
Abstract
The α-l-rhamnosidase (rha1) gene was homologously expressed in Aspergillus niger strains CCTCC 206047 and CCTCC 206047ΔpyrG, using hygromycin B and auxotrophic as selection markers. The engineered A. niger strains RHA001-1 and RHA003-1 were screened, yielding α-l-rhamnosidase activities of 20.81 ± 0.56 U/mL and 15.35 ± 0.87 U/mL, respectively. The copy numbers of the rha1 gene in strains RHA001-1 and RHA003-1 were found to be 18 and 14, respectively. Correlation analysis between copy number and enzyme activity in the A. niger strains revealed that α-l-rhamnosidase activity increased with the copy number of the rha1 gene. Recombinant α-l-rhamnosidase was utilized for the enzymatic debittering of Ougan juice, and its process conditions were optimized. Furthermore, the primary bitter substance neohesperidin (2.22 g/L) in Ougan juice was converted into hesperetin 7-O-glucoside (1.47 g/L) and hesperidin (0.143 g/L). This study presents a novel approach for the production of food-grade α-l-rhamnosidase and establishes a technical foundation for its application in the beverage industry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
α-l-rhamnosidase (E.C. 3.2.1.40) belongs to the glycoside hydrolase (GH) family, which can specifically hydrolyze l-rhamnose at the non-reducing terminus of a variety of glycoside compounds, such as naringenin, hesperidin, and rutin (Gao et al. 2021; Huang et al. 2016; Sun et al. 2021; Wang et al. 2020). α-l-rhamnosidase is a crucial enzyme in biotechnological applications, particularly in the beverage, food, and pharmaceutical industries (Fang et al. 2019; Peng et al. 2021; Xu et al. 2019).
α-l-rhamnosidase is widespread in many animals, plants, and microbes and was first discovered as a complex enzyme in celery seeds (Hall 1938; Qian et al. 2013; Shin et al. 2016; Suzuki 1962). α-l-rhamnosidase from microorganisms is the most widely used enzyme, and its production is mainly dependent on microbial fermentation. Various microbial taxa such as Bacillus sp. (Lyu et al. 2016), Lactobacillus sp. (Ferreira-Lazarte et al. 2021), and Mycobacterium sp. (Wu et al. 2018) among Aspergillus sp. (Li et al. 2022; Lyu et al. 2019) and Penicillium sp. (Xie et al. 2022) and Rhizoctonia sp. (Monti et al. 2004), as well as Pichia angusta X349 (Yanai and Sato 2000), Cryptococcus albidus (Borzova et al. 2017), and Clavispora lusitaniae (Singh et al. 2015) in yeast, and many other microbial taxa are capable of producing α-l-rhamnosidase.
To meet the demand for the industrialized production of α-l-rhamnosidase, the construction of engineered strains that efficiently express the recombinant protein of α-l-rhamnosidase through genetic engineering technology has become the main direction of current research. The α-l-rhamnosidase gene was amplified from the genomes of Aspergillus niger M 2018240 (Wang et al. 2019) and Aspergillus oryzae RIB40 (Ishikawa et al. 2017), and P. pastoris GS115 was used as the host. The optimal recombinant strains obtained had p-nitrophenyl-α-l-rhamnopyranoside (pNPR) hydrolyzing activities of 0.57 U/mg and 5.4 U/mg, respectively. Aspergillus-derived α-l-rhamnosidase showed good catalytic activity toward naringin. The α-l-rhamnosidase gene from A. tubingensis (Li et al. 2019) and A. niger JMU-TS528 (Li et al. 2016) were expressed in P. pastoris GS115, and the hydrolytic activities of the recombinant enzymes to naringin were 120 U/mg and 711.9 U/mL, respectively. In addition, Aspergillus-derived α-l-rhamnosidase genes are predominantly heterologously expressed in P. pastoris as a eukaryotic host and homologous expression is seldom involved. A. niger is a significant supplier of α-l-rhamnosidase, which the FDA classifies as generally regarded as safe (GRAS), so α-l-rhamnosidase expressed by A. niger can be used in food additives. After homologous expression of α-l-rhamnosidase in A. niger 3.350, the pNPR hydrolyzing activity of the target protein was only 0.658 U/mL after fermentation in a 5-L tank (Ye et al. 2022). Therefore, it is of great significance to further investigate the homologous expression of α-l-rhamnosidase from Aspergillus sp. to improve the enzyme activity.
Ougan (Citrus reticulata cv. Suavissima) is a traditional citrus species in Zhejiang province, China. Ougan is rich in vitamin C and a variety of minerals, with high nutritional value, and functions in cooling and detoxifying, resolving phlegm, and relieving cough (Zhang et al. 2014). Ougan has a strong bitter taste, it has been shown that the main bitter substances in Ougan are neohesperidin and naringin (Li et al. 2017; Yadav et al. 2013; Ye et al. 2011). Bitter substances seriously affect the taste of Ougan and lead to its low competitiveness in the citrus market; therefore, removing the bitter substances in Ougan is a focus of attention. α-l-rhamnosidase can hydrolyze bitter neohesperidin into non-bitter hesperetin 7-O-glucoside, which effectively reduces the bitterness of Ougan juice. In addition, pectinase produced by A. niger during fermentation breaks down pectin to clarify the juice, and β-d-glucosidase further hydrolyzes hesperidin monoglucoside to hesperidin.
