
Abstract
Bovicola caprae is an important obligate ectoparasite of goats worldwide including India. The present study aimed at the molecular confirmation, phylogenetics and population structure analyses of B. caprae infesting goats of three different agro-climatic locations in India, by targeting the mitochondrial cytochrome C oxidase subunit 1 (cox1) genetic marker. The phylogenetic tree exhibited the presence of two different lineages of B. caprae. The sequences generated herein clustered in lineage 2 along with the GenBank™ archived sequences from China and Iran. The sequences generated herein also showed the circulation of sub-lineages of B. caprae in India based on the analysis of pairwise genetic distances between sequences and median-joining haplotype network. The population structure analyses revealed low nucleotide (0.00353 ± 0.00291 and 0.02694 ± 0.00363) and high haplotype (0.667 ± 0.314 and 0.618 ± 0.104) diversities for the present study isolates as well as for the complete dataset, respectively, which evinced a recent demographic expansion. High genetic differentiation (FST value = 0.97826) and low gene flow (Nm = 0.00556) were also recorded in the different lineages/populations. In conclusion, the present study addressed the research gap and provided the first insight into the phylogenetics of the goat louse B. caprae and highlighted the circulation of sub-lineages of the ectoparasite in India.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In India, goat farming is an integral part of the livestock industry as it is associated with the socio-cultural, economic, agricultural and even religious beliefs of humans (Ajith et al. 2017a, b). The basic purpose behind goat farming is low input and investment and good output in the form of milk, wool and meat (Moudgil et al. 2017). The parasitic (ecto- and endoparasitic) diseases are considered to be one of the major constraints in maintaining the good health of the traditional transhumance and organized goat flocks (Pilarczyk et al. 2021).
Lice infestation in goats, commonly called caprine pediculosis is considered to be a serious problem in goat flocks especially reared in extensive grazing systems (Ajith et al. 2019). Primarily based on feeding habits, goat lice can be classified into two major subgroups; chewing or biting lice including the members of the order Mallophaga and sucking lice consisting of the members of the order Anoplura (Ajith et al. 2019). Goats in India are primarily parasitized by a biting louse, Bovicola caprae and a sucking louse, Linognathus africanus (Ajith et al. 2017b). Biting or chewing louse of goats, B. caprae is an important obligate ectoparasite of goats worldwide including India (Ajith et al. 2017a). However, no serious efforts have been put in for cladistics as well as population structure analysis to ascertain the origin and dispersal of this economically important ectoparasite.
Caprine chewing lice infestation primarily induces irritation, self-excoriation, alopecia, severe pruritus and papulocrustous dermatitis (Taylor et al. 2016; Ajith et al. 2017b). The initial clinical manifestation in the form of severe irritation is attributed to hypersensitivity reactions caused by the antigens present in lice saliva (Ajith et al. 2017b). In line with the pathogenesis associated with other ectoparasites, lice are also incriminated to transmit various Rickettsia species in animals and humans (Fournier et al. 2002; Hornok et al. 2010).
In the recent past, extensive studies had been carried out on human head and body lice populations with the help of multiple genetic markers (Ghavami et al. 2020). The researchers targeted both phylogeography as well as demographic dynamics of Pediculus humanus capitis (Amanzougaghene et al. 2019; Yingklang et al. 2021). Also, mitochondrial genetic markers played a pivotal role in establishing the different lineages of human head louse based on the dispersal or prevalence in different geographical regions of the world (Ghavami et al. 2020; Yin et al. 2023). However, any such study had not been performed on chewing lice populations of domesticated animals, despite their utmost economic importance. Additionally, understanding the population changes (expansion, a sudden contraction or a bottleneck) based on neutrality tests is also of significant importance to ascertain the evolutionary patterns, spatiotemporal dynamics and genetic exchange (gene flow, migration, genetic drift and genetic selection pressure) in different populations (Yin et al. 2023). The information thus retrieved could be employed for the implementation of the most effective control strategies.
It is also established that agro-climatic regions, breeds, immune status, hygiene and rearing systems significantly impact the distribution and prevalence of lice in goats (Ajith et al. 2017a). Hence, the present study is the first attempt to shed light on the molecular confirmation, genetic diversity and population structure of B. caprae infesting goats of three different agro-climatic locations in India based on mitochondrial cytochrome C oxidase subunit 1 (cox1) genetic marker.
