
Abstract
Objective
To investigate the functions of eIF3b in chronic myelogenous leukemia (CML).
Methods
The expression of eIF3b was inhibited by transfecting aspecifically designed shRNA into the CML cell lines of TK-6 and K562. The CCK8 assay was conducted to determine cell viability, and flow cytometry was used to examine the change in the cell cycle and cell apoptosis. RNAsequencing was applied to screen the candidate targets of eIF3b to identify the underlying mechanisms of eIF3b.An in vivo tumour xenograft mouse model was established by injecting shRNA transfected cells into the NCG mice. The tumour size and body weight of mice were monitored every other day. The mice were sacrificed 2 weeks after the tumour cell injection. The expression of eIF3b and target genes in the tumour tissues were determined by immunohistochemical staining and Western blotting.
Results
The group with inhibited expression of eIF3b led to about 50% lower cell viability compared with that of the control group (P < 0.05). Flow cytometry suggested that the percentage of increase in apoptotic cells was eight times higher than those in control group for TK-6 and K562 cells (P < 0.05). However, the difference between the cell amounts in the S phase for the experiment and control groups was not significant. After RNAsequencing and further validation via qPCR, C3G was screened as the potential target of eIF3b involved in the cell proliferation and apoptosis of CML cell lines. Subsequent in vivo analysis proved that the inhibition of eIF3b suppressed tumour formation and decreased C3G expression, thereby indicating that C3G was the potential target of eIF3b.
Conclusion
eIF3b is correlated with the cell proliferation and cell apoptosis of CML. Moreover, eIF3b regulation most probably occurs via regulating the expression of C3G.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myelogenous leukemia (CML) accounts for 15%–20% of adult leukemia and is a disorder that manifests as uncontrolled myeloid cell proliferation (Gu et al. 2012). As a feature of CML, the uncontrolled proliferation of myeloid cells chiefly occurs in the bone marrow and blood (Ghosh et al. 2014). CML is the so-called Philadelphia chromosome that is acquired from abnormal chromosome translocation and usually involves the BCR–ABL fusion oncogene (Burke and Carroll 2010). The BCR–ABL fusion gene-encoded protein is involved in potential pathways that disrupt tyrosine kinase activity and regulates cell proliferation, differentiation, apoptosis and genomic stability (Gregor et al. 2019). The conventional treatment for CML includes chemotherapeutic agents, such as hydroxyurea and busulfan. However, the side effects of chemical medicines are vital and limit their application (Massaro et al. 2018). Knowledge of the molecular mechanisms underlying CML is important in the development of new therapeutic treatments. Nevertheless, such mechanisms are still unclear.
Given that the uncontrolled myeloid cell proliferation in CML is similar to that of cancerous cells during their development and progression, the genes commonly involved in cancerous development maybe involved in CML as well. In addition, the mRNA translational regulation can promote the tumourous development in angiogenesis, transformation and cell metastasis and invasion (Silvera et al. 2010). Therefore, translation initiation is the crucial step for protein synthesis from mRNA. Eukaryotic translation initiation factors (eIFs) are involved in the regulation of this process (Jackson et al. 2010). eIF overexpression is commonly detected in cancerous cells. For example, eIF4A, eIF4E and eIF5A overexpression are detected in several human cancers and have been considered as the potential targets for anticancer therapies (Bhat et al. 2015; Pettersson et al. 2014; Sridharan et al. 2019; Zheng et al. 2019). The eIF3b subunit is overexpressed in cancers, such as prostate and bladder cancers, and is correlated with bad prognosis (Wang et al. 2013). A recent study has indicated thateIF3b is correlated with osteosarcoma. The silencing of eIF3b induces cell apoptosis and suppresses cell growth in U2OS cells (Choi et al. 2017). As CML commonly occurs in the bone marrow, eIF3b mayalso be involved in the development and progress of CML.
In this study, TK-6 and K562 cells were applied, and the expression of eIF3b was inhibited using shRNA andsheIF3b. Cell viability and cell apoptosis were determined to understand the functions of eIF3b. The differentially expressed genes during this process were screened using RNA-seq, and the expression of relevant genes was further validated using NCG mice on the basis of the subcutaneous tumour formation assay. This study aimed to investigate the correlation between CML and eIF3b and the underlying regulation mechanisms.