The main production mode of α-l-rhamnosidase is microbial fermentation. In the fermentation process of strains, the fermentation yield of original strains is low, many extracellular enzymes are produced, and purification is difficult, so it is an inevitable trend to obtain strains with high expression of this enzyme by modern molecular biology technology. This study from NCBI database retrieval to A. niger α-l-rhamnosidase gene (GenBank: XM_001389049.1), the rha1 gene was amplified from the genome of A. niger CCTCC 206047, and two recombinant plasmids were constructed. Homologous expression of α-l-rhamnosidase was achieved in A. niger CCTCC 206047 and CCTCC 206047ΔpyrG strains using PEG-mediated protoplast transformation with hygromycin B and auxotrophic as selection markers, respectively. The crude enzyme of A. niger engineered strain was applied to the enzymatic debittering of Ougan juice, and the optimal process conditions for enzymatic de-bittering of Ougan juice were explored.
Materials and methods
Microbial strains and plasmids
A. niger CCTCC 206047, A. niger CCTCC 206047ΔpyrG strain, and plasmid pCAMBIA-PglaA-TcbhI-hph-PtrpC are kept in our laboratory. Takara Biotechnology (Dalian, China) provided the pMD19-T vector, while Tsingke Biotechnology (Hangzhou, China) provided the E. coli DH5α.
Chemicals
The SYRB Green Realtime PCR Master and ClonExpress MultiS One Step Cloning Kit were purchased from Vazyme (Nanjing, China). The Column Fungal Genomic DNA Extraction Kit was bought from Sangon (Shanghai, China). Zhejiang Senma Ecological Agriculture Development Co., Ltd (Wenzhou, China) was the source of Ougan (Citrus reticulata cv. Suavissima) fruit. Hesperidin, hesperetin, hesperetin 7-O-glucoside, and p-Nitrophenyl-α-l-rhamnose were purchased from Aladdin Co., Ltd (Shanghai, China).
Sequence analysis of α-l-rhamnosidase gene
Based on published protein profiles of α-l-rhamnosidase and commercial α-l-rhamnosidase, the rha1 gene (GeneBank: XM_001389049.1) from A. niger was retrieved from GeneBank (Ye et al. 2022). The α-l-rhamnosidase sequences from different Aspergillus were analyzed for homology using Blast in NCBI (https://www.ncbi.nlm.nih.gov). MEGA-X software (https://www.megasoftware.net) was used for sequence alignment and phylogenetic tree construction based on the neighbor-joining method. The signal peptide of the rha1 gene was predicted using the SignalP 5.0 program (https://services.healthtech.dtu.dk/ services/SignalP-5.0) (Nielsen 2017).
Construction of recombinant plasmids
After activation of A. niger CCTCC 206047 on PDA solid medium, a small number of spores were inoculated into the DPY medium and cultured at 30 °C for two days. Mycelia were collected and the genome was extracted according to the instructions of the Column Fungal Genomic DNA Extraction Kit (Sangon Biotech, Shanghai, China).
The genomic DNA of A. niger CCTCC 206047 was used as a template to amplify the rha1 gene using the specific primers rha1-F and rha1-R (Table S1). The PCR products were used as templates to amplify a fragment in addition to its signal peptide and with the homology arm of plasmid pCAMBIA-PglaA-TcbhI-hph-PtrpC using primers Sig-rha1-F and Sig-rha1-R (Table S1). The plasmid pCAMBIA-PglaA-TcbhI-hph-PtrpC was used as the template, and the linearized fragment of the plasmid was reverse amplified with primers pCAMBIA-F and pCAMBIA-R (Table S1). It was then ligated with the rha1 gene according to the instructions of the ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China) and transformed into E. coli DH5α competent cells. Single colonies were chosen and identified by PCR and sequencing, following overnight culture at 37 °C. The correct plasmid was named pCAMBIA-rha1.
The genomic DNA of A. niger CCTCC 206047 was used as the template to amplify the pyrG gene (GenBank: XM_001395395.2) with specific primers and to generate the homology arm of the plasmid pCAMBIA-rha1. The constructed plasmid pCAMBIA-rha1 was used as the template, and the linearized plasmid pCAMBIA-rha1 was reverse amplified using primers pCAMBIA-rha-F and pCAMBIA-rha-R (Table S1). The pyrG gene was ligated with pCAMBIA-rha1, and the recombinant product was transformed into E. coli DH5α. The positive recombinant plasmid screened using PCR and sequencing was named pCAMBIA-rha1-pyrG.
