
Abstract
The influence of chronic diseases on various facets of macrophage cellular senescence is poorly understood. This study evaluated the impact of chronic hyperglycemia on the induction of cellular senescence and subsequent immunosurveillance functions in RAW264.7 macrophages. Macrophages were cultured under normal glucose (NG; 5 mM), high glucose (HG; 20 mM), and very high glucose (VHG; 40 mM) conditions and assessed for markers of cellular senescence. Hyperglycemia induced strong upregulation of SA-β-gal activity, and loss of PCNA and Lamin B1 gene expression while markers of cell cycle arrest generally decreased. Non-significant changes in SASP-related proteins were observed while ROS levels slightly decreased and mitochondrial membrane potential increased. Protein concentration on the exosome membrane surface and their stability appeared to increase under hyperglycemic conditions. However, when macrophages were exposed to the secretory media (SM) of senescent preadipocytes, a dramatic increase in the levels of all inflammatory proteins was recorded especially in the VHG group that was also accompanied by upregulation of NF-κB and NLRP3 gene expression. SM treatment to hyperglycemic macrophages activated the TLR-2/Myd88 pathway but decreased the expression of scavenger receptors RAGE, CD36, and Olr-1 while CD44 and CXCL16 expression increased. On exposure to LPS, a strong upregulation in NO, ROS, and inflammatory cytokines was observed. Together, these results suggest that primary markers of cellular senescence are aberrantly expressed under chronic hyperglycemic conditions in macrophages with no significant SASP activation. Nonetheless, hyperglycemia strongly deregulates macrophage functions leading to impaired immunosurveillance of senescent cells and aggravation of inflamm-aging. This work provides novel insights into how hyperglycemia-induced dysfunctions can impact the potency of macrophages to manage senescent cell burden in aging tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Macrophages are among the first cells to recognize external infectious agents as well as damaged cells and are thus crucial for initiating and resolving inflammatory immune responses. Several age-related changes in macrophage phenotype and effector functions have been documented under the umbrella term of ‘immunosenescence’ (Clark et al. 2020; De Maeyer & Chambers 2021; Lloberas et al. 2019). In addition, macrophage-specific immunological and metabolic dysfunctions are strongly implicated in the increased predisposition of the elderly to age-related inflammatory pathologies and impaired physiological processes such as wound healing (Duong et al. 2021; van Beek et al. 2019). Evidence is now emerging that macrophages are critical players involved in the recognition and clearance of senescent cells during aging (Kale et al. 2020; Li et al. 2020). It is widely accepted that an increase in senescent cell burden may directly contribute to tissue dysfunctions with advancing age which could at least be partially attributed to their decreased clearance by immune cells such as macrophages and NK cells (Ovadya et al. 2018; Pignolo et al. 2020; Prata et al. 2018). Moreover, it is being increasingly rationalized that cellular senescence could also be an active regulator of macrophage effector functions which might influence their ability to counter accumulating senescent cells especially since tissue-resident macrophages are in direct contact with aging non-immune cells (Sharma 2021). However, cellular senescence in macrophages is rather poorly characterized and studies have shown conflicting reports regarding the presence and significance of markers of cellular senescence in aging macrophages (Hall et al. 2017, 2016; Sharma et al. 2022a, b). Further, there is sporadic and limited knowledge as to how different tissue-resident macrophages are influenced by cellular senescence both in aging and disease.
Diabetes-associated hyperglycemia is a chronic condition with multiple deleterious effects such as the increased risk of chronic kidney disease, retinopathy, neuropathy, and oxi-inflammatory stress in general (Giri et al. 2018). Recent studies have also demonstrated that hyperglycemia can induce premature cellular senescence both in vitro and in vivo, and therefore augment accelerated aging (Bertelli et al. 2022; Jiang et al. 2022; Marino et al. 2022). Macrophage/monocyte cell lines such as RAW264.7 and THP-1 have been previously used as cellular models of stress-induced cellular senescence (Mytych et al. 2017; Wang et al. 2020; Xu et al. 2020). Although hyperglycemia has been documented to induce immune dysfunctions, however, whether such conditions can also influence various aspects of macrophage cellular senescence is little understood. Therefore, in the present work, we first determined whether long-term exposure to hyperglycemic conditions induces cellular senescence, SASP, and cellular dysfunctions in the murine RAW264.7 macrophage cell line. We then investigated whether such hyperglycemia-induced changes in macrophages also affected their ability to respond to the senescence-associated secretory phenotype (SASP) of senescent preadipocyte cells as well as lipopolysaccharide (LPS) in vitro. Our results demonstrate that hyperglycemia induced differential expression of cellular senescence markers in macrophages although strong deregulations in downstream effector functions were apparent.