Materials and Methods
Study Area and Sample Collection
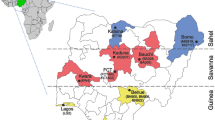
The present study targeted three geographically distant locations from three different states; Himachal Pradesh, Haryana and Rajasthan (Fig. 1). The study involved Palampur, Himachal Pradesh (32.1109° N, 76.5363° E), located in the north-western Himalayas at an elevation of 1472–2350 m above mean sea level, with an annual average rainfall of 1578 mm. The study involved Fatehabad, Haryana (29.5132° N, 75.4510° E), which is an arid zone, located at an elevation of 207–222 m above mean sea level, with an annual average rainfall of 272 mm. The study also involved the alluvial plain zone of Rajasthan state, i.e. Pali (25.7781° N, 73.3311° E), located at an elevation of 149–1099 m above mean sea level with an annual average rainfall of 525 mm. The lice were collected from local breeds of goats of Himachal Pradesh (Gaddi breed), Haryana and Rajasthan, non-invasively with the help of a pooter and forceps. The collected lice (n = 10 from each location) were preserved in 70% ethanol for further analysis.

Map showing the sampling sites of the study area; Palampur (red rectangle) of Himachal Pradesh, Fatehabad (violet rectangle) of Haryana and Pali (blue rectangle) of Rajasthan states (Color figure online)
Genomic DNA Extraction, PCR Amplification and Sequencing
The collected lice specimen (n = 10) from the goats of each location were subjected to genomic DNA extraction after cleaning lice twice with normal saline. The genomic DNA was extracted by using Qiagen DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The retrieved genomic DNA was stored at − 20 °C till further molecular analysis. The amplification of DNA sequences was performed by targeting mitochondrial cytochrome C oxidase subunit 1 (cox1) genetic marker through PCR by using the primers; forward: 5’-GGAGGATTTGGAAATTGATTAGTTCC- 3’ and reverse: 5’- CCAGGAAGAATAAGAATATAAACTTC- 3’ (Yong et al. 2003). The 25 µL PCR reaction mixture comprised 12.5 μL of master mix (GoTaq Green Master Mix, Promega, Madison, WI, USA), 2.0 μL of each primer (forward and reverse) (10 pmol), 3 μL of genomic DNA template and 5.5 μL of nuclease-free water. The amplification of genomic DNA was carried out in a thermal cycler (Bio-Rad T100™ Thermal cycler, USA) by using the following conditions: initial denaturation (94 °C for 3 min), 40 cycles each of denaturation (94 °C for 30 s), annealing (47 °C for 30 s) and extension (68 °C for 1 min), and then final extension (68 °C for 3 min). The amplified products were electrophoresed through 1.25% agarose gel along with a 100 bp marker (DNAmark™ 100 bp, G-Biosciences, USA) and then visualized under a gel documentation system. To check the specificity of the primers, the genomic DNA of ixodid tick Rhipicephalus microplus was used as negative template control in all the amplification reactions. The amplicons of ~ 520 bp size corresponding to partial amplification of the cox1 gene sequence were subjected to custom sequencing (Biokart India Pvt. Ltd., Bengaluru, Karnataka). Sanger sequencing based on chain termination PCR was performed on the amplified products by using the aforementioned forward and reverse primers. The custom- sequenced products were analyzed for misread sequences, which were eventually aligned using BioEdit version 7.0.5.3 (Hall 1999). The NCBI BLAST algorithm (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to ascertain the identity of each nucleotide sequence. The consensus sequences generated herein were deposited in the GenBank™ under the accession numbers LC768866-68.
Phylogenetic Analysis
The phylogenetic analysis of the sequences generated in the present study was performed with the GenBank™ archived sequences of Bovicola sp. (Table 1), with Molecular Evolutionary Genetic Analysis (MEGA) 11.0.10 software by constructing a phylogenetic tree using Maximum-likelihood method (Tamura et al. 2021). To infer the evolutionary history of B. caprae and for the construction of the phylogenetic tree, the best substitution model was found to be Kimura 2-parameter model (Kimura 1980) by using MEGA 11.0.10. Bootstrap analysis was performed using 1000 replicates and the distance scale was estimated at 0.20. The GenBank™ sequence corresponding to the ixodid tick R. microplus (LC715165) was used as an outgroup to root the tree. A maximum composite likelihood model was employed to estimate the evolutionary divergence based on pairwise genetic distances between the sequences generated herein with the GenBank™ archived B. caprae sequences (Tamura et al. 2004) by using MEGA 11.0.10. The comparative sequence analysis between the present study sequences and the GenBank™ archived B. caprae sequences from Iran (MZ817000) and China (MF927687) was performed to analyze the variations in the sequences (Hall 1999) by using MEGA 11.0.10.