Materials and methods
Cell culture
TK-6 and K562 cells were obtained from the Shanghai Cell Bank (Shanghai, China) and cultured at 33 °C with 10% CO2. High-glucose DMEM culture medium with 10% fetal bovine serum (Gibco, USA) was employed. The cells were cultured, amplified and passaged. After 3 days, the cells were digested and pelleted by centrifugation. Cell morphology was observed using a light microscope, and the suspension had a concentration of 1 × 106/ml.
Lentivirus preparation and transfection
The full length of the eIF3b was synthesised and cloned to the pcDNA3.1(+) vector together with the inverted repeat sequence. The antisense sequences targeted eIF3b, which was also synthesised and cloned into the pcDNA3.1(+) vector to generate shRNA. An empty vector pcDNA3.1(+) was used as the control. 293T cells were applied and seeded in a 10 cm plate. When the cells grew to 80–90% of the plate area, the cells were harvested by trypsin and amplified in a 15 cm plate. pcDNA3.1(+) vector-eIF3b/shRNA and package vectors (pGag/Pol, pRev and pVSV-G) were mixed in a 1.5 ml culture medium without serum and incubated at room temperature for 5 min. The RNAi-Mate (300 µl)was prepared, diluted in a 1.5 ml culture medium, mixed with the vector mixture and incubated for 20–25 min to form the transfection complex. The culture medium in the cells was removed and replaced by 8 ml medium without the serum. The transfection complex was added to the cells, and the mixture was incubated for 4–6 h at 37 °C with 5% CO2. The culture medium was removed and replaced with DMEM medium with serum and incubated at 37 °C and 5% CO2 for 72 h. The cell culture supernatant was aspirated and centrifuged at 4000 rpm for 4 min. The supernatant was filtered with a 0.45 µm filter and centrifuged at 20,000 rpm for 2 h. The virus solution was stored at − 80 °C before use. The TK-6 and K562 cells were seeded in 96-well plates at a density of 3 × 104 cells/well. The virus solution was diluted 10 × gradient into five different concentrations with the DMEM medium. The supernatant with culture medium in each well was discarded and supplemented with 100 µl of the virus solutions in different concentrations for each well. Instead of the virus solution, saline was used as control. The cells were incubated at 37 °C and 5% CO2 for 24 h. The cell culture supernatant was replaced with freshly prepared DMEM medium (100 µl). The cells were subsequently incubated for 72 h. The FACS flow cytometry was applied to detect the fluorescence. The titre of the virus was calculated in accordance with the dilution ratio. RT-PCR was performed to detect the expression of eIF3b.
Colony formation assay
A total of 400 TK-6 and K562 cells were counted and then seeded into 6-well plates incubated at 37 °C and 5% CO2 for 14 days. The cells were then treated with methanol for 15 min for fixation and then stained with 0.05% crystal violet. Colonies > 50 were scored.
Cell cycle apoptosis assay
The cells were seeded in 6-well plates at a density of 5 × 105cells/well overnight at 37 °C. The cells were pelleted and washed with PBS. The cell pellets were then fixed with 70% ethanol on ice, followed by resuspension in 500 μl propidium iodide solution to stain the cells. The cell suspensions were detected using the FACSCalibur flow cytometer (BD Biosciences, USA) at 488and 620 nm for excitation and emission wavelengths, respectively. A total of 10,000 gated events were collected using the CellQuest Pro software (BD Sciences), and the flow cytometry data were analysed for the cell cycle by using the WinList and ModFit from the Verity Software House (Topsham, USA). For the cell apoptosis assay, 1 × binding buffer was used to resuspend the cells and 5 μl propidium iodide staining solution and 5 μl fluorochrome-conjugated Annexin V were added. The samples were tested using the FACSCalibur flow cytometer, and the percentage of the apoptotic cells was measured.
