
Abstract
The genus Hypochaeris was likely introduced into South America by a unique long-distance dispersal event from an ancestral Northwest African species, with subsequent radiation and diversification throughout the temperate zones of the continent. Hypochaeris catharinensis is an endemic species of Southern Brazil growing mostly in high-elevation grasslands with dry, rocky, and shallow soils. Here, we used nine microsatellite loci and a total sample of 441 individuals to assess genetic variability patterns within and among 13 H. catharinensis populations. The results showed a total of 215 alleles in the nine loci tested, the expected and observed heterozygosities average were 0.31 and 0.43, respectively, and allelic richness varied from 4.09 to 7.23. The levels of allelic richness showed that genetic variability is maintained by gene flow, thus ensuring that these populations are not genetically isolated. We found no correlation between geographic and genetic distances, with high levels of genetic variability within, rather than among, populations. The low levels of genetic structure in the species are consistent with recent evolutionary processes of the Hypochaeris genus in South America; after adaptive radiation and species differentiation, dispersal to similar habitats led to the establishment of new populations. Along with initial founder effects, the existence of gene flow explains the current genetic variation across the H. catharinensis populations. In addition, historic environmental changes reported for Southern Brazil may explain the current clustered distribution of the species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypochaeris L. (Asteraceae) has a disjunct geographic distribution across the Old World and South America. The genus consists of approximately 60 species, with 15 occurring in the Mediterranean and Europe, two in Asia, and approximately 45 in the temperate region of South America. Evolutionarily, Hypochaeris is a very young genus in South America (Samuel et al. 2003; Stuessy et al. 2003). A molecular clock analysis proposed that the South American group of species diverged during the Pleistocene, between 0.25 and 1.1 million years ago (Tremetsberger et al. 2005, 2013). The disjunct distribution of the genus suggests that a probable ancestor from the Old World, similar to the present-day H. angustifolia (Lit. & Maire) Maire from Northwest Africa, arrived in South America via a long-distance dispersal event (Tremetsberger et al. 2006). Establishment followed by adaptive radiation, isolation, and eventually speciation allowed the genus to colonize new habitats throughout the temperate zones of South America (Samuel et al. 2003; Stuessy et al. 2003; Ruas et al. 2005; Tremetsberger et al. 2005, 2006; Weiss-Schneeweiss et al. 2008). Studies on the phylogenetic relationships of the genus show limited divergence, with the South American species generating a monophyletic group (Cerbah et al. 1998; Samuel et al. 2003; Tremetsberger et al. 2006; Reck et al. 2011), further supporting the recent history of this group of species in South America.
Long-distance dispersal events and colonization with rapid speciation were suggested as a way to allow Hypochaeris species to occupy a range of habitats in South America. Dispersion followed by geographic isolation and selection within different ecological zones enabled the development of high levels of morphological divergence, indicating a pattern of speciation by adaptive radiation (Stuessy et al. 2003). Population genetic studies based on molecular markers support the assumption that isolation by distance and founder events shaped the geographic distribution patterns of the South American complex of Hypochaeris species (Muellner et al. 2005; Tremetserger et al. 2009; Urtubey et al. 2009; Rodrigues et al. 2016). Furthermore, biogeographic events and climate changes also played important roles in the distribution and isolation of populations. A study of Hypochaeris palustris (Phil.) De Wild., for example, showed that Pleistocene refugia, possibly in unglaciated areas, and recolonization routes influenced the postglacial distribution of the species in the southern Andes (Muellner et al. 2005). Similarly, a study on Hypochaeris incana (Hook. & Arn.) Macloskie demonstrated how Pleistocene refugia defined the postglacial geographic distribution of Patagonian and sub-Antarctic species (Tremetserger et al. 2009). The authors report that in some areas, populations of H. incana were mostly diploid with high genetic diversity and connected by high levels of gene flow, whereas other areas contained mainly tetraploid individuals occurring as isolated and highly differentiated populations with low genetic diversity. In contrast, for the ecologically restricted species Hypochaeris lutea (Vell.) Britton, that occurring only in moist environments as swamps and wetlands, Rodrigues et al. (2016) found no evidence of climatic pressure on the species’ genetic structure and distribution.
Several Hypochaeris species in South America have either endemic or widespread distribution (Bortiri 1999; Cabrera et al. 2000; Azevêdo-Gonçalves and Matzenbacher 2007). Some species are capable of colonizing a wide variety of habitats from sea level to over 5000 m (Tremetsberger et al. 2006; Urtubey et al. 2009), while others are restricted to coastal or high-elevation zones or are capable of growing in grasslands or modified environments (Reck et al. 2011; Rodrigues et al. 2016). In Brazil, the Hypochaeris genus is represented by approximately 12 species, most of which occur in Rio Grande do Sul State (Ruas et al. 2005; Azevêdo-Gonçalves and Matzenbacher 2007; Rodrigues et al. 2016).