Plasmid transformation and screening of transformants
Transformation of A. niger CCTCC 206047 using hygromycin B as a selection marker. When transforming the CCTCC 206047ΔpyrG strain, auxotrophic was used as a selection marker, and the selection marker gene pyrG encoded orotidine 5′-phosphate decarboxylase. Strains with deletion of pyrG are unable to synthesize uracil/uridine and should be supplemented with additional uracil/uridine for normal growth. After the pyrG gene was transferred to the CCTCC 206047ΔpyrG strain, the recipient strain returned to a phenotype and could grow on basal media.
A. niger CCTCC 206047 and A. niger CCTCC 206047ΔpyrG strains were inoculated into DPY medium and cultured for 24 h to prepare protoplasts. The linearized plasmids pCAMBIA-rha1 and pCAMBIA-rha1-pyrG carrying the pyrG gene were randomly integrated into the genomes of A. niger and CCTCC 206047ΔpyrG using the PEG-mediated protoplast transformation method, with hygromycin B and auxotrophic as selection markers. After culturing at 30 °C, the transformants with A. niger CCTCC 206047 as the host were transferred to PDA solid medium containing hygromycin B, while the transformants with A. niger CCTCC 206047ΔpyrG as the host were transferred to non-resistant PDA solid medium and cultured at 30 °C for 4 days. Transformant spores were inoculated into DPY medium and incubated at 30 °C for 2 days. The mycelium was collected and the genome was extracted and identified using PCR and sequencing to screen for positive transformants.
Cultivation of A. niger engineered strain
Positive transformants were inoculated on PDA solid medium and cultured at 30 °C for four days. The spores were then washed with sterile water to prepare a spore suspension containing 107 spores/ml. After inoculation in the seed media, the spore suspension was grown for 24 h at 30 °C and 220 rpm. The 2% (v/v) seed solution was transferred to the fermentation medium and incubated at 30 °C and 220 rpm for 96 h to produce α-l-rhamnosidase. The supernatant of the fermentation broth was collected by centrifugation at 12,000 rpm for 10 min at the end of fermentation. The enzyme activity and copy number of α-l-rhamnosidase in the fermentation broth were determined and SDS-PAGE was performed.
α-l-Rhamnosidase activity assay
pNPR was used as a substrate to determine the rha activity (Yadav et al. 2010). 10 μL pNPR (10 mM) was added to 480 μL phosphate citrate buffer (pH 4.5) and preheated at 60 °C for 3 min, after which 10 μL of enzyme solution or inactivated enzyme solution (control group) was added for 10 min, and the reaction was stopped by the addition of 500 μL of 1 M sodium carbonate solution. The release of p-nitrophenol (p-NP) was measured at 405 nm, three parallel in each group. The amount of enzyme required to produce 1 μmol of p-NP per minute was defined as one unit (U).
Gel electrophoresis
The homogeneity and molecular weight of the purified enzyme were estimated using sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE). The separation gel had a concentration of 12% acrylamide, while the concentration gel had 5% acrylamide. Proteins were stained with Coomassie Brilliant Blue R-250. A 180 kDa Prestained Protein Marker (Vazyme Nanjing) was used as the molecular weight standard.
Determination of rha1 gene copy number
The copy number of the rha1 gene of the recombinant A. niger strains was determined using the double standard curve method of real-time quantitative PCR (q-PCR) with gpdA as the reference gene. The standard plasmids T-Vector-gpdA and T-Vector-rha1 were constructed, and the plasmid copy number was calculated according to the following formula: copies/μL = (6.02 × 1023) × (ng/μL × 10–9) / (DNA length × 660). The two standard plasmids were diluted to 109–104 copies/μL by the serial dilution method. The standard plasmid was subjected to q-PCR using RT-gpdA-F, RT-gpdA-R, RT-rha1-F, and RT-rha1-R as primers; the specific sequences of the primers are shown in Table S1. The standard curve was plotted with the copy number of the standard plasmid along the X-axis and the measured Cp value along the Y-axis. The genome of the positive transformants was used as the template for qPCR to calculate the copy number of rha1 in the recombinant A. niger strain.
Preparation conditions of Ougan juice and its analytical methods
The seed was peeled and removed before the pulp was squeezed, the crude juice was centrifuged at 12,000 rpm for 5 min, and the supernatant was the juice from Ougan.
Ougan juice was prepared by peeling and removing the seed, the Ougan pulp was squeezed, the crude juice was centrifuged at 12, 000 rpm for 5 min, and the supernatant was the Ougan juice. Determination of flavonoid content and transmittance of ougan juice.
The Ougan juice was appropriately diluted and filtered through a 0.22 μm membrane, and then a high-performance liquid chromatography (HPLC) system (Waters2695, China) was used for the quantitative analysis of flavonoids in Ougan juice. HPLC was performed on a UItimate® AQ_C18 column (3 μm, 250 × 4.6 mm, Shanghai, China) with a flow rate of 1 mL/min and an injection volume of 10 μL; mobile phase A was 0.1% formic acid and mobile phase B was 100% acetonitrile. Gradient elution was performed using 85%A at 0–1 min, 75%A at 1–4 min, 60%A at 4–14 min, 50%A at 14–24 min, and 85%A at 24–36 min. The contents of neohesperidin and its hydrolyzed products, hesperetin 7-O-glucoside and hesperidin, were determined by HPLC at 283 nm.