CCK-8 assay for cell viability detection
The cells were suspended, seeded in a 96-well plate at a density of 2 × 103 cells/well and incubated at 37 °C in a 5% CO2-humidified incubator. Each well was filled with a culture medium to obtain a final volume of 100 µl. After incubation for 24–96 h, 10 µl of 5 mg/ml CCK-8 (Dojindo, Japan) was added to each well. The cells were incubated with the solution for 4 h. Absorbance was measured at 450 nm by using a luminometer (BioTek uQuant, USA).
RNAsequencing
The total RNA from the specimens and cells was purified using the RNeasy Mini Kit (QIAGEN, Germany). RNA integrity was evaluated on the basis of the RIN value by using the Agilent Bioanalyzer 2100 (Agilent, CA, USA). RNA cleanup was conducted using the RNA Clean XP Kit (Beckman Coulter, CA, UA), and the DNA residue was removed using the RNase-free DNase Set (QIAGEN, Germany). The concentration and the 260/280 ratio of the RNA were determined using the NanoDrop 2000 (Thermo Fisher, USA). Linear RNA was digested by the RNase R before library preparation so that the circular RNA remained (Epicenter, USA). The circular RNA (1 μg) was used for library preparation following the manufacturer’s instruction for the VAHTSTM mRNA-seq v2 Library Prep Kit (Vazyme, Nanjing, China). The RNA was fragmented, and the double-strand cDNA was synthesised. End-polishing and adapter ligation were performed for the circular RNA. The ligated cDNA was amplified using universal PCR to obtain a sufficient library for sequencing. The Agilent Bioanalyzer 2100 was used to evaluate the quality of the library. RNAsequencing was conducted on the Illumina Hiseq 4000. The data were then assembled and annotated with the corresponding symbols of transcripts. The differentially expressed mRNAs were screened using the R software in accordance with the q value < 0.05 and foldchange > 2.0.
GO and pathway analysis
The DAVID (Database for Annotation, Visualisation and Integrated Discovery) was used to annotate the potential functions in various signalling pathways of the differentially expressed mRNA. The functional annotation of parental genes was then predicted using the GO functional annotation. A scatter plot was used to present the results of the GO analysis. KEGG pathway annotation was employed to show the relevant pathways of the differentially expressed mRNAs.
RT-PCR
circRNAs and genes were verified using RT-PCR. The M-MLV Reverse Transcriptase (Promega, USA) was used to synthesise cDNA. The PCR reaction was prepared using the GoTaq qPCR Master Mix (Promega, USA) and performed on the ABI 7500 system (Applied Biosystem, USA). GAPDH was employed as the housekeeping gene for comparative quantification.
Western blotting
Cells were lysed in 1% SDS lysis buffer. BCA assay was performed to determine the protein concentration. To separate the protein, 10% SDS–PAGE was used. The protein was then transferred onto nitrocellulose membranes. Nonfat milk in PBS was used to block the membrane at room temperature for 1 h. The membrane was incubated overnight at 4 °C with the primary antibody (Abcam, UK). After several washes with PBS, the membranes were incubated in the blocking buffer with a secondary antibody coupled to horseradish peroxidase for 2 h at room temperature. The complexes were formed on the membrane. Then, the membrane was detected using the ECLplus (Amersham Biosciences/GE Healthcare, Velizy, France).
In vivo experiments
Male NCG mice (Charles River Laboratories, Saint Constant, Canada) aged 6–8 weeks were housed in a pathogen-free environment. The animal experiment was approved by the Yijishan Hospital. For the subcutaneous tumour model, a 2 × 106 cell suspension in 100 μl was injected subcutaneously in the abdomen of the NCG mice. The control mice were injected with saline. Tumour growth was measured every 2 days by using a dial calliper. The animals were sacrificed after 2 weeks, and the tumours were subsequently collected. Part of the tissues was fixed in formalin and embedded in paraffin. The remaining tissues were stored at – 80 °C for the Western blotting assay.