Hypochaeris catharinensis Cabrera is a perennial herb endemic to the steppes or high-elevation grasslands of Santa Catarina and northern Rio Grande do Sul States, but it has been also reported in southern Paraná State (Cabrera 1963; Veloso and Góes-Filho 1982). Its ecological restriction to the Araucaria Plateau has led to a process of evolutionary endemism (Azevêdo-Gonçalves and Matzenbacher 2007), with the species restricted mostly to high-elevation zones (up to 1400 m) with dry, rocky, and shallow soils resulting from basalt weathering (Fig. 1). These ecological factors have led to the development of a horizontal rhizome which is characteristic of this species, and the development of rosette and oblanceolate leaves that favor photosynthesis (Azevêdo-Gonçalves and Matzenbacher 2007). Little has been published on the breeding systems of H. catharinensis, but Amplified Fragment Length Polymorphism (AFLP) data (Reck et al. 2011) suggest that the species reproduces preferably through cross-fertilization in addition to rhizome budding. Although the occurrence of self-incompatibility has been reported for some Hypochaeris species (Nettancourt 1977; Lane 1996), to date this mechanism has not been confirmed for H. catharinensis.

a–fHypochaeris catharinensis (E. Urtubey, C. Ruas and P. Ruas 350): a typical community of Araucaria angustifolia forests with H. catharinensis (area between Bom Jardim da Serra and São Joaquin, Santa Catarina state); b typical plant; c capitulum during anthesis (E. Urtubey, C. Ruas and P. Ruas 358); d oblanceolate basal leaves; E. Madure capitulum (E. Urtubey, C. Ruas and P. Ruas 356). Photographs by E. Urtubey
Although Reck et al. (2011) detected a close phylogenetic relationship between H. catharinensis and H. lutea (Lutea group), no studies have assessed the genetic structure of H. catharinensis. As such, we studied 13 populations of H. catharinensis using nine microsatellite loci to evaluate the genetic variability and structure of the species and elucidate the biogeographic history of H. catharinensis in South America and the rapid speciation process inland. For this purpose, we considered two biogeographic patterns in the genus; (1) species influenced by the Pleistocene glaciations, with posterior recolonization from ancient glacial refuge areas and (2) species no influenced by the glaciations, but by successive colonization events by long-distance dispersal.
Materials and methods
Sampling
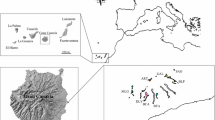
The study included 13 H. catharinensis populations (Table 1) from across the species’ range in the Brazilian states of Rio Grande do Sul (five populations), Santa Catarina (seven populations), and Paraná (one population) (Fig. 2). Leaf tissues were collected and stored in silica gel (Table 1), with a minimum distance of three meters between sampled individuals in each population. Voucher specimens were deposited in the Herbarium of Londrina State University (FUEL) (Table 1).

Map with collection sites of Hypochaeris catharinensis populations in Santa Catarina and Rio Grande do Sul in Brazil. Each acronym indicates the localization of a population according to Table 2
DNA extraction and amplification
Genomic DNA was extracted from each individual according to the 2% CTAB protocol (Doyle and Doyle 1987) with the following modifications: after precipitation with isopropanol and subsequent centrifugation, the DNA pellet was washed with 70% ethanol, dried at room temperature, and resuspended in TE buffer. The quality and concentration of total DNA were evaluated using a Scandrop spectrometer (Analytik Jena, Germany). For the PCR reactions, we used nine pairs of microsatellite primers (Table 2) obtained from a microsatellite library developed for H. catharinensis (Chaves et al. 2015). For amplification, we used indirect fluorescent labeling of the fragments with a three-primer system (Schuelke 2000) where the forward primers were tagged with M13 sequences (TGTAAAACGACG GCCAGT) at the 5′ end. The reactions occurred in a volume of 10 µL containing: 4.5 µL of 1 × GoTaq Green Master Mix (Promega, Madison, WI, USA); 0.08 µL and 0.32 µL of forward and reverse primers (5 pM each), respectively; 0.32 µL of the M13 primer (5 pM) labeled with each of the fluorophores 6-FAM, HEX, or NED with 2 μL (30 ng) of genomic DNA; and nuclease-free water to adjust the volume to 10 µL. PCRs were performed in a PTC 200 thermocycler (MJ Research, Inc. USA) in a touchdown program. In brief, the initial annealing temperature was 65 °C for each primer, with PCR cycles comprised of: 1 cycle of 94 °C for 4 min followed by 12 cycles of 94 °C for 30 s, 65 °C for 30 s, and 72 °C for 30 s, with the temperature in each PCR cycle decreasing 1 °C from annealing per cycle until reaching 55 °C; 29 additional cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; one cycle of 94 °C for 30 s, 54 °C for 30 s, and 72 °C for 30 s; 7 cycles of 94 °C for 30 s, 53 °C for 45 s, and 72 °C for 45 s; and a final extension at 60 °C for 40 min. The PCR products were resolved by capillary electrophoresis in an ABI 3500×L Genetic Analyzer (Applied Biosystems, California, USA) with the molecular weight standard GeneScan 600 LIZ Size Standard (Life Technologies, California, USA), in the molecular multi-user laboratory of Londrina State University. Allele sizes were determined based on comparison with the molecular weight marker GeneScan-600 LIZ Size Standard (Life Technologies, California, USA) using the software GeneMapper version 4.1 (Chatterji and Pachter 2006).
