) or initially established during the Plio-Pleistocene transition by glacial cycles may explain the differences in the levels of genetic diversity (examples in Franco et al. 2017; Silva et al. 2018). In addition, U. pectinifera populations occur on the westernmost side of the distribution of this genus, with a larger range outside the protected areas, and are therefore most prone to habitat degradation by humans. The relatively low diversity of the individuals at the Datas locality (Upp-1; HE: 0.027; AR: 1.083), which occurs outside the protected areas, is also related to the small number of individuals. This finding highlights the consequences of anthropogenic disturbance, as previously mentioned by Schulz and Machado (2000). Similarly, the EBK approach predicted a heterogeneous level of genetic diversity from moderate to high among the populations in unprotected and protected areas, respectively (Fig. 1). However, the maintenance of a positive FIS in U. gummifera and U. pectinifera is a sign of habitat fragmentation (Lowe et al. 2015). It is worth noting that our results confirm the occurrence of recent bottleneck in all Uebelmannia populations. These results and the low Ne estimates obtained here mostly reflect the scenario of habitat disturbance and fragmentation (Frankham 2002; Allendorf and Luikart 2006). Contemporary Ne values are the most useful estimators in conservation and wildlife management for predicting the extinction risk of populations (Luikart et al. 2010). Thus, we are confident that these estimates might be useful for conservation assessments of U. buiningii (the species with the narrowest geographical distribution), as well as for U. pectinifera and U. gummifera.
The high molecular variance among Uebelmannia groups is mostly reflected in the taxonomic groups and subspecies, but it was most significant under the scenario of extreme population structure, with nearly each locality forming a distinct cluster. These results are in agreement with the naturally fragmented distribution of Cactaceae in CR due to constant fires, restricting these species to patches on rocky outcrops (Taylor and Zappi 2004). The low or absence of gene flow estimated among Uebelmannia populations by STRUCTURE explains the scope of drift-driven genetic differentiation and suggests that each genetic cluster might be an evolutionarily significant unit (Crandall et al. 2000). Similar reports of high population genetic differentiation and low gene flow have been described for Pilosocereus cactus species, which have a broader distribution in the Cerrado biome (Bonatelli et al. 2014; Khan et al. 2018a). However, our results contradict those regarding the microendemic cactus Pilosocereus aureispinus, in which all populations in CR were clustered into one unique genetic group (Khan et al. 2018b). Considering that Uebelmannia species are self-incompatible (Teixeira et al. 2018), the high genetic structure may also be attributed to the low abundance of pollinators and potential mates together with the low seed production rates and aggregated distribution. Thus, the prediction of effective cross-pollination in the CR species under OCBIL theory seems to not be applicable to Uebelmannia. In addition to the high genetic structure and low gene flow, although we did not find evidence of IBD, we did find high local genetic differentiation among the similarly distributed U. buiningii and U. gummifera, showing important fine-scale differentiation, which is in agreement with other species with restricted ranges (Moreira et al. 2010). For instance, even though U. gummifera populations are separated by a few kilometers, all of them exhibit private alleles, a common characteristic found in isolated populations (Bocanegra-González et al. 2018). In addition to the natural differentiation among patches, anthropogenic barriers caused by habitat loss and illegal collection seem to be important factors shaping the extreme population structure in this rare and endangered genus.