Median-Joining Network
The relationships between B. caprae haplotypes were estimated through median-joining haplotype network analysis by using PopART software (http://popart.otago.ac.nz) (Leigh and Bryant 2015). In total, eleven GenBank™ archived B. caprae sequences including the present study sequences were involved in the haplotype network analysis. The misidentified B. caprae sequences available in the GenBank™ were not included in the median-joining haplotype network analysis.
Population Dynamics and Genetic Differentiation Indices
Demographic dynamics or population structure analysis involving population diversity indices [number of isolates, number of mutations, average number of pairwise nucleotide differences (K), nucleotide diversity (π), number of haplotypes (H) and haplotype diversity (Hd)] and neutrality indices (Fu’s Fs, Fu and Li’s F, Fu and Li’s D and Tajima’s D), were assessed using DnaSPv6 software (Rozas et al. 2017). DnaSPv6 software was also used for estimating Wright’s F statistics estimating pairwise genetic difference (FST), the average number of pairwise nucleotide differences (Kxy) and nucleotide substitution per site (Dxy). The gene flow (Nm) between the populations was assessed using the Arlequin 3.5.2 software (Nehra et al. 2022). The values of genetic differentiation were defined as high, moderate, low and negligible based on FST values greater than 0.25, between 0.25 and 0.15, between 0.15 and 0.05, and less than 0.05, respectively (Low et al. 2015). Whereas the level of gene flow was considered as high, intermediate and low, if the values of Nm were recorded greater than 1, between 0.25 and 0.99, and less than 0.25, respectively (Low et al. 2015).
Results
Phylogenetic Analysis
To address the research gap, the present study aimed at genetic diversity and population structure of B. caprae infesting goats of three different agro-climatic locations in India based on the mitochondrial cox1 genetic marker. The BLAST analysis revealed 99.61–100 and 96.13–99.39% nucleotide homology within the sequences generated herein and with the GenBank™ sequences from China and Iran, respectively. The phylogenetic tree demonstrated that the present study sequences along with the GenBank™ archived sequences from China (MF927687) and Iran (MZ817000) clustered in lineage 2, however, arranged in the sister groups at high bootstrap values (Fig. 2). Whereas, all other sequences from Iran formed a sister lineage 1 (Fig. 2). Both the lineages were supported by high bootstrap values (Fig. 2). Additionally, B. caprae sequences from Turkey (KP256539-40) sorted with Bovicola sp. retrieved from Galapagos hawk in the USA.

Maximum-likelihood evolutionary tree inferred from partial mitochondrial cox1 gene sequence. The percentage of trees in which the associated taxa clustered together/bootstrap values are indicated on each node. The bar represents 0.20 substitutions per site. Rhipicephalus microplus (LC715165) is used as an out-group species to root the tree
The evolutionary divergence studies based on genetic distances revealed no differences between the sequences from Fatehabad (LC768867) and Pali (LC768868), India (Table 2). However, the sequence generated from Palampur, India (LC768866) exhibited slight variation from the aforementioned present study sequences. In addition, the sequences from Iran, belonging to different lineages depicted comparatively high values ranging from 0.552 to 1.5439 (Table 2). The sequences generated herein showed nucleotide polymorphisms at different positions with respect to the sequences from Iran and China (G180A, A187T, C253T, G577T, G669A, G672T, C678T, C690T) (Fig. 3). The sequence from Palampur, India also exhibited substitutions (C253T, G577T) as to other two sequences generated herein from Fatehabad and Pali, India (Fig. 3).