Data analysis
The microsatellite alleles were organized in a data matrix according to size and subsequently analyzed using several computer programs. The presence of null alleles was estimated using the Micro-Checker program, version 2.2.3 (Van-Oosterhout et al. 2004), with a confidence level of 0.05. Genetic diversity was measured by the number of alleles per population (A), the number of effective alleles (E), observed heterozygosity (Ho), and expected heterozygosity (He) calculated in Popgene v.1.3.2 software (Yeh et al. 1999). The exact tests for the Hardy–Weinberg Equilibrium (HWE), allelic richness (R), private alleles (P), and inbreeding coefficient (FIS) with Bonferroni correction (95%, α = 0.05 and 2340 randomizations) were estimated using Fstat v. 2.9.3.2 (Goudet 2002). Unlike the sample size for each population, the allelic richness was calculated using the rarefaction method and the program HP-rare (Kalinowski 2005). The relatedness coefficient was measured using the Coancestry software (Wang 2011).
The analysis of molecular variance (AMOVA) considered one group of populations and three groups of populations (K = 3) according to Structure cluster. The genotypic data were estimated using 1000 permutations for AMOVA and to determine pairwise significant differences. For the Mantel test and correlation between distance matrices, we considered 10,000 permutations, and for the exact test of population differentiation we used 10,000 steps in a Markov chain with 100,000 dememorization steps, at a significance level of 0.01. AMOVA, index coefficients (FST; FSC; FCT;), and linkage disequilibrium were calculated using Arlequin version 3.11 (Excoffier et al. 2005). The index coefficient FST tested the permutation rate among populations and groups; FSC tested the permutation rate among populations within groups; and FCT tested the permutation rate of populations among groups.
A hierarchical clustering and principal component analysis (PCA) were completed using the adegenet package (Jombart 2008) in the R Development Core Team software (2011). Hierarchical clustering is achieved by cutting the tree at a certain height and pooling the few remaining branches into the defined clusters, or by cutting the tree based on genetic distances and indirectly defining genetic clusters. The PCA uses allelic frequencies to calculate eigenvalues which represent the amount of genetic diversity measured by the multivariate method or the variance of the corresponding principal component. To test whether the studied populations underwent a recent genetic bottleneck effect, the Bottleneck software (Cornuet and Luikart 1996; Piry et al. 1999) was applied considering the following evolutionary models for microsatellite loci: Infinite Allele Model (IAM), Two-Phase Model (TPM), and Stepwise Mutation Model (SMM). The probabilities based on heterozygosity excess/deficiency were obtained with the Wilcoxon sign-rank test (Luikart et al. 1998). This test provides relatively high power for analyses using a small number of loci.
The estimated rate of migration was calculated using BayesAss v.1.3 software (Wilson and Rannala 2003), and the possible correlation between geographic and genetic distances was calculated using the Pearson correlation test in the Bioestat 5.0 software (Ayres et al. 2007). To identify the number of clusters (K) of genetically similar populations, we used the software Structure v. 2.3.3 (Hubisz et al. 2009), considering an admixture model with correlated allele frequencies, a set burn-in length of 10,000 interactions and Markov chain Monte Carlo, and simulation runs with a minimum of K = 1 to a maximum of K = 16 (the number of sampled populations + 3) with 20 replications per K (Evano et al. 2005). The optimal number of clusters was estimated with the ∆K method (Evano et al. 2005) in the Structure Harvester Web site (Earl and VonHoldt 2012).
Results
We successfully genotyped nine microsatellite loci for 13 H. catharinensis populations (Table 3). PCR amplification yielded a total of 215 alleles in 441 individuals, with the number of alleles ranging from 12 (Hcat19) to 35 (Hcat24b) and an average of 24 alleles per locus. The locus Hcat6 was monomorphic in four populations (P-5, P-7, P-8, and P-9), while locus Hcat19 was monomorphic in two populations (P-9 and P-10). The average number of alleles per population ranged from 4.78 (P-11) to 9.56 (P-4), with an overall average of 6.10 alleles. The number of effective alleles ranged from 1.94 (P-6) to 3.76 (P-12), and private alleles ranged from 1 (P-3, P-11) to 24 (P-4). The allelic richness varied from 4.09 to 7.23 for populations P-3 and P-4, respectively, with an average of 4.97 (Table 3). After rarefaction, the allelic richness was similar, ranging from 4.47 (P-1) to 5.44 (P-8) (Supplementary material s6). The number of null alleles was significant (α = 0.05) for populations P-1, P-2, P-4, and P-12 (Table 3). However, it is unlikely that these null alleles will influence the calculation of genetic parameters (Carlsson 2008).