Conservation implications
This is the first conservation genetics study on the microendemic and endangered Uebelmannia confined to CR landscapes. The results discussed here are useful for designing in situ and ex situ conservation and management guidelines for Uebelmannia. The moderate-to-high levels of genetic diversity in the face of extreme population fragmentation and isolation suggest that most populations have genetic potential for conservation strategies. However, this picture may be a delayed response to the environmental changes, highlighting the urgency in conservation strategies (Aavik et al. 2019). Despite the apparently nondepauperated genetic diversity found in this taxon, the choice of populations for in situ conservation often relies on the stability of habitat for the maintenance of long-term populations and the absence of inbreeding in these populations (Bocanegra-González et al. 2018). Given the assumptions regarding the habitat conditions, most populations with high genetic diversity occur at the borders of protected areas and are subjected to human disturbance and overharvesting. Furthermore, signs of inbreeding were identified for nearly all population genetic clusters (except Ub-1 and Ub-2). Nevertheless, the high levels of genetic variation associated with the population structuring at the fine scale and the presence of private alleles show the evolutionary potential of these populations in terms of long-term persistence, mitigating the negative effects of inbreeding for in situ management. In this sense, we suggest the in situ conservation of the U. buiningii and U. gummifera population clusters (Ub-1, Ub-2, Ub3, Ugg-1, Ugg-2, Ugg-3, and Ugg-4) at the border of Serra Negra State Park and U. pectinifera (Upp-2, Upp-3, Upp-4, Uph-1, and Uph-4) nearest to Biribiri State Park and Sempre Vivas National Park. In particular, our data suggest that the most compelling results will come from expanding the northwestern border of Serra Negra State Park, where most of the genetic and taxonomic variation in Uebelmannia occurs. Furthermore, as another vital component of the conservation of Uebelmannia, we suggest seed collection from all genetic clusters identified here. Seed collection planning will be most effective if accompanied by storage techniques for the long-term preservation of germplasm, such as cryopreservation, and considering updated guidelines for sampling effort required to preserve population genetic variation and reduce the effects of germination failures (Hoban 2019). Even in cases in which populations have low diversity or common alleles, we still suggest management actions until the adaptive responses of such populations to the environment and human degradation are clear. However, such actions can be hampered by the reduced number of individuals in populations near roadsides or far from natural parks, a scenario found in most of the U. pectinifera genetic clusters. For these clustered populations, we strongly suggest ex situ conservation for the subsequent reintroduction of plants into their natural habitats. Finally, we highlight the robustness of the extreme population genetic clusters obtained here according to a wide range of statistics using microsatellite markers. Thus, we suggest that eventual translocation of individuals should be performed with caution due to the apparent risk of outbreeding depression in Uebelmannia populations. We also suggest that further investigations should be conducted using a genome-wide multilocus approach to improve the understanding of these genetic clusters and provide insight into the development of future translocations plans.
References
Allendorf FW, Luikart G (2006) Conservation and the genetics of populations. Wiley Blackwell, Oxford
Aavik T, Thetloff T, Träger S, Hernández-Agramonte IM, Reinula I, Pärtel M (2019) Delayed and immediate effects of habitat loss on the genetic diversity of the grassland plant Trifolium montanum. Biodivers Conserv 28:3299–3319
BFG (2018) Brazilian Flora 2020: Innovation and collaboration to meet Target 1 of the Global Strategy for Plant Conservation (GSPC). Rodrigúesia 69:1513–1527
Bitencourt C, Rapini A (2013) Centres of endemism in the Espinhaço Range: identifying cradles and museums of Asclepiadoideae (Apocynaceae). Syst Biodivers 11:525–536