Materials and methods
Cell lines and treatment protocols
RAW264.7 murine macrophage cells were obtained from the National Centre For Cell Science (NCCS), Pune, India. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HiMedia, India) at respective glucose concentrations and supplemented with 10% fetal bovine serum (FBS) and 100 μg/mL of penicillin–streptomycin (15,140,122; Invitrogen, USA) at 37 ºC in a 5% CO2 incubator. A long-term cell culture model of hyperglycemia was established as described previously (Suzuki et al. 2021). Briefly, cells were cultured for seven consecutive days in three separate groups based on glucose levels in the media. Normoglycemia (NG) group representing 5 mM glucose, hyperglycemia (HG) group representing 25 mM glucose or very high glucose (VHG) group representing 40 mM glucose were prepared by supplementing with d-glucose (TC130; HiMedia, India) to desired concentrations (Suzuki et al. 2021). Further, cells were initially acclimatized to normoglycemic conditions by passaging twice before the beginning of the actual experimental groups. For experiments involving the secretome of senescent preadipocytes, murine 3T3-L1 cell line (obtained from NCCS, Pune, India) was cultured and maintained in DMEM media supplemented with 10% FBS and 100 μg/mL of penicillin–streptomycin (15,140,122; Invitrogen, USA) at 37ºC in a 5% CO2 incubator. Cellular senescence was induced in preadipocytes using acute and repeated hydrogen peroxide (H2O2) exposure as we have reported previously (Kumar et al. 2019). Briefly, cells were stimulated with H2O2 at a sublethal concentration of 150 µM for 3 h and for three consecutive days. After another three days of incubation, the culture supernatant of the senescent preadipocytes was collected, filtered, aliquoted, and stored at−80ºC till further use (Kumar et al. 2019, 2020). This supernatant was referred to as the senescent media (SM), and whenever required in the present study, it was directly used to stimulate RAW264.7 cells for 24 h at 37 ºC in a 5% CO2 incubator (Kumar et al. 2020). In experiments involving lipopolysaccharide (LPS), respective experimental cell groups representing different glucose concentrations, were stimulated with 1 μg/mL LPS (Merck; L2630) for 24 h as we have noted previously (Diwan et al. 2023) followed by various cellular, biochemical, and molecular analyses.
Intracellular ROS production
Levels of reactive oxygen species (ROS) in RAW 264.7 cells were detected using 2′,7′-dichlorofluorescin diacetate (DCFH-DA) (Merck, D6883) redox probe as previously described (Kim & Xue 2020). Briefly, after respective treatments, cells were washed with PBS and incubated with DCFH-DA solution (10 μM) for 30 min at 37 °C in a CO2 incubator. Cells were again washed twice with PBS and subject to cell lysis using RIPA buffer. The cell lysate was then centrifuged at 16,000 × g for 15 min at 4 °C and the supernatant was collected. The supernatant (100 μL) was then transferred to a black 96-well plate and the fluorescence intensity was immediately measured using a Varioskan Lux microplate reader at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. ROS levels in each sample are expressed relative to fluorescence intensities.
NO production assay
Nitric oxide (NO) levels were quantified in the culture supernatants using the Griess reagent assay kit (Cat.#G2930) (Promega, Madison, WI, U.S.A) according to the manufacturer’s protocol. Briefly, 50 µL of the sample was added to a 96-well plate, followed by the addition of 50 μL of sulfanilamide solution (1% sulfanilamide in 5% phosphoric acid) and subsequent incubation for 10 min at RT, protected from light. Afterward, 50 μL of NED solution (0.1% N-1-napthylethylenediamine dihydrochloride in water) was added to each well, and the mixture was incubated for another 10 min at RT, protected from light. Finally, the absorbance was measured at 540 nm using the Varioskan Lux microplate reader. The nitrite concentration in the samples was quantified relative to the standard sodium nitrite.
Mitochondrial membrane potential assessment
The effect of hyperglycemia on macrophage mitochondrial membrane polarization was assessed by using a commercially available kit (Abcam Inc., ab113852) as per the manufacturer’s instructions. In this method, TMRE (tetramethylrhodamine, ethyl ester) is used to label active mitochondria. TMRE is a cell-permeant, positively charged dye that readily accumulates in active mitochondria due to their relative negative charge. Depolarized or inactive mitochondria have decreased membrane potential and fail to sequester TMRE. FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone) which is an ionophore was also used in this assay as a positive control that depolarizes the mitochondrial membrane thereby reducing TMRE sequestration and therefore positive signal. Briefly, cells were incubated with TMRE and FCCP (in the appropriate control group) for 30 min followed by washing with PBS and analyzed with a microplate reader at Ex/Em 549/575 nm. Results are also expressed as relative to FCCP that indicate the magnitude of depolarization induced by hyperglycemia vis-à-vis standard FCCP-induced depolarization.