Comparative multiple sequence alignment of the consensus sequences of the mitochondrial cox1 gene of B. caprae revealed nucleotide polymorphisms in present study isolates (LC768866-68) at different positions (G180A, A187T, C253T, G577T, G669A, G672T, C678T, C690T) with respect to the sequences from Iran (MZ817000) and China (MF927687). The sequence from Palampur, India (LC768866) also exhibited substitutions (C253T, G577T) with respect to other two sequences generated in the present study from Fatehabad (LC768867) and Pali (LC768868), India
Median-Joining Network Analysis
In totality, 11 sequences of B. caprae yielded three different haplotypes (Table 3). Haplotype 3 (Hap_3) containing 6 sequences was the predominant haplotype, which belonged to lineage 1 (Fig. 4). However, there was no central haplotype recorded in the dataset. All the sequences of lineage 1 belonged to Iran. Whereas, lineage 2 comprised haplotypes 1 and 2 with sequences from India, Iran and China. The sequence generated herein from Palampur, Himachal Pradesh formed Hap_1, which separated from other sequences generated in the present study (Hap_2) with two mutational steps. The sequences of lineage 1 were separated from the sequences of lineage 2 by 18 mutational steps (Fig. 4).

The median-joining haplotype network of B. caprae from different countries based on the partial mitochondrial cox1 gene sequence. Each circle depicts a unique haplotype and the circle size is relative to haplotype frequency. Nucleotide differences/haplotype substitutions/mutations are denoted by the hatch marks/bars across the lines connecting the haplotypes with each bar representing a single nucleotide variation. A color code to the country of origin is given (Color figure online)
Population Structure Analysis
In total, 11 valid/true sequences of B. caprae were included for population structure analysis. The overall dataset exhibited a low nucleotide (0.02694 ± 0.00363) and a high haplotype diversity (0.618 ± 0.104). Similarly, the sequences generated in the present study also evinced a very low nucleotide (0.00353 ± 0.00291) and a high haplotype diversity (0.667 ± 0.314). The results of the neutrality tests revealed significantly positive values for Tajima’s D (2.22502; p < 0.05) and Fu and Li's F (1.60455; p < 0.05) and a non-significant positive value for Fu and Li's D (1.13877; p > 0.10) and Fu’s Fs statistic (8.819).
Gene Flow and Genetic Differentiation Indices
The details of gene flow and genetic differentiation indices are given in Table 4. The low values of inter-population nucleotide differences (Da = 0.04762) and an average number of nucleotide substitutions per site between the populations (Dxy = 0.04868) depicted high genetic structuring. The pairwise FST value between the lineages was observed to be 0.97826, indicating a very high genetic differentiation. The Nm value recorded between the lineages was 0.00556, which was indicative of very low gene flow between the populations.
Discussion
In this study, firstly, the molecular confirmation and cladistics of goat louse B. caprae were performed using the cox1 mitochondrial genetic marker. Secondly, we assessed phylogeography, genetic diversity and demography dynamics of B. caprae in India in relation to the isolates reported from different parts of the world. In the past, various studies involving classical light microscopy and scanning electron microscopy had been performed to identify B. caprae morphologically (Sebei et al. 2004). To the best of our knowledge, this is the first study to address the research gap associated with phylogenetic characterization and demographic dynamics of B. caprae infesting goats in India.
The detailed molecular studies of human louse based on mitochondrial markers resulted in six deeply divergent lineages distributed throughout the globe (Amanzougaghene et al. 2019). However, the perusal of the literature revealed that no such study had been conducted on goat louse B. caprae. In the present study, we also targeted the mitochondrial gene to assess the phylogeography of B. caprae because the mitochondrial genome is maternally inherited and non-recombining (Ascunce et al. 2013). Whereas, inbreeding in lice of different lineages/clades or haplogroups in sympatric environments may result in the interchange in nuclear genes (Ascunce et al. 2013). Thus, mitochondrial markers had been proved critical in determining evolutionary patterns and similarities between lice species distributed in different geographic regions/locations. The phylogenetic analysis in the present study also revealed the presence of two lineages or haplogroups of B. caprae based on the mitochondrial cox1 gene sequence. However, B. caprae sequences involved in the present study did not exhibit geographic isolation. The finding could be attributed to the availability of a limited number of sequences of B. caprae in the GenBank™ and hence, the inclusion of only available sequences from Asian countries in the present study. It is also evident from the phylogenetic findings that the co-occurrence of both the lineages (1 and 2) in Iran may allow interbreeding and interchange in nuclear genes (Ascunce et al. 2013). Furthermore, B. caprae sequences reported from Turkey acted like an outgroup and clustered with Bovicola sp. recorded from a hawk, indicating the misidentification of the lice species involved.