The absorbance of Ougan juice at 680 nm was determined using an ultraviolet–visible spectrophotometer (distilled water as control), and the transmittance was calculated. The larger the transmittance, the higher the clarity, which was calculated using the following equation:
where, T-transmittance, A-absorbance value.
Research on enzymatic debittering process of Ougan juice
The optimum debittering conditions for Ougan juice were investigated using a crude enzyme solution of recombinant α-l-rhamnosidase. Ougan juice (10 mL) was added to a 15 mL centrifuge tube and preheated in a metal bath at 60 °C, and then different volumes (0%, 0.01%, 0.05%, 0.1%, 0.15%, and 0.2% v/v) of α-l-rhamnosidase enzyme solution were added. Enzymatic hydrolysis at different temperatures for different times. The temperature was varied from 30 to 70 °C, and samples were taken every ten min of reaction to determine the enzyme activity until the reaction reached 80 min. The reaction liquid was boiled at the end of the reaction, the contents of neohesperidin, hesperetin 7-O-glucoside, and hesperidin were detected by HPLC, and the transmittance of the Ougan juice was determined using an ultraviolet–visible spectrophotometer.
Statistical analysis
Data values were expressed as mean ± standard deviation (SD). All data were analyzed for normality prior to statistical testing by Origin®2021 (OriginLab Corporation). The Student's t-test was applied for comparisons between the two groups. The values of several groups were analyzed using one-way ANOVA. Statistical significance was defined as P-value < 0.05.
Results and discussion
Sequence analysis of α-l-rhamnosidase gene
Phylogenetic analysis of the amino acid sequences of α-l-rhamnosidases using MEGA-X software showed that the eight α-l-rhamnosidases were divided into four branches: rha1 and rha7, rha4 and rha6, rha3 and rha2, and rha5 and rha8 (Fig. 1). rha1 shares 96% identity with r-Rha1 from A. niger JMU-TS528 (Li et al. 2016) and 99% identity with Rha from A. niger CCTCC M 2018240 (Wang et al. 2019). Similar to most α-l-rhamnosidases from fungi, α-l-rhamnosidase (rha1) from A. niger also exhibits low homology with α-l-rhamnosidases from bacteria (Miyata et al. 2005). Amino acids 1–17 in the N-terminus of rha1 are its signal peptides. Previous studies have shown that the glaA signal peptide contributes to the expression and secretion of α-galactosidase in A. niger (Xu et al. 2018). Therefore, the signal peptide of rha1 itself was removed when constructing the recombinant plasmid, and the glaA signal peptide wasused in this study.

Phylogenetic tree analysis of eight α-l-rhamnosidase
Construction of recombinant plasmids
The results of 1% agarose gel electrophoresis showed that the length of the rha1 gene and linearized plasmid pCAMBIA-PglaA-TcbhI-hph-PtrpC were approximately 2000 bp and, respectively (Fig. 2A and B), which is consistent with the expected size. The ligated products of the rha1 gene and plasmid pCAMBIA-PglaA-TcbhI-hph-PtrpC were transformed into E. coli DH5α, and the positive transformants were screened by PCR and sequencing with a fragment of 885 bp (Fig. 2C), indicating that the pCAMBIA-rha1 plasmid had been successfully constructed.

Construction of recombinant plasmids. Lane M: DNA marker. A Electrophoresis results of rha1 gene. B Electrophoretic results of the linearized plasmid pCAMBIA-PglaA-TcbhI-hph-PtrpC. Lane 1: The linearized plasmid. C Colony PCR electrophoresis results of E. coli DH5α transformants of recombinant plasmid. Lane 1–10: Colony PCR bands of plasmid pCAMBIA-rha1 transformants. D Electrophoresis results of the pyrG gene. Lane 1: pyrG gene. E Electrophoresis results of the linearized plasmid pCAMBIA-rha1. Lane 1: pCAMBIA-rha1 linearized plasmid. F Colony PCR electrophoresis results of E. coli DH5α transformants of recombinant plasmid. Lane 1–10: Colony PCR bands of plasmid pCAMBIA-rha1-pyrG transformants
The results of 1% agarose gel electrophoresis showed that the length of the pyrG gene and pCAMBIA-rha1 linearized plasmid were 2000 bp and more than 8000 bp, respectively (Fig. 2D and E), which is consistent with the expected size. The ligated products of pyrG and plasmid pCAMBIA-rha1 were transformed into E. coli DH5α, and positive transformants were screened by PCR and sequencing (Fig. 2F). The plasmid, pCAMBIA-rha1-pyrG, was successfully constructed.