Senescence-associated β-galactosidase activity
The activity of β-galactosidase was assessed using β-galactosidase assay kit (Cell Signaling Technologies, USA, #9860) as per the manufacturer’s protocol. Briefly, at the end of the experiment, cells were washed with PBS to remove culture media and then fixed with a fixative solution. Cells were again washed and treated with a staining solution followed by overnight incubation in the dark at 37 ºC in the absence of CO2. The next day, cells were observed and imaged for the appearance of blue color as a marker of senescence-associated β-galactosidase activity. Several images were taken, and cells were manually counted for the enumeration of percent SA-β-gal positive cells.
Estimation of cytokines
Cytokines viz., IL-6, TNF-α, IL-1-β, and IL-10 were estimated in cell culture supernatants using commercially available sandwich ELISA kits (ELISA MAX™ Deluxe set, BioLegend, San Diego, U.S.A) while MCP-1 was estimated using another sandwich ELISA kit (Thermo Fisher Scientific). Briefly, after respective treatments, the cell culture supernatants were collected, centrifuged, aliquoted, and stored at -80 ºC until further assessment. Sandwich ELISA was performed according to the manufacturer’s protocol and the plate was read on Varioskan Lux microplate reader. Results are expressed as picograms/mL for each tested cytokine.
Quantitative real-time gene expression analysis
Total cellular RNA was isolated using the TRI-reagent (Sigma-Aldrich, USA, Cat. #T9424). Briefly, after respective treatments, cells were homogenized in TRI-reagent, and total RNA was isolated as per the manufacturer’s protocol (Diwan & Sharma 2022). The quality and quantity of the isolated RNA were determined, and the RNA was aliquoted and stored at -80 °C until further analysis. qRT-PCR was performed using CFX96 Touch Real-Time PCR Detection System (BioRad Inc.). In brief, 50 ng of RNA template was used per reaction using the Thermo Scientific Verso SYBR Green 1-Step qRT-PCR Low ROX Kit (Cat. #AB-4106/C) as per the manufacturer’s protocol. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression was utilized as a housekeeping control to quantify relative mRNA expression using the ΔΔCt method as previously described (Diwan & Sharma 2022). The list of primers used is provided in Table 1.
Exosomes isolation and characterization
Exosomes were isolated from cell culture media using the total exosome isolation reagent kit (Thermo Fisher Scientific, USA; Cat. No. 4478359) as per the manufacturer’s instructions. Briefly, after respective treatments, cells were washed twice and cultured in serum-free DMEM media for 24 h in a CO2 incubator at 37 ºC following which the culture supernatant was collected and centrifuged at 2000 × g for 30 min. The pellet was discarded, and the supernatant was mixed with the exosome isolation reagent (1:0.5). The mixture was subsequently incubated overnight at 4 ºC followed by centrifugation at 10,000 × g for 1 h. The supernatant was then discarded and pelleted exosomes were suspended in PBS solution and stored at −80 ºC till further analyses. Total exosomal surface membrane protein levels were determined using the Bradford method as described previously (Dash et al. 2021). Exosome size and mean zeta potential were determined using Litesizer 500 (Anton Paar GmbH, Austria) particle analyzer.
SDS-PAGE and Western blotting
Total cellular proteins were isolated, quantified, and subject to SDS-PAGE as described previously (Sharma, Kumar, et al., 2022). Briefly, isolated proteins (40 μg) were subjected to SDS-PAGE using gel at 10% concentration followed by transfer to polyvinylidene difluoride membrane (GE Healthcare Life Sciences, Europe) using a semidry trans-blotter (Bio-Rad, USA). Non-fat dried milk (5%) was used for subsequent blocking of the membranes at room temperature and after washing, the membranes were probed with the anti-p16Ink4a (PA5-20,379; Thermo Fisher Scientific, USA) and anti-β-actin (MA1-140; Thermo Fisher Scientific, USA) antibodies overnight at 4 °C with gentle shaking. HRP-conjugated secondary antibody (Cat no. 170–6515/16; Bio-Rad, USA) was used for further incubation of membranes at room temperature for 1 h. After washing, protein bands were detected using Clarity Western ECL Substrate (Bio-Rad, USA), and relative protein expression of p16Ink4a was quantified against β-actin using the Gel- Analyzer software (version 19.1).
Statistical analyses
Data are expressed as mean ± S.D (n = 3). Significant differences among the groups were determined using one-way ANOVA followed by Tukey’s post-hoc test. Differences between means were considered statistically significant at p ≤ 0.05.