Bocanegra-González KT, Thomas E, Guillemin M, Carvalho D, Gutiérrez JP, Alcázar Caicedo C, Moscoso Higuita LG, Becerra LA, González MA (2018) Genetic diversity of Ceiba pentandra in Colombian seasonally dry tropical forest: Implications for conservation and management. Biol Conserv 227:29–37
Bonatelli IAS, Perez MF, Peterson AT, Taylor NP, Zappi DC, Machado MC, Koch I, Pires AHC, Moraes EM (2014) Interglacial microrefugia and diversification of a cactus species complex: phylogeography and palaeodistributional reconstructions for Pilosocereus aurisetus and allies. Mol Ecol 23:3044–3063
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Conceição AA, Rapini A, Carmo FF, Brito JC, Silva GA, Neves SPS, Jacobi CM (2016) Rupestrian grassland vegetation, diversity, and origin. In: Fernandes GW (ed) Ecology and conservation of mountaintop grasslands in Brasil. Springer International, Switzerland, pp 105–123
Corander J, Sirén J, Arjas E (2008) Bayesian spatial modeling of genetic population structure. Comput Stat 23:111–129
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genet 144:2001–2014
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15:290–295
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Duforet-Frebourg N, Blum MGB (2014) Non-stationary patterns of isolation-by-distance: inferring measures of local genetic differentiation with Bayesian kriging. Evolution 68:2745–2745
Excoffier L, Heckel G (2006) Computer programs for population genetics data analysis: a survival guide. Nat Rev 7:745–758
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fernandes GW (2016) The megadiverse rupestrian grassland. In: Fernandes GW (ed) Ecology and cnservation of mountaintop grasslands in Brasil. Springer International, Switzerland, pp 3–11
Franco FF, Silva GAR, Moraes EM, Taylor N, Zappi DC, Jojima CL, Machado MC (2017) Plio-Pleistocene diversification of Cereus (Cactaceae, Cereeae) and closely allied genera. Bot J Linn Soc 183:199–210
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Giulietti AM, Pirani JR, Harley RM (1997) Espinhaço Range region, eastern Brazil. In: Davis SD, Heywood VH, Herrera-Macbryde O, Villa-Lobos J, Hamilton AC (eds) Centres of plant diversity: a guide and strategy for their conservation. IUCN Publication Unit, Cambridge, pp 397–404
Goettsch B, Hilton-Taylor C, Cruz-Piñón G, Duffy JP et al (2015) High proportion of cactus species threatened with extinction. Nat Plants 1:15142
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www2.unil.ch/popgen/softwares/fstat.htm. Updated from Goudet (1995)
Guillot G, Motrier F, Estoup A (2005) Geneland: a computer package for landscape genetics. Mol Ecol Notes 5:712–715
Hoban S (2019) New guidance for ex situ gene conservation: Sampling realistic population systems and accounting for collection attrition. Biol Conserv 235:199–208
Hernández-Hernández T, Hernández HM, De-Nova JA, Puente R, Eguiarte LE, Magallón S (2011) Phylogenetic relationships and evolution of growth form in Cactaceae (Caryophyllales, Eudicotyledoneae). Am J Bot 98:44–61
Hopper SD (2009) OCBIL theory: towards an integrated understanding of the evolution, ecology and conservation of biodiversity on old, climatically buffered, infertile landscapes. Plant Soil 322:49–86
IUCN (2018) IUCN Red List of Threatened Species. Version 2013.2. Available at http://www.iucnredlist.org. Accessed 20 Aug 2018
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602
Jombart T (2008) adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Khan G, Godoy MO, Franco FF, Perez MF, Taylor NP, Zappi DC, Machado MC, Moraes EM (2018a) Extreme population subdivision or cryptic speciation in the cactus Pilosocereus jauruensis? A taxonomic challenge posed by a naturally fragmented system. Syst Biodivers 16:188–199
Khan G, Ribeiro PM, Bonatelli IAS, Perez MF, Franco FF, Moraes EM (2018) Weak population structure and no genetic erosion in Pilosocereus aureispinus: a microendemic and threatened cactus species from eastern Brazil. PLoS ONE 13(4):e0195475
Krivoruchko K (2012) Empirical Bayesian Kriging. ArcUser Fall, Redlands
Lowe A, Cavers S, Boshier D, Breed M, Hollingsworth P (2015) The resilience of forest fragmentation genetics—no longer a paradox—we were just looking in the wrong place. Heredity 115:97–99
Loyola R, Machado N, Vila-Nova D, Martins EM, Martinelli G (2014) Áreas Prioritárias para Conservação e Uso Sustentável da Flora Brasileira Ameaçada de Extinção. Andrea Jakobsson. Estúdio Instituto de Pesquisa Jardim Botânico do Rio de Janeiro, Rio de Janeiro