Transformation of A. niger with α-l-rhamnosidase
A. niger was transformed using PEG-mediated protoplast transformation. The positive control with A. niger CCTCC 206047 as the host did not contain resistant drugs, while the positive control with the CCTCC 206047ΔpyrG strain as the host was supplemented with uracil nucleosides, so the positive control grew normally. The negative controls received screening pressure for adding hygromycin B or not supplementing the uracil nucleoside, and failed to grow the mycelium. The experimental group was transferred to A. niger CCTCC 206047 and CCTCC 206047ΔpyrG strains containing hygromycin B and carrying the pyrG gene, respectively, and the transformants were able to grow normally on the screening plates. Single colonies on the transformation plates were selected for passaging.
Twenty-three transformants of the A. niger CCTCC 206047 strain were selected for genomic validation, of which five were positive transformants (Fig. 3A), with a positive transformation rate of only 21.7%. In contrast, ten transformants from the CCTCC 206047ΔpyrG strain were selected for genomic validation, of which nine were positive transformants (Fig. 3B), with a 90% positive transformation rate. The results of PCR and sequencing were consistent with expectations, indicating that two strains of A. niger CCTCC 206047 and CCTCC 206047ΔpyrG with homologous expression of α-l-rhamnosidase were successfully constructed and named RHA001 and RHA003, respectively.

Electrophoresis results of genomic PCR of A. niger transformants. Lane M: DNA marker. A Lane 1–23: PCR electrophoresis results of transformants from A. niger CCTCC 206047 strain. Lane 24: Control A. niger CCTCC 206047. B Lane 1–10: PCR electrophoresis results of transformants from A. niger CCTCC 206047ΔpyrG strain. Lane 11: Control A. niger CCTCC 206047ΔpyrG strain
Enzyme activity of recombinant α-l-rhamnosidase
Transformants were selected from the stable genetically recombinant A. niger strains RHA001 and RHA003 for inoculation into PDA solid media, numbered RHA001-1 to RHA001-3 and RHA003-1 to RHA003-6, respectively. The activity of α-l-rhamnosidase was determined after fermentation. The results showed that The enzyme activity of RHA001-1 in the three recombinant A. niger strains (RHA001-1 to RHA001-3) was the highest at 20.81 ± 0.56 U/mL, which was 6.29-fold higher than that of the host A. niger CCTCC 206047. Among the six recombinant A. niger strains (RHA003-1 to RHA003-6), RHA003-1 had the highest enzyme activity 15.35 ± 0.87 U/mL, which was 7.04-fold higher than that of the CCTCC 206047ΔpyrG strain.
Ye et al. expressed the synthetic rha1 gene in A. niger 3.350 strain, and the activity of α-l-rhamnosidase was 0.471 U/mL (Ye et al. 2022), whereas the enzyme activity of the α-l-rhamnosidase from the A. niger engineered strain constructed in this study was much higher than this level. This may be because of the following two factors. First, the A. niger used in this study is an excellent strain that secretes α-l-rhamnosidase, which has a strong ability to secrete and express endogenous α-l-rhamnosidase. Second, in this study, the rha1 gene was amplified from A. niger genomic DNA, and the retention of the two introns contained in the rha1 gene was beneficial for gene expression, which was consistent with previously reported findings (Zhu et al. 2020). The presence of introns may make the secondary structure more stable, thereby protecting the precursor mRNA from degradation in the nucleus, and ultimately increasing the expression level of the enzyme.
SDS-PAGE analysis of recombinant α-l-rhamnosidase
The results of SDS-PAGE showed that the bands of the fermentation broth of the recombinant A. niger strains RHA001 and RHA003 became thicker at about 100 kDa, but the theoretical molecular weight of rha1 was about 70 kDa (Fig. 4A and B). The analysis results of NetNGlyc 1.0 software showed 13 potential N-glycosylation sites in the Rha1 gene. This suggests that post-translational N-glycosylation modification of fungal proteins results in a larger molecular weight of Rha1. The study results of Li et al. also reported that the apparent molecular weight of A. niger α-l-rhamnosidase was high after recombinant expression in Pichia pastoris (Li et al. 2016). Subsequently, the protein band was identified by mass spectrometry, and the results showed that the band contained Rha1. Among the six recombinant A. niger strains, RHA001-1 and RHA003-1 had deeper bands of the target protein, indicating that the protein expression levels of these two engineered strains were higher, and the identification results were consistent with the results of enzyme activity.

SDS-PAGE analysis of fermentation broth of A. niger engineered strains. Lane M: protein marker. A SDS-PAGE of A. niger RHA001 fermentation broth. Lane 1: Control A. niger CCTCC 206047. Lane 2–4: RHA001-1, RHA001-2, RHA001-3 fermentation supernatant. B SDS-PAGE of A. niger RHA003 fermentation broth. Lane 1: Control A. niger CCTCC 206047ΔpyrG strain. Lanes 2–4: RHA003-1, RHA003-2, RHA003-3 fermentation supernatant
Analysis of the copy number of the rha1 gene
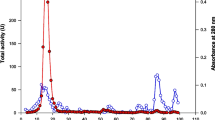
The copy number of A. niger engineered strains were determined by the double standard curve method of q-PCR, the copy number of the rha1 gene of A. niger CCTCC 206047 was 1, while A. niger engineered strains were all more than 2 (Table S2). Correlation analysis of copy number and enzyme activity of A. niger engineered strains revealed that α-l-rhamnosidase activity was positively correlated with the copy number of the rha1 gene, and the engineered strain RHA001-1, with the highest enzyme activity, also had the highest copy number, which was 18 (Fig. 5).