Results
Differential effects of hyperglycemia on markers of cellular senescence
We first assessed the cytocompatibility of prolonged hyperglycemia exposure in RAW264.7 macrophages using trypan blue exclusion assay and observed no cytotoxic effects of high glucose since more than 90% of cells were viable regardless of the treatment (Fig. 1A). Next, various cellular and molecular markers of cellular senescence were measured in macrophages after exposure to hyperglycemic conditions for seven consecutive days. Increased SA-β-gal activity is considered a canonical marker of cellular senescence. In our study, we observed SA-β-gal expression in all tested groups but a very strong and ubiquitous increase in SA-β-gal activity was apparent in macrophages cultured under both the hyperglycemic conditions (i.e., HG and VHG) as compared to the NG group (Fig. 1B-E). On the other hand, p53 and p21 gene expression significantly decreased by nearly 50% in HG and VHG groups as compared to the control while a non-significant albeit decreasing trend was also evident in p16Ink4a and Rb expression (Fig. 2A-D). However, protein expression of p16Ink4a demonstrated a clear decrease on account of both HG and VHG treatments (Fig. 2G). VHG-treated cells reported a strong decline in the expression of Lamin B1 while PCNA expression declined significantly in both HG and VHG groups as compared to the NG control (Fig. 2 E–F).

Effect of long-term hyperglycemic conditions on SA-β-gal activity. (A) Cell viability analysis (B-D) Representative images of SA-β-gal activity. (E) % SA-β-gal positive cells. Values are mean ± S.D (n = 3). *Represents significant difference among different groups; **p ≤ 0.01, ****p ≤ 0.0001

Influence of hyperglycemia on primary markers of cellular senescence. Relative quantitative gene expression analysis of (A) p53 (B) p21WAF1 (C) p16Ink4a (D) Rb (E) PCNA (F) Lamin B1. (G) Relative p16Ink4a protein expression. *Represents significant difference among different groups; * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
Lack of a robust SASP activation in hyperglycemic macrophages
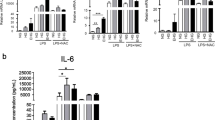
To further characterize whether prolonged hyperglycemia induces the development of cellular senescence-dependent SASP in macrophages, levels of various cytokines and expression of different inflammatory transcription factors were determined. Although MCP-1 levels recorded a mild yet significant increase in the HG-treated group, non-significant changes in the levels of IL-1β, IL-6, and IL-10 were also recorded as compared to the remaining groups (Fig. 3A-E). Further, no significant change in any of the tested cytokines was observed between NG and VHG groups. In addition, gene expression of inflammatory transcription factor NLRP3 was significantly higher (over 3 folds) in the HG-treated group only while the expression of NF-κB did not change significantly in either of the tested groups (Fig. 3F-G).

Impact of long-term hyperglycemia on protein levels of cytokines. (A) MCP-1 (B) TNF-α (C) IL-6 (D) IL-1β (E) IL-10. Relative quantitative gene expression analysis of (F) NF-κB (G) NLRP3. Values are mean ± S.D (n = 3). *Represents significant difference among different groups; * p ≤ 0.05, **p ≤ 0.01
Impaired redox activity in macrophages treated with hyperglycemic conditions
The effect of long-term hyperglycemic conditions on intracellular oxi-inflammatory stress in macrophages was determined. Measurement of intracellular ROS levels revealed a slight yet significant decrease in both HG and VHG groups as compared to NG (Fig. 4A). However, no significant changes could be observed in macrophage NO production among any of the tested groups (Fig. 4B). On the other hand, both HG and VHG-treated cells observed a strong increase in mitochondrial membrane potential as revealed by increased fluorescence of sequestered TMRE dye relative to the NG group (Fig. 4C). Further, when analyzed relative to FCCP-induced depolarization, VHG treated group appeared to be the most resistant to depolarization of mitochondrial transmembrane potential (Fig. 4D).

Effect of long-term hyperglycemia on macrophage redox homeostasis. (A) Intracellular ROS levels (B) NO production (C) Mitochondrial membrane potential (D) Mitochondrial membrane potential relative to FCCP. Values are mean ± S.D (n = 3). *Represents significant difference among different groups; * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001
Hyperglycemia affects macrophage exosome surface membrane proteins
Bradford’s reagent is used to quantify the levels of surface membrane proteins in exosomes since this reagent specifically binds to proteins present on the exosome membrane. Cells cultured in HG and VHG conditions induced near dose-dependent over 3 folds significant increase in the concentration of surface exosome membrane proteins (Fig. 5A). However, no significant effect of hyperglycemia on exosome diameter could be observed although the exosome mean zeta potential appeared to be significantly more negative and thus more stable in macrophages exposed to 40 mM glucose levels (VHG) (Fig. 5B-C).