The pairwise genetic distances among sequences revealed high values further insinuating diverged lineages. The finding supported the observation of the evolutionary tree evincing the diverged lineages of the sequences recorded from Iran to the sequences generated herein. On the contrary, the sequences of the present study exhibited extremely low values within themselves, hence alluding absence of diverged lineages. However, the sequence recorded from Palampur, India also exhibited slight variation, which was indicative of the presence of a sub-lineage of B. caprae. The observation was further supported by the nucleotide polymorphism recorded during the comparative sequence analysis. The finding could be attributed to the fact that the endemicity of the parasites might result in nucleotide substitutions or polymorphisms or mutations due to anthropogenic pressure such as transhumance or trading of animals (Solano et al. 2016).
The phylogenetic or evolutionary tree analysis is inadequately explanatory for the variations identified in the sequences included in the study. Owing to the capability of median-joining haplotype network analysis to adjudge even a single nucleotide polymorphism, it is considered to be a better approach for studying the genetic diversity and relationship among the sequences (Dumaidi et al. 2020). Hence, cladogram analysis was performed in the present study to identify the misidentified/misannotated B. caprae sequences in the GenBank™. In the present study, the haplogroup corresponding to lineage 2 consisted of sequences from India, China and Iran with the presence of two haplotypes; Hap_1 and Hap_2. Hap_1 represented the sequence from Palampur, India; whereas, Hap_2 comprised the sequences from Fatehabad and Pali, India generated herein and from Iran and China. However, no such differentiation was observed during phylogenetic analysis. In haplogroup/lineage 2, the haplotypes differed with 1–2 mutational steps or nucleotide variations, which indicated the common origin of the sequences due to recent population expansion or genetic bottleneck or selective sweep (Dumaidi et al. 2020). In the median-joining haplotype network analysis, no central haplotype was recorded. The finding further indicated the uncertainty of the existence of a widespread haplotype of B. caprae infesting goat populations (Ohiolei et al. 2019). Since, only a few sequences of B. caprae corresponding to the mitochondrial cox1 gene had been submitted to the GenBank™, which eventually restricted the identification of a central haplotype. Detailed geographical studies involving an extensive DNA analysis are a prerequisite for resolving the issue of the identification of a widespread/central or ancestral haplotype (Ohiolei et al. 2021).
The population structure analysis revealed a low nucleotide and a high haplotype diversity for the complete data set as well as for the sequences generated herein, which indicated a recent demographic expansion and low gene flow followed by genetic drifts or bottlenecks (Moudgil et al. 2022). The recent demographic expansion might have resulted in coalescence, which eventually lead to low nucleotide diversity. Also, the amassed mutations might have increased haplotype diversities (Moudgil et al. 2023). The positive neutrality test values recorded in the present study were suggestive of high polymorphism experienced by the populations. The finding could be incriminated to any factor which had endangered the survival of the lice population such as the use of insecticides/pediculicides (Phadungsaksawasdi et al. 2021).
Lice are not capable of self-dispersion. Anthropogenic short/long-range host mobility especially under transhumance systems could be considered the sole reason behind lice dispersion, which would eventually influence their population structure. The findings of the present study associated with a high value of FST and a low value of Nm also supported the aforementioned assertion. High genetic differentiation in lice populations could be associated with the biological factors (including the host specificity) and behavioral traits (one-host feeding) of lice.
The present study provided the first molecular insight into the phylogenetic relationship and population structuring of B. caprae infesting goats of different agro-climatic locations in India with respect to the goat louse species prevalent in the world. The negative values of the neutrality indices for the complete dataset indicated population expansion of the ectoparasite. Furthermore, the sequences generated herein also showed the circulation of sub-lineages of B. caprae in India based on the analysis of pairwise genetic distance between sequences and median-joining haplotype network. The present pilot study is of utmost significance considering the importance of caprine pediculosis and would prove beneficial in designing effective control strategies. Additionally, the results of the present study would also lay the basis for future comprehensive studies involving both biting and sucking lice infesting goats of a wider study area.
Code Availability
Not applicable.