The average observed and expected heterozygosities were 0.39 and 0.44, respectively. The values of observed heterozygosity ranged from 0.31 (P-12) to 0.43 (P-10), and the expected heterozygosity varied from 0.36 (P-6) to 0.53 (P-4). For most of the studied populations, observed heterozygosity was lower than expected, except for populations P-9 and P-7 that showed similar values of observed and expected heterozygosities, and populations P-3, P-6 and P-10 that showed higher values for observed heterozygosity. The exact test showed that all populations deviated significantly from the Hardy–Weinberg equilibrium, while no significant linkage disequilibrium was found between any pairwise combination of loci (Table 3).
The inbreeding coefficient (FIS) was positive and significant for eight populations (P-1, P-2, P-4, P-5, P-6, P-8, P-12, P-13) (Table 3). The AMOVA revealed that most of the genetic variation is within populations (77.08%) rather than among populations (22.92%) (Table 4). The AMOVA for K = 3 structured groups (Fig. 3) exhibited high variance among groups (23.21%) and low variance among populations within groups, thus confirming the clusters established through Bayesian analysis. Different magnitudes of gene flow were observed in all populations (Table 5). The highest migration rates were found for P-1, with migrants from populations P-8 (0.289), P-11 (0.014), and P-4 (0.015), and for P-2, with migrants from P-8 (0.263), P-11 (0.022), and P-1 (0.016). Population P-8 had the highest (0.901) rate of non-migrants.

Delta K values for the Hypochaeris catharinensis populations from Structure Harvester program, based on the Bayesian analysis
The pairwise FST values ranged from 0.0267 between populations P-13 and P-8, which are 158 km apart, to 0.365 between P-8 and P-10, located 175 km apart (Supplementary material s1). The Pearson correlation test showed no correlation between genetic and geographic distances (r = − 0.6205; P = 0.1887). The Wilcoxon test (Supplementary material s2), using evolutionary models IAM, SMM, and TPM for microsatellite loci, indicated that five populations of H. catharinensis underwent recent decreases in effective size. Twelve populations showed deficiencies in heterozygosity for the TPM model and thirteen for the SMM model, with population P-10 exhibiting heterozygosity deficiency only in the SMM model.
The number of population clusters (K) generated by the Bayesian analysis was K = 3 (Figs. 3 and 4b), with 38% admixture of individuals. The hierarchical clustering (Fig. 4a) and the PCA analysis (Supplementary material s3) agree with the result for the Bayesian analysis of K = 3 (Fig. 3). Most populations (P-1, P-2, P-3, P-4, P-6, P-7, P-9 and P-11) have limited differentiation and belong to one sub-structured group; P-10 and P-12 are structured as a second group anchored in this first group; and populations P-5, P-8, and P-13 compose a separate, well-define group. A sharp decrease in the PCA eigenvalues can also indicate relevant structural boundaries among populations. The relatedness coefficient ranged from − 0.61 to 0.26 (Supplementary material s5), with P-8 showing negative or no relatedness values across all populations, while for P-3, P-6, P-9, and P-11 the relatedness coefficient was positive and significant for most populations.

A groupment analysis, a hierarchical clustering using Euclidean distances and b Bayesian clusters for K = 3 groups formed by the analysis in thirteen Hypochaeris catharinensis populations, in X-axis are the allele frequencies and Y-axis are the population’s name
Discussion
Despite its restricted distribution and both vegetative and outcrossing reproduction, our SSR data showed moderate variability for H. catharinensis, as indicated by the average number of alleles per locus (A), average effective alleles (E), and allelic richness (R) (Table 3, Supplementary material s4; s6). Similar results were also reported for Hypochaeris tropicalis Cabrera, a highly endemic species with a distribution restricted to the southwest of Rio Grande do Sul State, Brazil. Using nine microsatellite loci, Paula (2015) showed that the number of alleles per locus for H. tropicalis ranged from 3 to 14, with an average of 6.2, and the number of effective alleles ranged from 1.7 to 1.9.
The analysis of molecular variance (AMOVA) showed that most genetic variability was within populations (77.08%) rather than among populations (22.92%) (Table 4). This pattern of genetic structure was also observed based on AFLP data studies for H. catharinensis (83.64/16.36; Reck 2010) and H. lutea (76.67/12.51; Rodrigues et al. 2016). Based on AFLP and SSR markers, populations of H. tropicalis presented a genetic variation of 91.06 and 97.08 within populations and 8.94 and 2.92 among populations, respectively (Paula 2015). These results are very different with those obtained for other Hypochaeris species, such as the Patagonian and sub-Antarctic species H. incana (67% among populations) that seems to have expanded its population in postglacial times (Tremetserger et al. 2009) and Hypochaeris tenuifolia (Hook. et Arn.) Griseb. (30.89% among populations), a species with a recent history of colonization from a refuge on the Lonquimay Volcano in Chile (Tremetsberger et al. 2003). The H. catharinensis populations studied herein showed high levels of genetic differentiation among populations (FST = 22.92) and groups (FST = 23.21); according to Wade and McCauley (1988), a decrease in heterozygosity and an increase in differentiation are expected in recently founded populations.