Influence of high glucose conditions on macrophage exosomes. (A) Surface exosomal membrane proteins levels (B) Exosome diameter (C) Mean zeta potential. Values are mean ± S.D (n = 3). *Represents significant difference among different groups at p ≤ 0.05
Exposure to SM augments pro-inflammatory response in hyperglycemic macrophages
On stimulation with SM of senescent preadipocytes, a dramatic change in the cytokine expression profile was evident in hyperglycemia-exposed macrophages as compared to their native expression (non-SM treated). In general, all measured cytokines recorded near the dose-dependent effect of hyperglycemia wherein a more exaggerated proinflammatory environment was evident in the VHG group as compared to the HG group (Fig. 6A-E). Further, all measured cytokines demonstrated a strong and significant difference between VHG and NG groups especially related to IL-6, IL-1β, and TNF-α levels (Fig. 6A-E). In addition, MCP-1, TNF-α, and IL-6 levels recorded a multifold increase in macrophages exposed to SM as compared to non-SM treated macrophages suggesting robust cytokine stimulation. However, the levels of intracellular ROS and NO production followed a similar trend as observed in non-SM-treated cells (Fig. 6F-G). Further mRNA analysis of pro-inflammatory transcription factor NF-κB also revealed a strong ( ̴ 50 folds) and significant upregulation in VHG treated macrophages (Fig. 6H). NLRP3, the downstream target of NF-κB, also recorded a dramatic increase in both HG and VHG treated groups as compared to the control NG group (Fig. 6I).

Analyses of activation status of hyperglycemia treated macrophages on account of SM exposure. Levels of (A) IL-6 (B) TNF-α (C) IL-1β (D) MCP-1 (E) IL-10 (F) ROS (G) NO. Relative gene expression of (H) NF-κB and (I) NLRP3. Values are mean ± S.D (n = 3). *Represents significant difference among different groups; * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
SM treatment impairs cell surface receptor response in VHG-treated macrophages
The effect of SM exposure on receptors involved in macrophage-mediated cell recognition and stimulation was assessed. A strong and significant multifold increase in TLR-2 and MyD88 gene expression was evident in VHG-treated macrophages as compared to either NG or HG-treated macrophages while no such effect on TLR-4 expression could be observed (Fig. 7A-C). On the other hand, the expression of various macrophage scavenger receptors was differentially influenced by exposure to SM. The receptor for advanced glycation end products (RAGE), oxidized low-density lipoprotein receptor 1 (Olr1), and CD36 demonstrated a strong decrease in both HG and VHG groups as compared to NG-treated cells (Fig. 7D-F). On the other hand, expressions of CD44, CXCL16, and CD206 receptors showed a significant increase in hyperglycemic cells exposed to SM as compared to NG cells (Fig. 7G-I).

Influence of hyperglycemia on expression of scavenger receptors in macrophages on account of SM exposure. Relative gene expression of (A) TLR-2 (B) MyD88 (C) TLR-4 (D) RAGE (E) OLR-1 (F) CD36 (G) CD44 (H) CD206 (I) CXCL16. Values are mean ± S.D (n = 3). *Represents significant difference among different groups; * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
Hyperglycemia exacerbates macrophage response to LPS stimulation
To further assess how hyperglycemia impairs effector functions in macrophages, respective cells were exposed to LPS for 24 h and then analyzed for various markers of stimulation. Intracellular ROS and NO production strongly and significantly increased in HG and VHG groups as compared to NG-treated cells (Fig. 8A-B). Similarly, proinflammatory and activator cytokines MCP-1 and TNF-α recorded a significant increase in the VHG group although no significant changes in IL-1β and IL-6 levels could be observed in any of the tested groups (Fig. 8C-F). On the other hand, levels of IL-10 registered a strong increase in both HG and VHG groups further indicating the deregulation of the inflammatory response (Fig. 8G). Moreover, except for MCP-1, a multifold increase in all tested parameters was evident in LPS-stimulated cells as compared to SM or non-SM-treated cells.