References
Ajith Y, Dimri U, Gopalakrishnan A, Devi G (2017a) A study on prevalence and factors associated with ectoparasitism in goats of two agro-climatic regions in India. J Parasit Dis 41(3):739–746. https://doi.org/10.1007/s12639-017-0881-y
Ajith Y, Dimri U, Singh SK, Gopalakrishnan A, Devi G, Verma MR, Joshi V, Alam S (2017b) Lice induced immuno-oxidative wreckage of goats. Vet Parasitol 242:24–30. https://doi.org/10.1016/j.vetpar.2017.05.019
Ajith Y, Dimri U, Gopalakrishnan A, Devi G (2019) A field study on the efficacy of ivermectin via subcutaneous route against chewing lice (Bovicola caprae) infestation in naturally infested goats. Onderstepoort J Vet Res 86(1):e1–e5. https://doi.org/10.4102/ojvr.v86i1.1635
Amanzougaghene N, Fenollar F, Davoust B, Djossou F, Ashfaq M, Bitam I, Raoult D, Mediannikov O (2019) Mitochondrial diversity and phylogeographic analysis of Pediculus humanus reveals a new Amazonian clade “F.” Infect Genet Evol 70:1–8. https://doi.org/10.1016/j.meegid.2019.02.006
Ascunce MS, Toups MA, Kassu G, Fane J, Scholl K, Reed DL (2013) Nuclear genetic diversity in human lice (Pediculus humanus) reveals continental differences and high inbreeding among worldwide populations. PLoS ONE 8(2):e57619. https://doi.org/10.1371/journal.pone.0057619
Dumaidi K, Qaraqe H, Al-Jawabreh A, Abu-Helu R, Samarah F, Al-Jawabreh H (2020) Genetic diversity, haplotype analysis, and risk factor assessment of hepatitis A virus isolates from the West Bank, Palestine during the period between 2014 and 2016. PLoS ONE 15(12):e0240339. https://doi.org/10.1371/journal.pone.0240339
Fournier PE, Ndihokubwayo JB, Guidran J, Kelly PJ, Raoult D (2002) Human pathogens in body and head lice. Emerg Infect Dis 8(12):1515–1518. https://doi.org/10.3201/eid0812.020111
Ghavami MB, Ghanbari M, Panahi S, Taghiloo B (2020) Diversity of mitochondrial genes and predominance of Clade B in different head lice populations in the northwest of Iran. Parasites Vectors 13(1):485. https://doi.org/10.1186/s13071-020-04364-z
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hornok S, Hofmann-Lehmann R, de Mera IG, Meli ML, Elek V, Hajtós I, Répási A, Gönczi E, Tánczos B, Farkas R, Lutz H, de la Fuente J (2010) Survey on blood-sucking lice (Phthiraptera: Anoplura) of ruminants and pigs with molecular detection of Anaplasma and Rickettsia spp. Vet Parasitol 174(3–4):355–358. https://doi.org/10.1016/j.vetpar.2010.09.003
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120. https://doi.org/10.1007/BF01731581
Leigh JW, Bryant D (2015) PopART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Low VL, Tay ST, Kho KL, Koh FX, Tan TK, Lim YA, Ong BL, Panchadcharam C, Norma-Rashid Y, Sofian-Azirun M (2015) Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasites Vectors 8:341. https://doi.org/10.1186/s13071-015-0956-5
Moudgil AD, Sharma A, Verma MS, Kumar R, Dogra PK, Moudgil P (2017) Gastrointestinal parasitic infections in Indian Gaddi (goat) breed bucks: clinical, hemato-biochemical, parasitological and chemotherapeutic studies. J Parasit Dis 41(4):1059–1065. https://doi.org/10.1007/s12639-017-0934-2
Moudgil AD, Nehra AK, Vohra S, Thakur SD, Sharma D (2022) Prevalence and phylogeography of Taenia hydatigena metacestodes from goats of India. Parasitology 149(9):1193–1198. https://doi.org/10.1017/S0031182022000713
Moudgil AD, Nehra AK, Vohra S (2023) Phylogeography and demographic dynamics of Rhipicephalus microplus from North India. Infect Genet Evol 112:105464. https://doi.org/10.1016/j.meegid.2023.105464
Nehra AK, Kumari A, Kundave VR, Vohra S, Ram H (2022) Molecular insights into the population structure and haplotype network of Theileria annulata based on the small-subunit ribosomal RNA (18S rRNA) gene. Infect Genet Evol 99:105252. https://doi.org/10.1016/j.meegid.2022.105252
Ohiolei JA, Luka J, Zhu GQ, Yan HB, Li L, Magaji AA, Alvi MA, Wu YT, Li JQ, Fu BQ, Jia WZ (2019) First molecular description, phylogeny and genetic variation of Taenia hydatigena from Nigerian sheep and goats based on three mitochondrial genes. Parasites Vectors 12(1):547. https://doi.org/10.1186/s13071-019-3807-y