The gene flow (Table 5) may explain the high level of genetic variability observed within populations than among populations of H. catharinensis. Moreover, the distribution of genetic variability indicates a predominance of outcrossing with effective pollen and propagule dispersal mechanisms (Kageyama et al. 2003).
Our analysis of FIS showed that populations with significant inbreeding values exhibited genetic variability (A, E, R) similar or slightly greater than populations with nonsignificant FIS (Table 3). Additionally, populations with significant heterozygosity deficiency according to all three models of allele mutation showed slightly higher or similar genetic variability to populations that were significant for only one or two of the models (Tables 3; Supplementary material s2).
The comparison of genetic parameters (A, E, R, and FIS) (Table 3), rate of migration (Table 5), and heterozygosity deficiency/excess (Tables 3 and 5; Supplementary material s2) showed that population P-4 exhibited the greatest average number of alleles (9.56) and allelic richness (7.13); however, it had a positive and significant FIS and a significant heterozygosity deficiency based on Wilcoxon test in the TPM and SMM models (Supplementary material s2). Yet, the P-4 population also showed a rate of migration of 0.296, supporting the idea that gene flow strongly counterbalances the effects of genetic bottlenecks and endogamy. On the other hand, P-8 showed an intermediate level of genetic variability with a positive and significant FIS (Table 3), as well as the lowest rate of migration (0.064; Table 5), heterozygosity deficiency for the TPM and SMM models (Supplementary material s2), and an absence of relatedness with all other populations (Supplementary material s5). These results suggest that the number of migrants was not sufficient to counterbalance endogamy and the effects of reduced population size in P-8.
Most H. catharinensis populations were significant for endogamy and exhibited heterozygosity deficiency in at least one mutational model. Nevertheless, the levels of diversity found within populations were maintained by gene flow (Table 5), suggesting that these populations are not genetically isolated and justifying the lack of linkage disequilibrium and fixed alleles in all populations. Likewise, the level of allelic richness provides additional evidence that the genetic variability of the studied H. catharinensis populations is being maintained by gene flow, which ensures that the populations are not genetically isolated. The allelic richness estimated in H. catharinensis varied from 4.09 to 7.23. In H. radicata, a European Hypochaeris species, the allelic richness varied from 3.30 to 3.42 with high values of genetic variability that were mostly maintained by gene flow (Mix et al. 2006). Previous studies have shown that high levels of allelic richness, even of neutral alleles, increase a species’ ability to evolve by making a larger proportion of the genotypic space accessible to fewer mutational events (Wagner 2008). Therefore, allelic richness is a strong indicator of the evolutionary potential of a population (Allendorf 1986; Caballero and Garcia-Dorado 2013; Allendorf et al. 2012) and this measure has been suggested as key factor in population studies (Petit et al. 1998; Foulley and Ollivier 2006).
The influence of the recent evolutionary history of the genus established by a founder event in South America (Tremetsberger et al. 2005, 2013) and a reduction in the populations size can be observed through the increase in excess homozygotes, higher frequency of a set of alleles, and moderate genetic differentiation (Tables 3 and 4; supplementary material s2; s4). Slatkin (1995) argued that with founder effects and genetic bottlenecks, alleles present in one copy immediately after the event may increase in frequency, reaching high frequencies due to strong genetic drift, while the population is still small.
We found no correlation between geographic and genetic distance among the H. catharinensis populations, suggesting that these populations do not follow a clear pattern of isolation by distance. Hypochaeris catharinensis exhibits an evolutionary process of endemism, probably due to ecological restrictions, since it is well adapted to particular conditions as higher elevation (up to 1400 m), cooler temperatures, and dry, rocky, and shallow soils (Azevêdo-Gonçalves and Matzenbacher 2007). The ecological requirements and adaptation to similar, although discontinuous, niches thus limit the establishment of individuals and shape the distribution pattern of the species. A clustered distribution and absence of correlation between genetic and geographic distances were also documented for H. lutea (Rodrigues et al. 2016) and for H. tropicalis (Paula 2015). The pairwise FST estimates revealed that the levels of genetic differentiation varied considerably among H. catharinensis populations. These values range from 0.027 between P-8 and P-13, which are 158 km apart, to 0.365 between P-10 and P-8, which are 175 km apart (Supplementary material s1), consistent with the values reported for H. lutea (Rodrigues et al. 2016). A different pattern was found for H. tropicalis, which showed low values of genetic differentiation (Paula 2015). Species with an island-like distribution that lack correlation between geographic and genetic distances and a geographic structure may be associated with repeated introduction to similar niches by an ancestral population through the establishment of the new populations (founder events) (Lopez-Sepulveda et al. 2015) and gene flow. Such a pattern can be attributed to the three species (H. catharinensis, H. lutea, H. tropicalis) endemic to Southern Brazil.