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW (2010) Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11:355–373
Martinelli G, Moraes MA (Orgs) (2013) Livro vermelho da flora do Brasil. Andrea Jakobsson: Jardim Botânico do Rio de Janeiro, Rio de Janeiro
Meirmans PG (2015) Seven common mistakes in population genetics and how to avoid them. Mol Ecol 24:3223–3231
Monteiro L, Machado N, Martins E, Pougy N, Verdi M, Martinelli G, Loyola R (2018) Conservation priorities for the threatened flora of mountaintop grasslands in Brazil. Flora 238:234–243
Moraes EM, Cidade FW, Silva GAR, Machado MC (2014) Polymorphic microsatellite markers for the rare and endangered cactus Uebelmannia pectinifera (Cactaceae) and its congeneric species. Genet Mol Res 13:10359–10366
Moraes EM, Perez MF, Teo MF, Zappi DC, Taylor NP, Machado MC (2012) Cross-species amplification of microsatellites reveals incongruence in the molecular variation and taxonomic limits of the Pilosocereus aurisetus group (Cactaceae). Genetica 140:277–285
Moreira RG, McCauley RA, Corte´s-Palome AC, Fernandes GW, Oyama K (2010) Spatial genetic structure of Coccoloba cereifera (Polygonaceae), a critically endangered microendemic species of Brazilian rupestrian fields. Conserv Genet 11:1247–1255
Morellato LPC, Silveira FAO (2018) Plant life in campo rupestre: new lessons from an ancient biodiversity hotspot. Flora 238:1–10
Mucina L (2018) Vegetation of Brazilian campos rupestres on siliceous substrates and their global analogues. Flora 238:11–23
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Beer E, Robinson S, Vásquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Putman AI, Carbone I (2014) Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol Evol 4:4399–4428
Ribeiro-Silva S, Zappi D, Taylor N, Machado M (2011) Plano de Ação para Conservação das Cactáceas. Série Espécies Ameaçadas nº 24. Instituto Chico Mendes de Conservação da Biodiversidade, ICMBio, Brasília
Rice W (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Schulz R, Machado M (2000) Uebelmannia and their environment. Schulz Publishing, Teesdale
Silva GAR, Antonelli A, Lendel A, Moraes EM, Manfrin MH (2018) The impact of early Quaternary climate change on the diversification and population dynamics of a South American cactus species. J Biogeogr 45:76–88
Silveira FAO, Negreiros D, Barbosa NPU, Buisson E, Carmo FF, Carstensen DW, Conceição AA, Cornelissen TG, Echternacht L, Fernandes GW, Garcia QS, Guerra TG, Jacobi CM, Lemos-Filho JP, Le Stradic S, Morellato LP, Neves FS, Oliveira RS, Schaefer CE, Viana PL, Lambers H (2016) Ecology and evolution of plant diversity in the endangered campo rupestre: a neglected conservation priority. Plant Soil 403:129–152
Taylor NP, Zappi DC (2004) Cacti of eastern Brazil. Royal Botanic Gardens, Kew, Richmond
Teixeira VD, Verola CF, Costa IR, Zappi DC, Costa GM, Silva SR, Costa MAPC, Aona LYS (2018) Investigating the floral and reproductive biology of the endangered microendemic cactus Uebelmannia buiningii Donald (Minas Gerais, Brazil). Folia Geobot. https://doi.org/10.1007/s12224-018-9315-6
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Zappi DC, Moro MF, Meagher TR, Nic Lughadha E (2017) Plant biodiversity drivers in Brazilian campos rupestres: insights from phylogenetic structure. Front Plant Sci 8:2141
Zappi D, Taylor N (2008) Diversidade e endemismo das Cactaceae na Cadeia do Espinhaço. Megadiversidade 4:111–116
Acknowledgements
We thank the Minas Gerais State Forestry Institute and the Chico Mendes Biodiversity Conservation Institute for the permit for sample collections. We also thank Gerardus Olsthoorn for supplying some of the samples used in this study and PROBIO II-ICMBio and Embrapa-Cenargen for providing support in our field trips. Finally, we thank two anonymous reviewers for their valuable comments that helped to improve the manuscript. This work was supported in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by David Hawksworth.
Electronic supplementary material
Below is the link to the electronic supplementary material. \
Rights and permissions
About this article

Cite this article
Silva, G.A.R., Khan, G., Ribeiro-Silva, S. et al. Extreme genetic structure in a relict cactus genus from campo rupestre landscapes: implications for conservation. Biodivers Conserv 29, 1263–1281 (2020). https://doi.org/10.1007/s10531-020-01934-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-020-01934-6