Correlation between rha1 gene copy number and α-l-rhamnosidase activity
At the same time, the results of this study also showed that increasing the copy number of the glaA gene in the A. niger genome can increase the expression of glucoamylase, and when the copy number is below 20, the copy number has a linear relationship with expression (Verdoes et al. 1993). The results of this study support this finding.
Effect of recombinant α-l-rhamnosidase on enzymatic debittering of Ougan juice
Ougan juice was subjected to HPLC before enzymolysis, and it was found that the main bitter substance in Ougan juice was neohesperidin (Fig. S1D), with a content of 2.22 g/L, and the transmittance of Ougan juice was 16.3%. Ougan juice was hydrolyzed by the crude enzyme solution of A. niger engineered strain RHA001-1, and the Ougan juice from the enzymolysis process was subjected to HPLC. The bitter substance, neohesperidin, was gradually hydrolyzed to hesperetin 7-O-glucoside and hesperidin during enzymatic hydrolysis (Fig. S1E). Because the crude enzyme solution also contained β-d-glucosidase, the bitter substance neohesperidin could not be completely hydrolyzed when too little enzyme solution was added. However, when too much enzyme solution is added, β-d-glucosidase hydrolyzes hesperetin 7-O-glucoside into hesperidin, reducing the content of the product, hesperetin 7-O-glucoside, thus affecting the debittering effect of Ougan juice. When the crude enzyme solution was added at 0.1%, neohesperidin was completely enzymatically hydrolyzed by α-l-rhamnosidase, and the debittering effect of Ougan juice was the best (Fig. 6A).

Effects of enzyme addition, enzymolysis temperature and enzymolysis time on the conversion of neohesperidin in Ougan juice. A Effect of crude enzyme solution addition on the conversion of neohesperidin in Ougan juice. B Effect of enzymolysis temperature on the conversion of neohesperidin in Ougan juice. C Effect of enzymolysis time on the conversion of neohesperidin in Ougan juice
The different optimal enzymatic hydrolysis temperatures for the ectoenzymes of A. niger directly affect enzyme activity, thus affecting the debittering effect of the Ougan juice. After enzymatic hydrolysis at 50 °C and 60 °C for 20 min, the neohesperidin content in Ougan juice was similar, but that of hesperetin 7-O-glucoside was lower in Ougan juice at 50 °C. This may be because the activity of β-d-glucosidase is higher at 50 °C, whereas the activity of α-l-rhamnosidase is higher at 60 °C. Therefore, the debittering effect of Ougan juice was best at 60 °C (Fig. 6B).
The transmittance of Ougan juice increased with increasing enzymolysis time (Fig. 6C), and the reason for this result is the presence of pectinase in the A. niger RHA001-1 enzyme solution. It has been shown that pectinase can prevent the flocculation of soluble solids by breaking down pectin. Thus, it improves juice transmission (Chen et al. 2023). The activity of pectinase in the A. niger RHA001-1 crude enzyme solution was measured at 1394.58 U/mL. After enzymatic hydrolysis for 50 min, the transmittance of the Ougan juice increased slightly, indicating that the clarification effect of pectinase on Ougan juice was more significant in the first 50 min. The neohesperidin was completely hydrolyzed at 60 min, and the contents of hesperetin 7-O-glucoside and hesperidin were 1.47 g/L and 0.143 g/L, respectively, and the transmittance of Ougan juice was 34.4% at this time. After 60 min, hesperetin 7-O-glucoside was further hydrolyzed to hesperidin by β-d-glucosidase; thus, 60 min was determined to be the optimal enzymatic hydrolysis time (Fig. 6C).
Conclusions
This study will rha1 gene was homologously expressed in A. niger CCTCC 206047 and CCTCC 206047ΔpyrG strains. Two recombinant strains with high expression of Rha1, RHA001-1 and RHA003-1, were successfully screened, and their enzyme activities were 20.81 ± 0.56 U/mL and 15.35 ± 0.87 U/mL, respectively. Enzyme activity was positively correlated with the rha1 gene copy number, and the rha1 gene copy number of the engineered strain RHA001-1 was as high as 18. The crude enzyme solution of A. niger engineered strain RHA001-1 was used for the enzymatic degradation of Ougan juice, and the bitter substance neohesperidin was converted into hesperetin 7-O-glucoside and hesperidin, which effectively improved the taste of Ougan juice. Homologous expression of α-l-rhamnosidase was achieved in A. niger, and the expressed α-l-rhamnosidase could be used as a food-grade enzyme. Moreover, the enzyme solution of the A. niger engineered strain was used to enzymatically hydrolyze Ougan juice to improve its taste, providing a reference for the application of A. niger enzyme solution in the food field.