Analyses of activation status of hyperglycemia treated macrophages on account of LPS exposure. (A) Intracellular ROS levels (B) NO production (C) MCP-1 (D) TNF-α (E) IL-1β (F) IL-6 (G) IL-10. Values are mean ± S.D (n = 3). *Represents significant difference among different groups; * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001
Discussion
Macrophages are evolved to sense external and internal cellular damage and unlike other cells, are highly plastic and dynamic in response to various stressors. Chronic diseases such as diabetes impair macrophage effector functions and deregulate the immune response. In addition, hyperglycemic conditions (20–200 mM glucose) have been demonstrated to induce premature cellular senescence in different types of non-immune cells such as endothelial cells and stem cells (Jiang et al. 2022; Wan et al. 2022; Yin et al. 2021). Our observations in the present study suggest that hyperglycemic conditions in macrophages may only have caused mild senescence at best since mixed results in terms of various markers of cellular senescence were noted. Notably, only SA-β-gal activity and PCNA expression showed characteristic changes akin to cellular senescence under both HG and VHG conditions while the expression of cyclins and their downstream effectors did not exhibit consistent results but appeared to be downregulated. It needs to be emphasized here that the nature and relevance of cellular senescence markers in macrophages is poorly understood and debatable. Moreover, there is no information as to how disease conditions such as chronic hyperglycemia might impact cellular senescence in macrophages. Previous studies have identified that senescent macrophages are typically characterized by increased expression of p16Ink4a, SA-β-gal activity, and the development of SASP (Kim et al. 2015; Liu et al. 2019; Sebastián et al. 2009; Talma et al. 2021; Yousefzadeh et al. 2021). However, there is also increasing evidence that SA-β-gal activity and p16Inka4a expression in macrophages could just be a type of p53-independent physiological stress response mechanism to various immune stimuli rather than the irreversible process of cellular senescence (Hall et al. 2017). Further, activated and maturing macrophages are also known to exhibit increased SA-β-gal expression although the accumulation of SA-β-gal + macrophages in injured and aged tissues has also been noticed (de Mera-Rodríguez et al. 2021; Frescas et al. 2017; Oishi & Manabe 2016). A recent study identified two subpopulations of high p16Inka4a expressing tissue-resident macrophages in both young and aged murine brains thereby strongly suggesting age-independent relevance and the presence of p16Inka4a positive macrophages (Talma et al. 2021). A high level of p16Ink4a expression was also observed in specific processes unrelated to senescence such as in monocyte differentiation (Cudejko et al. 2011). In addition, conflicting reports concerning the role of p16Ink4a activation and inflammatory aggravation relevant to the SASP of senescent macrophages have also been noted (Cudejko et al. 2011; Murakami et al. 2012). Taken together, it is apparent that the canonical markers of senescence, including but not limited to p16Inka4a and SA-β-gal activity, might play a more exhaustive and unexpected role in macrophages that may not be directly related to macrophage senescence. Our observations in this study corroborate this notion and further assert that primary senescence markers in macrophages could also be differentially influenced in diseased conditions although the exact underlying mechanisms are not clear. In terms of hyperglycemia and macrophages, a recent study observed that short-term (24 h) exposure to high glucose (30 mM) induced SA-β-gal activity and activation of p16Ink4a and p21 senescence pathways in RAW264.7 macrophages (Zhang et al. 2019). Our results are in partial agreement with this work, but it needs to be emphasized that given the known ambiguous functional relevance of primary senescence markers in macrophages, characterization of senescence in these cells after long-term exposure, such as tested in the present study, and with more exhaustive analysis could be a more meaningful approach for understanding any impending senescence in macrophages.
Studies describing the effects of hyperglycemia on macrophage ROS levels have been inconsistent. While there is evidence that high glucose may increase macrophage ROS production, there are also studies that report no such effect. Corroborating our findings on ROS levels, previous studies have observed that exposure to high glucose did not induce an overt increase in resting macrophage ROS levels but only exaggerated their ROS response when stimulated with external threats such as LPS (Suzuki et al. 2021; Zhang et al. 2021). Similarly, another study noted that long term sensitization of bone marrow derived macrophages (BMDM) with high glucose condition impaired phagocytic response due to decreased production of ROS associated with oxidative burst (Pavlou et al. 2018). It also needs to be emphasized here that unlike PAMPs such as LPS, glucose is a relatively mild stressor, and since macrophages are more robustly equipped to handle oxidative stress as compared to other cells (Virág et al. 2019); it may not be unusual to note the lack of any hyperglycemia-intracellular oxidative stress in macrophages especially under resting conditions. Moreover, since mitochondria is the chief site of cellular ROS production, we noticed a significant increase in mitochondrial membrane polarization in HG and VHG-treated macrophages. This also corroborates previous observations wherein, unlike other cells, macrophages could maintain and even increase their mitochondrial membrane potential in response to prevalent stress (Jones & Divakaruni 2020; Mills et al. 2016). In addition, sustained exposure to high glucose did not affect macrophage mitochondrial respiration (Pavlou et al. 2018) thereby again validating our observations that high glucose may have little to no adverse effect on mitochondrial functions in resting macrophages including on ROS production A strong correlation between increased intracellular ROS levels and decreased mitochondrial membrane potential is also known and oxidative stress has been demonstrated to induce rapid depolarization of mitochondria membrane (Park et al. 2011). Considering this and based on our observations on ROS and mitochondrial membrane potential in the present work, it appears that hyperglycemia induced a more stable mitochondrial membrane potential that may have also contributed to the apparent decrease in intracellular ROS levels.