Immunohistochemistry
Tissue samples were embedded in paraffin. Immunohistochemistry was performed to detect the protein expression. The specimens were fixed in formaldehyde, embedded in paraffin and serially sectioned at 5 μm. The sections were routinely dewaxed and immersed in 0.01 mol/lcitrate buffer (pH 6.0).The antigen was repaired by microwave. Then, freshly prepared 3% (v/v) H2O2 was added with distilled water, and the solution was incubated at room temperature for 10 min to deactivate the endogenous peroxidase. Normal goat serum blocking solution was added to the above solution, and the resulting solution was incubated for 20 min at room temperature. Primary antibodies were added at a concentration of 1:50 at 4 °C overnight and then rewarmed at 37 °C for 1 h. Biotinylated goat antimouse secondary antibody was added, and then the solution was incubated at 37 °C for 45 min. Biotinylated horseradish peroxidase-labelled streptomycin was added, and the solution was incubated at 37 °C for 30 min. Diaminobenzidine (DAB) was used for colour development. The sections were counterstained with haematoxylin, dehydrated and mounted. Instead of the primary antibody, PBS was used as a negative control.
Statistical analysis
When data corresponded to normal distribution, comparisons were performed using independent t-tests and one- and two-way ANOVA. The significance was established at P < 0.05. The SPSS and the GraphPad Prism software were used for the statistical analysis.
Results
eIF3b expression level is correlated with cell viability and cell proliferation
The eIF3b shRNAs were designed and transfected into TK-6 cells to suppress the expression of eIF3b. The performance of the shRNAs was determined using qPCR. The shRNAs, namely, sheIF3b#1 and sheIF3b#2, exhibited high efficiency in suppressingeIF3b expression. In particular, sheIF3b#1 and sheIF3b#2 inhibited eIF3b expression by − 50% and − 70%, respectively, compared with the blank group (Fig. 1a).Between the two shRNAs, sheIF3b#2 had the best inhibition efficiency at 70% and was used for the downstream assays.

eIF3b expression is positively correlated with cell viability. a Performance test of the specifically designed shRNA targeted on eIF3b; b CCK8 assays determined the cell viability after sheIF3b transfection; c Cell colony assays estimated the cell proliferation when the expression of eIF3b was inhibited by shRNA
Correlation of eIF3b expression with cell viability and cell proliferation
The correlation between eIF3b expression and cell viability was investigated using the CCK8 assay. sheIF3b was transfected to TK-6 and K562, followed by the CCK8 assay. The OD value can indicate cell viability (Fig. 1b). sheIF3b can suppress eIF3b expression to 30% compared with the normal cells. The group with inhibited eIF3b expression induced by eIF3b led to approximately 50% of the cell viability from 24 to 72 h after eIF3b transfection to TK-6 and K562 cells compared with the normal group. Thus, the expression of eIF3b was positively correlated to cell viability. Meanwhile, the capacity for cell proliferation was determined using the colony formation assay. The normal TK-6 and K562 cells and those transfected with sheIF3b were employed for the assay. The normal TK-6 and K562 cells formed approximately 80 colonies. However, after sheIF3b transfection, the number of colonies dropped to 20, which was 25% that of the normal cells (Fig. 1c). Therefore, decreased cell viability was attributed to the weak capacity for cell proliferation.
eIF3b inhibition promotes cell apoptosis and arrests cell cycle progression
The cell apoptosis and cell cycle were determined by flow cytometry assays. The TK-6 and K562 cells transfected with empty vector and sheIF3b shRNA were employed for the assays. In the cell apoptosis assay, the cells transfected with sheIF3b had an 8–10% increase in apoptotic cells compared with those transfected with the empty vector (Fig. 2a). The difference was statistically significant (P < 0.05). For the cell cycle, the TK-6 and K562 cells transfected with sheIF3b were fewer at the S phase, but more cells were retained at the G1 and G2 phases relative to the control group. Therefore, cell proliferation was inhibited (Fig. 2b). Moreover, decreased eIF3bexpression induced the inhibition of cell proliferation by inducing cell apoptosis and retaining the cells in the G0/G1 and G2/M phases.