Data availability
The data are available from the corresponding author on reasonable request.
References
Borzova N, Gudzenko O, Varbanets L (2017) Purification and characterization of a naringinase from Cryptococcus albidus. Appl Biochem Biotechnol 184:953–969
Chen X, Xu Y, Wu J, Yu Y, Zou B, Li L (2023) Effects of pectinase pre-treatment on the physicochemical properties, bioactive compounds, and volatile components of juices from different cultivars of guava. Foods 12:330
Fang X, Dong Y, Xie Y, Wang L, Wang J, Liu Y, Cao F (2019) Effects of β-glucosidase and α-rhamnosidase on the contents of flavonoids, ginkgolides, and aroma components in ginkgo tea drink. Molecules 24:2009
Ferreira-Lazarte A, Plaza-Vinuesa L, De las Rivas B, Villamiel M, Muñoz R, Moreno FJ (2021) Production of α-rhamnosidases from Lactobacillus plantarum WCFS1 and their role in deglycosylation of dietary flavonoids naringin and rutin. Int J Biol Macromol 193:1093–1102
Gao X, Feng T, Liu E, Shan P, Zhang Z, Liao L, Ma H (2021) Ougan juice debittering using ultrasound-aided enzymatic hydrolysis: impacts on aroma and taste. Food Chem 345:128767
Hall DH (1938) A new enzyme of the glycosidase type. Nature (London) 142:150–150
Huang G, Lv M, Hu J, Huang K, Xu H (2016) Glycosylation and activities of natural products. Mini Rev Med Chem 16:1013–1016
Ishikawa M, Shiono Y, Koseki T (2017) Biochemical characterization of Aspergillus oryzae recombinant α-l-rhamnosidase expressed in Pichia pastoris. J Biosci Bioeng 124:630–634
Li L, Yu Y, Zhang X, Jiang Z, Zhu Y, Xiao A, Chen F (2016) Expression and biochemical characterization of recombinant α-l-rhamnosidase r-Rha1 from Aspergillus niger JMU-TS528. Int J Biol Macromol 85:391–399
Li D, Tang Y, Lin J, Cai W (2017) Methods for genetic transformation of filamentous fungi. Microb Cell Fact 16:168
Li L, Gong J, Wang S, Li G, Gao T, Jiang Z, Li Q (2019) Heterologous expression and characterization of a new clade of Aspergillus α-l-rhamnosidase suitable for citrus juice processing. J Agric Food Chem 67:2926–2935
Li Q, Ge L, Zheng D, Zhang X, Zhao L (2022) Screening and characterization of a GH78 α-l-rhamnosidase from Aspergillus terreus and its application in the bioconversion of icariin to icaritin with recombinant β-glucosidase. Enzyme Microb Technol 153:109940
Lyu W, Wu Y, Liu Y, Lyu Z (2016) Draft genome sequence of Bacillus Litoralis c44, isolated from Chinese scholar tree (sophora japonica) forest soil. Genome Announc 4:e01059-e1116
Lyu Y, Zeng W, Du G, Chen J, Zhou J (2019) Efficient bioconversion of epimedin C to icariin by a glycosidase from Aspergillus nidulans. Biores Technol 289:121612
Miyata T, Kashige N, Satho T, Yamaguch T, Aso Y, Miake F (2005) Cloning, sequence analysis, and expression of the gene encoding Sphingomonas paucimobilis fp2001 α-l-rhamnosidase. Curr Microbiol 51:105–109
Monti D, Pisvejcová A, Kren V, Lama M, Riva S (2004) Generation of an alpha-L-rhamnosidase library and its application for the selective derhamnosylation of natural products. Biotechnol Bioeng 87:763–771
Nielsen H (2017) Predicting secretory proteins with SignalP. In: Kihara D (ed) Protein function prediction. Springer, Newyork, pp 59–73
Peng C, Li R, Ni H, Li LJ, Li QB (2021) The effects of α-L-rhamnosidase, β-d-glucosidase, and their combination on the quality of orange juice. J Food Process Preserv 45:86–96
Qian S, Wang H, Zhang C, Yu H (2013) Isolation and characterization of dioscin-α-l-rhamnosidase from bovine liver. J Mol Catal B Enzym 97:31–35
Shin NR, Moon JS, Shin SY, Li L, Lee YB, Kim TJ, Han NS (2016) Isolation and characterization of human intestinal Enterococcus avium EFEL009 converting rutin to quercetin. Lett Appl Microbiol 62:68–74
Singh P, Sahota PP, Singh RK (2015) Evaluation and characterization of new α-L-rhamnosidase-producing yeast strains. J Gen Appl Microbiol 61:149–156
Sun J, Li W, Liao H, Li L, Ni H, Chen F, Li Q (2021) Adding sorbitol improves the thermostability of α-l-rhamnosidase from Aspergillus niger and increases the conversion of hesperidin. J Food Biochem 46:14055
Suzuki H (1962) Hydrolysis of flavonoid glycosides by enzymes (rhamnodiastase) from Rhamnus and other sources. Arch Biochem Biophys 99:476–483