Ohiolei JA, Yan HB, Li L, Li WH, Wu YD, Alvi MA, Zhang NZ, Fu BQ, Wang XL, Jia WZ (2021) A new molecular nomenclature for Taenia hydatigena: mitochondrial DNA sequences reveal sufficient diversity suggesting the assignment of major haplotype divisions. Parasitology 148(3):311–326. https://doi.org/10.1017/S003118202000205X
Phadungsaksawasdi K, Sunantaraporn S, Seatamanoch N, Kongdachalert S, Phumee A, Kraivichian K, Sawaswong V, Payungporn S, Brownell N, Siriyasatien P (2021) Molecular analysis of mitochrondrial cytb of Pediculus humanus capitis in Thailand revealed potential historical connection with South Asia. PLoS ONE 16(9):e0257024. https://doi.org/10.1371/journal.pone.0257024
Pilarczyk B, Tomza-Marciniak A, Pilarczyk R, Bombik E, Seremak B, Udała J, Sadowska N (2021) A comparison of the prevalence of the parasites of the digestive tract in goats from organic and conventional farms. Animals 11(9):2581. https://doi.org/10.3390/ani11092581
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Sebei PJ, McCrindle CM, Green ED, Turner ML (2004) Use of scanning electron microscopy to confirm the identity of lice infesting communally grazed goat herds. Onderstepoort J Vet Res 71(2):87–92. https://doi.org/10.4102/ojvr.v71i2.269
Solano D, Navarro JC, León-Reyes A, Benítez-Ortiz W, Rodríguez-Hidalgo R (2016) Molecular analyses reveal two geographic and genetic lineages for tapeworms, Taenia solium and Taenia saginata, from Ecuador using mitochondrial DNA. Exp Parasitol 171:49–56. https://doi.org/10.1016/j.exppara.2016.10.015
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101(30):11030–11035. https://doi.org/10.1073/pnas.0404206101
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38(7):3022–3027. https://doi.org/10.1093/molbev/msab120
Taylor MA, Coop RL, Wall RL (2016) Veterinary parasitology, 4th edn. Blackwell Publishing, Oxford, pp 503–504
Yin F, Guo C, Tian Z, Li D, Mu D, Liu H, Guan G, Yin H, Li F (2023) Analysis of genetic diversity and population structure of Babesia gibsoni. Front Vet Sci 10:1147958. https://doi.org/10.3389/fvets.2023.1147958
Yingklang M, Banjong D, Haonon O, Intuyod K, Dangtakot R, Thongpon P, Pinlaor P, Laha T, Siriyasatien P, Pinlaor S (2021) Phylogeography and demographic history of Thai Pediculus humanus capitis (Phthiraptera: Pediculidae) revealed by mitochondrial DNA sequences. Infect Genet Evol 91:104825. https://doi.org/10.1016/j.meegid.2021.104825
Yong Z, Fournier PE, Rydkina E, Raoult D (2003) The geographical segregation of human lice preceded that of Pediculus humanus capitis and Pediculus humanus humanus. C R—Biol 326(6):565–574. https://doi.org/10.1016/s1631-0691(03)00153-7
Acknowledgements
Authors are thankful to the Director of Research and the Dean, College of Veterinary Sciences, Lala Lajpat Rai University of Veterinary and Animal Sciences for providing the necessary funds and facilities to carry out the research. The authors are also thankful to the RKVY-RAFTAAR for enriching the department with equipments which could be utilized during the research.
Funding
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
A. D. M. was involved in sample collection, investigation, methodology, phylogenetics, and drafting the manuscript. A. K. N. was involved in sample collection, investigation, and laboratory work. A. S. helped in collection of samples and reviewed the manuscript. S.P. helped in collection of samples and reviewed the manuscript. S. V. was involved in supervision and editing. All the authors contributed to the review.
Corresponding author
Ethics declarations
Conflict of interest
The authors report there are no competing interests to declare.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Ethical Approval
The research work was carried out with the permission (VCC/IAEC/2022/1679-1705) of Institutional Animal Ethics Committee of Lala Lajpat Rai University of Veterinary and Animal Sciences, Hisar, Haryana, India.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moudgil, A.D., Nehra, A.K., Sharma, A. et al. First Insight into the Phylogenetic Diversity of Bovicola caprae Infesting Goats of Different Agro-climatic Locations in India. Biochem Genet (2024). https://doi.org/10.1007/s10528-024-10886-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10528-024-10886-3