If chronic stress persists, cellular senescence often results in the development of SASP which is characterized by increased secretion of various pro-inflammatory cytokines and growth factors. In this regard, similar to our observations on ROS levels, we noted no significant inflammatory aggravation in macrophages even on sustained exposure to hyperglycemic conditions. This signifies that hyperglycemia-induced senescence in macrophages might lack any ostensible pro-inflammatory network or SASP and its quantifiable effects thereof. Corroborating these results, a recent study observed that 24 h exposure to high glucose (25 mM) did not augment any changes in TNF-α and IL-6 protein expression in resting BMDM while a strong upregulation of these cytokines was only apparent when stimulated with LPS (Sousa et al. 2023). Similarly, it was noted that long term hyperglycemia exposure did not influence a pro-inflammatory phenotype in naïve BMDM in vitro (Pavlou et al. 2018). Further, hyperglycemia appeared to only induce a pro-inflammatory cytokine response in macrophages under acute treatment conditions that, however, declined with chronic glucose exposure thereby suggesting time-dependent differential effects of hyperglycemia (Moganti et al. 2017). However, these observations are contrary to previous studies albeit in non-immune cells wherein hyperglycemic conditions could induce premature cellular senescence as well as the SASP (Bertelli et al. 2022; Marino et al. 2022). Combined with our observations of primary markers of senescence, this further signifies the uniqueness of macrophages related to the development and characterization of stress-induced cellular senescence. On the other hand, the concentration of surface membrane proteins in exosomes appeared to dose-dependently increase under hyperglycemia in tested macrophages. This could either suggest increased numbers of exosomes themselves or a cumulative increase in exosome surface membrane protein expression. Moreover, quantification of membrane proteins is also considered an indirect measure of exosome concentration in biological samples (Théry et al. 2006). Previously, an increase in levels of exosomes and other extracellular vesicles in various senescent cells has been noted (Oh et al. 2022) while a recent study in senescent RAW264.7 macrophages also observed increased levels of exosomes (Hattori et al. 2021). In addition, we also noted that the exosomes of the VHG group were slightly more physically stable (mean zeta potential) as compared to HG and NG groups which might suggest robust and effective activation of exosome synthesis in VHG-treated macrophages.
To understand the effects of hyperglycemia on macrophage effector functions vis-à-vis senescence, macrophages were exposed to the SM of senescent preadipocytes. We hypothesized that exposure to SM of other senescent cells might exacerbate the activation of hyperglycemia-treated macrophage thereby indicating whether these cells are functionally compromised while sensing the microenvironment of other senescent cells. This should allow for a better understanding of how hyperglycemia-induced senescence in macrophages is implicated in their compromised immunosurveillance. In this regard, contrary to the ambiguous observations related to cellular senescence markers in this study; exposure to SM induced ostensible signs of near dose-dependent and robust inflammatory aggravation in HG and VHG-treated macrophages as determined by levels of various pro-inflammatory cytokines as well activation of inflammatory transcription factors and their target gene (NF-κB/NLRP3). Since SASP consists of pro-inflammatory molecules and acts to recruit and activate the immune cells to the location of senescent cells, our results indicate that chronic hyperglycemic conditions might exaggerate the macrophage inflammatory activity in response to SASP and thus potentially contribute to inflamm-aging. This is also evidenced by the observations that the apparent increase in all measured pro-inflammatory cytokines after SM treatment was multifold higher than HG and VHG alone (non-SM) treated macrophages. It is important to consider here that adipose tissue is among the first to develop increased senescent cell burden with age and also exhibits increased age-dependent inflammatory aggravation which is attributed to altered immune activities (Sharma, Kumar, et al., 2022; Zhang et al. 2023). It is therefore reasonable to speculate that diabetes in advanced age might sensitize pro-inflammatory aggravation in macrophages during their response to the SASP of senescent fat cells which could thus directly contribute to inflamm-aging in the adipose tissue. These observations are in agreement with a recent study that identified increased SA-β-gal + senescent macrophages in obese human adipose tissue that could induce pro-inflammatory response and insulin resistance under hyperglycemic conditions (Matacchione et al. 2022). Further, chronic exposure to the SASP of senescent cells has been documented to induce pro-inflammatory and tumorigenic activation in various non-immune cells (Ortiz-Montero et al. 2017; Parvizi et al. 2021). Surprisingly though, NO or intracellular ROS levels did not change in SM-exposed macrophages and appeared similar to hyperglycemic exposure (non-SM treated cells). This may indicate that SM stimulation itself is not sufficient to induce intracellular oxidative stress in macrophages but rather sensitizes to increased activation of pro-inflammatory secretory cytokines through ROS-independent mechanisms as also noted previously (Bylund et al. 2007).