The Bayesian analysis showed that the studied populations are grouped into three clusters (K = 3; Figs. 3 and 4b). The recent evolution and colonization pattern of H. catharinensis possibly helped to shape these groups. Historic environmental changes can inform our understanding of the current genetic structure of H. catharinensis. During the Pleistocene, large areas of grassland occurred in the Southern Brazilian highlands, but by the middle of the Holocene the grasslands had decreased significantly (Behling et al. 2004). The dominance of grasslands is attributed to the cold and dry glacial environment that shifted to the hot and dry climatic conditions of the upper Holocene. In the late Quaternary, a gradual increase in rainfall resulted in forest expansion (Rambo 1954, 1956), shaping the current mosaic of grassland interspersed with Araucaria Forest (Pillar and Vélez 2010). As shown above, the geographic distribution of H. catharinensis consists of clusters in high-elevation grasslands in Southern Brazil. This pattern is similar to that of other taxa occupying the Brazilian highlands, the majority of which exhibit geographic isolation processes due to island-like distribution (Fregonezi et al. 2013) that is characteristic of the process of forest expansion-retraction over the grasslands.
Conclusion
The moderate genetic variability present in H. catharinensis is likely maintained by gene flow, thus ensuring that the populations do not become isolated. Our analysis suggests that all populations of H. catharinensis were established by a founder event and present an island-like distribution caused by repeated introduction into similar niches from an ancestral population. The H. catharinensis populations studied herein are structured in three groups and have intermediate levels of differentiation. Environmental changes that occurred since the Pleistocene and the discontinuous patchwork that is characteristic of forest expansion-retraction over the grasslands likely helped to shape the structure of the species.
References
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5:181–190. https://doi.org/10.1002/zoo.1430050212
Allendorf FW, Luikart GH, Aitken SN (2012) Conservation and the genetics of populations, 2nd edn. Wiley, West Sussex
Ayres M, Ayres JRM, Ayres DL, Santos AS (2007) Bioestat: 2,0—aplicações estatísticas nas áreas das ciências biológica e médicas. Sociedade Civil, Mamirauá, Belém
Azevêdo-Gonçalves CF, Matzenbacher NI (2007) O gênero Hypochaeris (Asteraceae) no Rio Grande do Sul, Brasil. Iheringia 62:55–87
Behling H, Pillar VD, Orlóci L, Buuermann SG (2004) Late quaternary Araucaria forest, grassland (Campos), fire and climate dynamics, studied by high-resolution pollen, charcoal and multivariate analysis of the Cambará do Sul core in southern Brazil. Palaeogeogr Palaeoclimatol 203:277–297. https://doi.org/10.1016/S0031-0182(03)00687-4
Bortiri E (1999) Flora Fanerogamica Argentina. Asteraceae, Lactuceae: Hypochaeris. Cordoba: CONICET. fasc. 63, pp. 1–25
Caballero A, Garcıa-Dorado A (2013) Allelic diversity and its implications for the rate of adaptation. Genetics 195:1373–1384. https://doi.org/10.1534/genetics.113.158410
Cabrera AL (1963) Estudios sobre o gênero Hypochaeris. Bol Soc Argent Bot 10:166–195
Cabrera AL, Crisci JV, Delucchi G, Freire SE, Giuliano DA, Iharlegui L, Katinas L, Sáenz AA, Sancho G, Urtubey E (2000) Catalogo ilustrado de las compuestas (Asteraceae) de la provincia de Buenos Aires, Argentina: Sistematica, Ecología y Usos. CONICET, Buenos Aires
Carlsson J (2008) Effects of null alleles on assignment testing. J Heredity 99:616–623. https://doi.org/10.1093/jhered/esn048
Cerbah M, Coulaud J, Siljak-Yakovlev S (1998) rDNA organization and evolutionary relationships in the genus Hypochaeris (Asteraceae). J Heredity 89:312–318. https://doi.org/10.1093/jhered/89.4.312
Chatterji S, Pachter L (2006) References based annotation with GeneMapper. Genome Biol 7:R29. https://doi.org/10.1186/gb-2006-7-4-r29
Chaves CL, Ruas CF, Ruas PM, Schneider AA, Rocha KR, Urtubey E, Ruas EA (2015) Isolation and characterization of twelve polymorphic microsatellite loci for Hypochaeris catharinensis (Asteraceae) and cross-amplification in related species. J Genet 94:e39–e42
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Doyle JJ, Doyle JL (1987) A rapid isolation procedure for small quantities of fresh leaf tissue. Phytoch Bull 19:11–15
Earl DA, VonHoldt BM (2012) Structure harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conservation Genet Resources 4:359–361
Evano G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molec Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin v. 3.1: an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Foulley J, Ollivier L (2006) Estimating allelic richness and its diversity. Livestock Sci 101:150–158