Flow cytometry assays for the estimation of cell cycle and cell apoptosis. a Cell apoptosis and b cell cycle were determined in TK-6 and K562 cells transfected with shCtrl and sheIF3b
Screening of differentially expressed genes by using whole-transcriptome sequencing
The cells transfected with the empty vector and sheIF3b were subjected to total RNA purification followed by whole-transcriptome sequencing. The gene expression profile was summarised, and the clustergram was presented as Fig. 3a. The genes also underwent GO and KEGG annotation. The results from the GO enrichment indicated that the genes were annotated with specific cell functions. Thus, the differentially expressed genes were significantly enriched in the protein transport and platelet activation function and in the secondary intracellular protein transport and extra cellular exosome related activity (Fig. 3b). The KEGG analysis revealed that the differentially expressed genes were enriched in the neurotrophin signalling pathway, glycine, serine and threonine metabolism and acute myeloid leukemia (Fig. 3c). In line with previous research, this work screened the genes involved in cell proliferation and apoptosis. The upregulated differentially expressed genes includedCDKN1B and FADD, and the downregulated differentially expressed genes included C3G, FN14, FZD5 and FosL1, all of which were chosen on the basis of the current research purpose. Amongst these genes, the downregulated differentially expressed genes that were positively correlated with the expression of eIF3b were chosen for the downstream validation.

RNA-seq in screening the target genes of eIF3b. a Clustergram of the RNA-seq results; b GO analysis to annotate the functions of the differentially expressed genes; c KEGG pathway analysis to annotate the involved pathways of the differentially expressed genes
Establishing the stable cells with eIF3b overexpression and inhibition
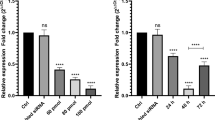
The eIF3b overexpression and the inhibition vectors were designed and transfected to the K562 cells to further validate the expression of the differentially expressed genes depending on whether the eIF3b was overexpressed or suppressed. The performance of the vectors was determined by RT-PCR. The empty vector induced no change in the expression of eIF3b. The overexpression vector, PLV-eIF3b, induced about 22-fold higher expression, whereas the inhibition vector, PLKO-eIF3b, induced 50% lower expression compared with the blank group (Fig. 4a). The performance of the vectors were fine and suitable for the downstream studies. The mRNA expression level of the differentially expressed genes were determined, and results corresponded to those from the whole-transcriptome sequencing, in which four candidate genes exhibited downregulated expression when eIF3b expression was inhibited (Fig. 4b). By contrast, the expression of eIF3b increased by 25-fold when the K562 cells were transfected with PLV-eIF3b.Accordingly, the expression levels of the four candidate genes, C3G, FZD5, FosL1 and FN14, increased significantly (P < 0.05; Fig. 4c). Amongst these genes, C3G, which was involved in cell proliferation and cell apoptosis processes, was further validated for protein levels using Western blotting. The K562 cells were used and transfected with an empty vector and sheIF3b. sheIF3b induced a 70% decrease in the protein level of eIF3b.The protein level of C3G decreased by 50% compared with that of the control group (Fig. 4d). The difference was statistically significant (P < 0.05).

Validation of the potential target genes of eIF3b. a Performance test of the overexpression and the suppression vectors of eIF3b. b Expression of the target genes in K562 cells transfected with the suppression vector of eIF3b. c Expression of the target genes in K562 cells transfected with the overexpression vector of eIF3b. d Western blotting revealed the protein level of eIF3b and C3G
eIF3b–C3G cascade regulates in vivo tumour formation
The K562 cells were used for the in vivo animal study. The cells were transfected with the Lv-eIF3b vector via the lentivirus to constantly suppress the expression of eIF3b. The control group was composed of the cells transfected with the empty virus. As validated by RT-PCR, the expression of eIF3b decreased to 25% compared with that of the control group (P < 0.001; Fig. 5). The cells of both groups were then subcutaneously injected to the abdomen of the NCG mice. The size and weight of the formed tumour in the mice were determined every 2 days. Based on the data recorded for 16 days, the size of the tumour in the eIF3b inhibition group was significantly smaller than that in the control group, and the significant difference was observed from the 12th day. However, the weight of the mice did not present any significant difference for both groups (Fig. 6a). The mice were sacrificed, and the tumours were collected after 2 weeks of K562 cell injection. For the comparison of the tumours in the two groups, the tumours of the eIF3b inhibition groups were smaller in size than those in the control group. The evidence above indicated that the expression level of eIF3b was positively correlated with tumour size during the oncological process and that the eIF3b expression suppressed the development of tumours.