Verdoes JC, Punt PJ, Schrickx JM, Van Verseveld HW, Stouthamer AH, Van Den Hondel CA (1993) Glucoamylase overexpression in Aspergillus niger: molecular genetic analysis of strains containing multiple copies of the glaA gene. Transgenic Res 2:84–92
Wang D, Zheng P, Chen P (2019) Production of a recombinant α-l-rhamnosidase from Aspergillus niger CCTCC M 2018240 in Pichia pastoris. Appl Biochem Biotechnol 189:1020–1037
Wang D, Zheng P, Chen P, Wu D (2020) Highly efficient enzymatic conversion of rutin to isoquercitrin and l-rhamnose using deep eutectic solvents. ACS Sustain Chem Eng 8:14905–14913
Wu T, Pei J, Ge L, Wang Z, Ding G, Xiao W, Zhao L (2018) Characterization of a α-l-rhamnosidase from Bacteroides thetaiotaomicron with high catalytic efficiency of epimedin C. Bioorg Chem 81:461–467
Xie J, Zhao J, Zhang N, Xu H, Yang J, Ye J, Jiang J (2022) Efficient production of isoquercitin, icariin and icariside II by a novel thermostable α-l-rhamnosidase PodoRha from Paenibacillus odorifer with high α-1, 6-/α-1, 2- glycoside specificity. Enzyme Microb Technol 158:110039
Xu Y, Wang YH, Liu TQ, Zhang H, Zhang H, Li J (2018) The glaA signal peptide substantially increases the expression and secretion of α-galactosidase in Aspergillus niger. Biotech Lett 40:949–955
Xu L, Liu X, Li Y, Yin Z, Jin L, Lu L, Xiao M (2019) Enzymatic rhamnosylation of anticancer drugs by an α-l-rhamnosidase from Alternaria sp. L1 for cancer-targeting and enzyme-activated prodrug therapy. Appl Microbiol Biotechnol 103:7997–8008
Yadav V, Yadav PK, Yadav S, Yadav KDS (2010) α-l-Rhamnosidase: a review. Process Biochem 45:1226–1235
Yadav S, Yadav RSS, Yadav KDS (2013) An α-l-rhamnosidase from Aspergillus awamori MTCC-2879 and its role in debittering of orange juice. Int J Food Sci Technol 48:927–933
Yanai T, Sato M (2000) Purification and characterization of an alpha-L-rhamnosidase from Pichia angusta X349. Biosci Biotechnol Biochem 64:2179–2185
Ye X-Q, Chen J-C, Liu D-H, Jiang P, Shi J, Xue S, Kakuda Y (2011) Identification of bioactive composition and antioxidant activity in young mandarin fruits. Food Chem 124:1561–1566
Ye H, Li X, Li L, Zhang Y, Zheng J (2022) Homologous expression and characterization of α-l-rhamnosidase from Aspergillus niger for the transformation of flavonoids. Appl Biochem Biotechnol 194:3453–3467
Zhang J, Wu Y, Zhao X, Luo F, Li X, Zhu H, Chen K (2014) Chemopreventive effect of flavonoids from Ougan (Citrus reticulata cv. Suavissima) fruit against cancer cell proliferation and migration. J Funct Foods 10:511–519
Zhu SY, Xu Y, Yu XW (2020) Improved homologous expression of the acidic lipase from Aspergillus niger. J Microbiol Biotechnol 30:196–205
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31600639) and Application Development Special Key Project of Chongqing (cstc2021jscx-jbgsX0002).
Supplementary Information
Supplementary Figure S1—Enzymatic hydrolysis of Ougan juice by α-L-rhamnosidase. (A) Standard of neohesperidin. (B) Standard of hesperetin 7-O-glucoside. (C) Standard of hesperidin. (D) Flavonoids contained in Ougan juice before enzymatic hydrolysis. (E) Flavonoids contained in Ougan juice during enzymatic hydrolysis.
Supplementary Table S1—The primers used in this study
Supplementary Table S2—Copy number of rha1 gene in Aspergillus niger engineered strains
Funding
This work was funded by National Natural Science Foundation of China, no. 31600639, jianyong zheng, Application Development Special Key Project of Chongqing, cstc2021jscx-jbgsX0002, Zhao Wang
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Ethical approval
Not applicable.
Consent to participate
The final manuscript has been seen and approved by all the authors.
Consent to publication
All the authors mutually agreed that the work should be published in Biotechnology Letters.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, F., Wang, X., Pan, L. et al. Homologous expression of Aspergillus niger α-l-rhamnosidase and its application in enzymatic debittering of Ougan juice. Biotechnol Lett (2024). https://doi.org/10.1007/s10529-024-03531-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10529-024-03531-x