Fregonezi JN, Turchetto C, Bonatto SL, Freitas LB (2013) Biogeographical history and diversification of Petunia and Calibrachoa (Solanaceae) in the Neotropical Pampas grassland. Bot J Linn Soc 171:140–153
Goudet J (2002) FSTAT: a program to estimate and test diversities and fixation indices (version 2.9.3.2). Available at: http://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 23 May 2016
Hubisz JM, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Molec Ecol Resources 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kageyama PY, Sebbenn AM, Ribas LA, Gandara FB, Castellen M, Perecim MB, Vencovsky R (2003) Diversidade genética em espécies arbóreas tropicais de diferentes estágios sucessionais por marcadores genéticos. Sci Forest 64:93–107
Kalinowski ST (2005) HP-Rare: a computer program for performing rarefaction on measures of allelic diversity. Molec Ecol Notes 5:187–189
Lane M (1996) Pollination biology of compositae. In: Caligari PDS, Hind DJN (eds) Compositae: biology and utilization, vol. 2. Royal Botanical Gardens, Kew, pp 61–80
Lopez-Sepulveda P, Takayama K, Greimler J, Crawford DJ, Peñailillo P, Baeza M, Ruiz E, Kohl G, Tremetsberger K, Gatica A, Letelier L, Novoa P, Novak J, Stuessy TF (2015) Progressive migration and anagenesis in Drimys confertifolia of the Juan Fernandez Archipelago, Chile. J Pl Res 128:73–90. https://doi.org/10.1007/s10265-014-0666-7
Luikart G, Allendorf FW, Cornuet JM, William BS (1998) Distortion of allele frequency distributions provides a test for recent population bottleneck. J Heredity 89:238–247. https://doi.org/10.1093/jhered/89.3.238
Mix C, Arens PFP, Rengelink R, Smulders MJM, Groenendael JMV, Ouborg NJ (2006) Regional gene flow and population structure of the wind-dispersed plant species Hypochaeris radicata (Asteraceae) in an agricultural landscape. Molec Ecol 15:1749–1758. https://doi.org/10.1111/j.1365-294X.2006.02887.x
Muellner AN, Tremetsberger K, Stuessy T, Baeza CM (2005) Pleistocene refugia and recolonization routes in the southern Andes: insights from Hypochaeris palustris (Asteraceae, Lactuceae). Molec Ecol 14:203–212. https://doi.org/10.1111/j.1365-294X.2004.02386.x
Nettancourt DE (1977) Incompatibility in angiosperms. Springer, Berlin
Paula GBN (2015) Estudos em Hypochaeris tropicalis Cabrera (Asteraceae) utilizando marcadores moleculares AFLP, SSR e ITS. MSc Thesis, Universidade Estadual de Londrina, Brazil
Petit RJ, Mousadik A, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conservation Biol 12:844–855. https://doi.org/10.1111/j.1523-1739.1998.96489.x
Pillar VP, Vélez E (2010) Extinção dos campos sulinos em unidades de conservação: um fenômeno natural ou um problema ético? Natureza Conserv 8:84–86. https://doi.org/10.4322/natcon.00801014
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: a program for detecting recent effective population size reductions from allele frequency data. J Heredity 90:502–503. https://doi.org/10.1093/jhered/90.4.502
R Development Core Team (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. Available at: http://www.R-project.org
Rambo B (1954) Análise histórica da flora de Porto Alegre. Sellowia 6:9–112
Rambo B (1956) A flora fanerogâmica dos aparados Riograndenses. Sellowia 7:35–298
Reck M (2010) Estudos moleculares em Hypochaeris catharinensis Cabrera (Asteraceae) utilizando marcadores AFLP. MSc Thesis, Universidade Estadual de Londrina, Brazil
Reck M, Benício LM, Ruas EA, Rodrigues LA, Ruas PM, Ortiz MA, Talavera S, Urtubey E, Stuessy T, Weiss-Schneeweiss H, Tremetsberger K, Michelan VS, Matzenbacher NI, Vanzela ALL, Terrab A, Samuel R, Ruas CF (2011) Karyotype and AFLP data reveal the phylogenetic position of the Brazilian endemic Hypochaeris catharinensis (Asteraceae). Pl Syst Evol 296:231–243. https://doi.org/10.1007/s00606-011-0490-7
Rodrigues LA, Ruas EA, Ruas PM, Reck M, Fiorin FG, Ortiz MA, Urtubey E, Matzenbacher NI, Ruas CF (2016) Population genetic structure of the South American species Hypochaeris lutea (Asteraceae). Pl Spec Biol 31:55–64. https://doi.org/10.1111/1442-1984.12084
Ruas CF, Vanzela ALL, Santos MO, Fregonezi JN, Ruas PM, Matzenbacher NI, Aguiar-Perecin MLR (2005) Chromosomal organization and phylogenetic relationships in Hypochaeris species (Asteraceae) from Brazil. Genet Molec Biol 28:129–139. https://doi.org/10.1590/S1415-47572005000100023
Samuel R, Stuessy TF, Tremetsberger K, Baeza CM, Siljak-Yakovlev S (2003) Phylogenetic relationships among species of Hypochaeris (Asteraceae, Cichorieae) based on ITS, plastid trnL intron, trnL-F spacer, and matK sequences. Amer J Bot 90:496–507. https://doi.org/10.3732/ajb.90.3.496
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Slatkin M (1995) A measure of population subdivision based on microsatellite allele Frequencies. Genetics 139:457–462