eIF3b expression was inhibited by LV-eIF3b

Tumour formation assays in the validation of the functions and target genes of eIF3b in NCG mice. a Tumour size and weight of the NCG mice during the tumour formation assays. b NCG mice were sacrificed, and the tumour tissues were collected. c The expression levels of eIF3b and C3G determined by immunohistochemical staining. d Western blotting determined the protein levels of eIF3b and C3G in the tumour tissues
The tumourous tissues were fixed in formalin and subjected to immunohistochemical staining to determine the expression levels of eIF3b and C3G. The eJF3b or C3G was stained brown after immunohistochemical staining. Both genes were expressed mainly in the cytoplasm. Overall, the tumourous slices in the eIF3b inhibition group presented lower expression levels for eIF3b and C3G compared with those in the control group (Fig. 6c).The purification of the total protein from the tumourous tissues and determination of the protein level of eIF3b and C3G by Western blotting were performed to further validate the results. Results were consistent with those of immunohistochemical staining. The protein levels of eIF3b and C3G in the eIF3b inhibition group decreased to 50% that of the control group (Fig. 6d). Therefore, the expression levels of eIF3b and C3G were positively correlated, and the eIF3b–C3G cascade maybe an important access for regulating tumour formation.
Discussion
In this study, the functions of eIF3b were targeted, and molecular assays were conducted for further validation. Results suggested that eIF3bregulatedthe cell viability, cell proliferation and cell apoptosis processes of the CML cell lines. The inhibition of eIF3b expression delayed cell proliferation and induced cell apoptosis. The RNA-seq suggested that the expression of C3G was positively correlated with the expression of eIF3b. Therefore, eIF3b may regulate the expression of C3G directly or indirectly, thereby regulating the relevant CML processes.
Based on a previous study on osteosarcoma, the tumour necrosis factor receptor superfamily member 21 (TNFTSF21) increases in expression when eIF3b has been silenced. TNFRSF21 is commonly known as Death receptor 6 (DR6), and its overexpression can induce cell apoptosis in various types of cells (Zeng et al. (2012); Mi et al. 2011). The main function of TNFRSF21 is to promote the activation of caspase-3for the initiation of cell apoptosis (Mi et al. 2011; Nikolaev et al. 2009). TNFRSF21 is over expressed in M17 neuroblastoma, Hela, human H4 neuroglioma and human cervical cancer cells (Zeng et al. 2012; Mi et al. 2011) but not in human breast cancer cells (Pan et al. 1998). Therefore, CML does not share completely similar underlying mechanism as osteosarcoma. The results from our prior study imply that the cell apoptosis is activated when the expression of eIF3b is inhibited. Therefore, other effectors, which may induce the cell apoptosis when eIF3b expression is silenced, may be included. Some studies have also posited the mechanism of eIF3b for enhancing cell proliferation and inhibiting cell apoptosis. The potential mechanism may entail reducing the apoptosis inhibitory protein, such as BCL2 expression, or activating the Akt pathway, thereby initialising cell proliferation (Wang et al. 2019; Xu et al. 2019; Tian et al. 2018).