Stuessy TF, Tremetsberger K, Müllner AN, Jankowicz J, Guo Y-P, Baeza CM, Samuel RM (2003) The melding of systematics and biogeography through investigations at the populational level: examples from the genus Hypochaeris (Asteraceae). Basic Appl Ecol 4:287–296. https://doi.org/10.1078/1439-1791-00160
Tremetsberger K, Stuessy TF, Samuel RM, Baeza CM, Fay MF (2003) Genetics of colonization in Hypochaeris tenuifolia (Asteraceae, Lactuceae) on Volcan Lonquimay, Chile. Molec Ecol 12:2649–2659
Tremetsberger K, Weiss-Shneeweiss H, Stuessy T, Samuel R, Kadlec G, Ortiz MA, Talavera S (2005) Nuclear ribosomal DNA and karyotypes indicate a NW African origin of South American Hypochaeris (Asteraceae, Cichorieae). Molec Phylogen Evol 35:102–116. https://doi.org/10.1016/j.ympev.2004.12.022
Tremetsberger K, Stuessy TF, Kadlec G, Urtubey E, Baeza CM, Beck SG, Valdebenito HA, Ruas CF, Matzenbacher NI (2006) AFLP Phylogeny of South American Species of Hypochaeris (Asteraceae, Lactuceae). Syst Bot 31:610–626
Tremetsberger K, Gemeinholzer B, Zetzsche H, Blackmore S, Kilian N, Talavera S (2013) Divergence time estimation in Cichorieae (Asteraceae) using a fossil-calibrated relaxed molecular clock. Organisms Diversity Evol 13:1–13. https://doi.org/10.1007/s13127-012-0094-2
Tremetserger K, Urtubeye E, Terrab A, Baeza CM, Ortiz MA, Talavera M, Nig CKO, Temsch EM, Khol G, Talavera S, Stuessy T (2009) Pleistocene refugia and polytopic replacement of diploids by tetraploids in the Patagonian and Subantarctic plant Hypochaeris incana (Asteraceae, Cichorieae). Molec Ecol 18:3668–3682. https://doi.org/10.1111/j.1365-294X.2009.04298.x
Urtubey E, Stuessy TF, Tremetsberge K (2009) Systematics of the south American Hypochaeris sessiliflora complex (Asteraceae, Cichorieae). Ann Missouri Bot Gard 96:685–714. https://doi.org/10.3417/2006136
Van-Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molec Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Veloso HP, Góes-Filho L (1982) Fitogeografia Brasileira, Classificação Fisionômico-Ecológica da Vegetação. Boletim Técnico Projeto RADAMBRASIL. Vegetação 1:1–86
Wade MJ, McCauley DE (1988) Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution 42:995–1005
Wagner A (2008) Robustness and evolvability: a paradox resolved. Proc Biol Sci 275:91–100. https://doi.org/10.1098/rspb.2007.1137
Wang J (2011) COANCESTRY: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Molec Ecol Resources 11:141–145
Weiss-Schneeweiss H, Tremetsberger K, Schneeweiss GM, Parker JS, Stuessy TF (2008) Karyotype diversification and evolution in diploid and polyploid South American Hypochaeris (Asteraceae) inferred from rDNA localization and genetic fingerprint data. Ann Bot (Oxford) 101:909–918. https://doi.org/10.1093/aob/mcn023
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Yeh FC, Yang RC, Boyle T (1999) Popgene: version 1.31. Microsoft Window-based freeware for population genetics analysis. Available at: https://sites.ualberta.ca/~fyeh/popgene.html
Acknowledgements
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ; nos. 201254/2003-4 and 201332/2003-5), Fundação Araucária de Amparo à Pesquisa do Estado do Paraná (no. 024/2007, protocol no. 9791), and CAPES—Coordenação de Aperfeiçoamento de Pessoal de Nível Superior through scholarships for Ph.D. students and grants (PNPD no. 2238/2009) to postdoctoral fellows.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Handling Editor: Christoph Oberprieler.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Information on Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Materials
Information on Electronic Supplementary Materials
Online Resource 1. Geographic distance in km (above diagonal) and pairwise FST values (below diagonal) among populations of Hypochaeris catharinensis (P = 0.01).
Online Resource 2. Wilcoxon test for mutation-drift disequilibrium and heterozygosity deficiency/excess (Hd/He) estimated for 13 populations of Hypochaeris catharinensis (P ≤ 0.05).
Online Resource 3. Principal component analysis and eigenvalues based on allelic frequencies of Hypochaeris catharinensis populations.
Online Resource 4. Allele frequency for each population of Hypochaeris catharinensis and overall loci.
Online Resource 5. Relatedness estimated coefficient for 13 populations of Hypochaeris catharinensis.
Online Resource 6. Allelic richness (R) and allelic richness using rarefaction method (HP-rare) for 13 populations of Hypochaeris catharinensis; N is the number of individuals in each population and MLG is the multilocus genotype using rarefaction.
Rights and permissions
About this article

Cite this article
Chaves, C.L., Ruas, E.A., Ruas, C.F. et al. Genetic diversity and structure of Hypochaeris catharinensis Cabrera (Asteraceae), an endemic species of Southern Brazil. Plant Syst Evol 305, 675–686 (2019). https://doi.org/10.1007/s00606-019-01598-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-019-01598-y