According to the RNA-seq profile analysis, C3G, Fn14, FZD5 and FosL1 were expressed positively with eIF3b. Thus, these genes maybe important targets that eIF3b regulates directly or indirectly to regulate the development and progress of CML. Evidence from prior research has confirmed that C3G is a key effector in regulating cell proliferation and apoptosis in CML. However, the evidence has also indicated that C3Gmayplay a dual regulatory role in CML cells via the modulation of apoptotic and survival pathways. The inhibition of C3G usually involves the enhancement of Akt phosphorylation and thereby presents a proliferative function (Voss et al. 2006). In other words, the inhibitory action on ERK and Akt is likely to be effective when C3G overexpression occurs, thereby inducing apoptosis. The theory above was not validated by our study because this work only showed the inhibitory effect on cell proliferation and induction of cell apoptosis when C3G is suppressed in expression. Thus, C3G may have performed other functions as a cell proliferation enhancement factor. As the previous studies described, C3G can participate in Rap1 activation. The C3G-mediated Rap1 activation is positively correlated with cell proliferation and survival in haematopoietic cells stimulated with IL-3 or expressing the BCR–ABL fusion protein via the activation of the PI3K–AKT and B-Raf–ERK pathways (Sequera et al. 2018; Priego et al. 2016). In this situation, the inhibition of C3G induces cell apoptosis. Therefore, the mechanism and functions of C3G are complicated. In most cases of malignant tumours, C3G–Rap1 contributes to the inhibition of proapoptotic pathways, such as the p38 MAPK pathway and the ERK and AKT cascades, and stimulates cell survival. Conversely, C3G performs its functions by the Rap1-independent mechanism involved in the phosphate activation of AKT Jiang et al. (2014). In our study, eIF3b inhibition decreased the mRNA and protein level of C3G, thereby inducing cell apoptosis and suppressing cell proliferation. Therefore, C3G was hypothesised to perform the C3G–Rap1 cascade mainly in CML cell lines and in theTK-6 and K562 cells. However, this hypothesis needs to be further validated.
In conclusion, our study confirmed that eIF3b is an important gene in the progress of CML. The potential regulatory target of eIF3b is C3G.Moreover, the eIF3b–C3G cascade has presented functions for regulating cell proliferation and cell apoptosis of CML cell lines. However, the mechanism for eIF3b regulation on C3G and the cell proliferation and apoptosis of CML cells remain unclear. Further studies are necessary to validate the functions of the eIF3b–C3G cascade. Actual clinical specimens from CML patients can be used to verify whether the mechanism proven in the present study is consistent. In addition, the genes that play an important role in the regulatory effects of eIF3b and C3G, such as Rap1, ERK and PI3K–AKT cascade, can be determined through mRNA and protein expressions to present the exact regulatory network in CML that is correlated witheIF3b.
References
Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I (2015) Targeting the translation machinery in cancer. Nat Rev Drug Discov 14:261
Burke BA, Carroll M (2010) BCR–ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia 24:1105–1112
Choi YJ, Lee YS, Lee HW, Shim DM, Seo SW (2017) Silencing of translation initiation factor eIF3b promotes apoptosis in osteosarcoma cells. Bone Jt Res 6:186–193
Ghosh D, Dey SK, Saha C (2014) Protective effect of black tea extract during chemotherapeutic drug induced oxidative damage on normal lymphocytes in comparison with cancerous K562 cells. Int J Sci Eng Res 5:437–447
Gregor T, Bosakova MK, Nita A, Abraham SP, Fafilek B, Cernohorsky NH, Rynes J, Foldynova-Trantirkova S, Zackova D, Mayer J, Trantirek L, Krejci P (2019) Elucidation of protein interactions necessary for the maintenance of the BCR-ABL signaling complex. Cell Mol Life Sci. https://doi.org/10.1007/s00018-019-03397-7
Gu Y, Chen T, Meng Z, Gan Y, Xu X, Lou G, Li H, Gan X, Zhou H, Tang J, Xu G, Huang L, Zhang X, Fang Y, Wang K, Zheng S, Huang W, Xu R (2012) CaMKII gamma, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood 120:4829–4839
Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113
Jiang X, Tang X, Zhang P, Liu G, Guo H (2014) Cyanidin-3-O-beta-glucoside protects primary mouse hepatocytes against high glucose-induced apoptosis by modulating mitochondrial dysfunction and the PI3K/Akt pathway. Biochem Pharmacol 90:135–144
Massaro F, Colafigli G, Molica M, Breccia M (2018) Novel tyrosine-kinase inhibitors for the treatment of chronic myeloid leukemia: safety and efficacy. Expert Rev Hematol 11:301–306
Mi S, Lee X, Hu Y, Ji B, Shao Z, Yang W, Huang G, Walus L, Rhodes K, Gong BJ (2011) Death receptor 6 negatively regulates oligodendrocyte survival, maturation and myelination. Nat Med 17:816–821
Nikolaev A, McLaughlin T, O’Leary DDM, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989
Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM (1998) Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett 431:351–356
Pettersson F, Del Rincon SV, Miller W Jr (2014) Eukaryotic translation initiation factor 4E as a novel therapeutic target in hematological malignancies and beyond. Expert Opin Ther Targets 18:1035–1048
Priego N, Arechederra M, Sequera C, Bragado P, Vazquez-Carballo A, Gutierrez-Uzquiza A, Martin-Granado V, Ventura JJ, Kazanietz MG, Guerrero C, Porras A (2016) C3G knock-down enhances migration and invasion by increasing Rap1-mediated p38alpha activation, while it impairs tumor growth through p38alpha-independent mechanisms. Oncotarget 7:45060–45078
Sequera C, Manzano S, Guerrero C, Porras A (2018) How Rap and its GEFs control liver physiology and cancer development C3G alterations in human hepatocarcinoma. Hepat Oncol 5:HEP05
Silvera D, Formenti SC, Schneider RJ (2010) Translational control in cancer. Nat Rev Cancer 10:254–266
Sridharan S, Robeson M, Bastihalli-Tukaramrao D, Howard CM, Subramaniyan B, Tilley AMC, Tiwari AK, Raman D (2019) Targeting of the eukaryotic translation initiation factor 4A against breast cancer stemness. Front Oncol 9:1311
Tian Y, Zhao K, Yuan L, Li J, Feng S, Feng Y, Fang Z, Li H, Deng R (2018) EIF3B correlates with advanced disease stages and poor prognosis, and it promotes proliferation and inhibits apoptosis in non-small cell lung cancer. Cancer Biomark 23:291–300
Voss AK, Krebs DL, Thomas T (2006) C3G regulates the size of the cerebral cortex neural precursor population. EMBO J 25:3652–3663
Wang H, Ru Y, Sanchez-Carbayo M, Wang X, Kieft JS, Theodorescu D (2013) Translation initiation factor eIF3b expression in human cancer and its role in tumor growth and lung colonization. Clin Cancer Res 19:2850–2860
Wang L, Wen X, Luan F, Fu T, Gao C, Du H, Guo T, Han J, Huangfu L, Cheng X, Ji J (2019) EIF3B is associated with poor outcomes in gastric cancer patients and promotes cancer progression via the PI3K/AKT/mTOR signaling pathway. Cancer Manag Res 11:7877–7891
Xu F, Zhang S, Liu Z, Gu J, Li Y, Wang L, Mao W, Zhu Q, Shou H, Ge D, Lu C (2019) TEX9 and eIF3b functionally synergize to promote the progression of esophageal squamous cell carcinoma. BMC Cancer 19:875
Zeng L, Li T, Xu DC, Liu J, Mao G, Cui MZ, Fu X, Xu X (2012) Death receptor 6 induces apoptosis not through type I or type II pathways, but via a unique mitochondria-dependent pathway by interacting with Bax protein. J Biol Chem 287:29125–29133
Zheng X, Gao L, Wang BT, Shen P, Yuan XF, Zhang LQ, Yang L, Zhang DP, Zhang Q, Wang XM (2019) Overexpression of EIF5A2 is associated with poor survival and aggressive tumor biology in gallbladder cancer. Histol Histopathol. https://doi.org/10.14670/HH-18-186
Funding
This study was supported by Natural Science Research Project of Colleges and Universities in Anhui Province (KJ2017A263).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that that there are no conflict of interest.
Research involving human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, L., Wei, Z., Chang, X. et al. eIF3b regulates the cell proliferation and apoptosis processes in chronic myelogenous leukemia cell lines via regulating the expression of C3G. Biotechnol Lett 42, 1275–1286 (2020). https://doi.org/10.1007/s10529-020-02878-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02878-1