Immunosurveillance by macrophages is key to their assessment and activation in response to tissue damage, endo-or exogenous threats, as well as senescent cells. Considering this, we next determined how cell surface markers such as scavenger receptors involved in the recognition of senescent cells are modulated in hyperglycemia-treated macrophages in response to SM exposure. TLR-2 is a recognition molecule not only for pathogens but also for ligands such as lipoproteins and heat shock proteins released by stressed tissues. Our study suggests that VHG stimulation induces a strong TLR-2 activation on exposure to SM which along with Myd88 activation can augment pro-inflammatory response as also noted in the present study. Further, a link between TLR-2, SASP, and cellular senescence was recently identified that indicates a central role of TLR-2 activation in augmenting the SASP (Hari et al. 2019). In addition, the downregulation of multiple genes involved in macrophage-mediated recognition and apoptosis of target cells was observed. Activation of RAGE is considered helpful for promoting the recognition and clearance of apoptotic cells by macrophages (Friggeri et al. 2011) and thus downregulation of RAGE expression under hyperglycemic conditions on account of SM treatment in this work indicates possible compromised effector functions and clearance of senescent cells. Similarly, another gene, i.e., CD36 involved in macrophage-mediated identification and phagocytosis of stressed cells was strongly downregulated which further suggested diminished effector functions of hyperglycemic macrophages in the response to SM. In agreement with these observations, a previous study noted that macrophage-mediated clearance of senescent cells can be enhanced by stimulating the expression of CD36 and RAGE (Li et al. 2020). Similarly, Olr1 is a major scavenger receptor for ox-LDL and apoptotic cells (Yu et al. 2015) and its downregulation in macrophages can result in inefficient recognition and clearance of senescent cells. On the other hand, an increase in CD44 expression was observed in macrophages which can directly increase macrophage homing and infiltration response thereby exacerbating their accumulation in response to SM. This is also supported by a strong increase in chemotactic cytokine MCP-1 levels in the present study in hyperglycemic macrophages exposed to the SM. Furthermore, CD44 recognizes hyaluronic acid as its major ligand which is also a part of the extracellular matrix as well as SASP. Overall, it is reasonable to assert that since macrophages already demonstrated an aggravated proinflammatory cytokine profile on account of SM exposure, their increased homing and accumulation (due to MCP-1/CD44) but diminished identification effector functions (as evidenced by decreased CD36, RAGE, and Olr1 expression) might actually result in more inflammatory aggravation and less effective resolution of senescent cells. It is also pertinent to consider here that the apparent changes in the cytokine profile or scavenger receptors in this study are a result of an indirect activation in response to SM exposure alone, and thus an even more aggravated state can be envisaged when macrophages come in direct contact with senescent cells in addition to the SM.
Stimulation of hyperglycemic macrophages with LPS resulted in a striking increase in intracellular ROS and NO production which was otherwise subdued during exposure to both hyperglycemia and SM. It is thus indicative of differential activation response pathways (such as NOX1) in macrophages governed by the type of stressor/stimulator (Moghadam et al. 2021). Our results are in agreement with previous studies demonstrating that hyperglycemic conditions alter LPS-mediated inflammatory pathways and other macrophage effector functions (Kim et al. 2022; Pavlou et al. 2018; Sousa et al. 2023). Although a mild effect of hyperglycemia on LPS-mediated cytokine response could be observed, LPS stimulation greatly exaggerated the production of cytokines including that of anti-inflammatory IL-10 but except for MCP-1 as compared to SM treatment. Thus, as far as extracellular aggravation of inflammation is concerned, hyperglycemic macrophages up to some extent appeared to respond alike when stimulated with either SM or LPS.
Conclusions
The present study suggests that chronic hyperglycemia in macrophages induces aberrant expression of primary markers of senescence and lacks SASP activation which together signifies the probable lack of an overt cellular senescence program under these conditions. This is not surprising given the fact that no ostensible signs of redox or mitochondrial stress could be observed in HG/VHG treated cells. Therefore, while interpreting cellular senescence in macrophages, caution should be exercised since it appears that not only the type of physiological stress, but also its duration can significantly impact the expression of cellular senescence markers. However, strikingly and most notably, this is not correspondingly translated into macrophage immunosurveillance response to the SASP of other senescent cells. Therefore, it indicates that regardless of the ambiguity observed in characterizing hyperglycemia-induced macrophage senescence per se, the effect of these cells on their ability to counter accumulating senescent cells is not only compromised but results in a strong pro-inflammatory environment that may be detrimental to the surrounding healthy cells. This provides novel insights into the disease-dependent loss of macrophage effector functions that may accelerate the accumulation of tissue senescent cells while also concurrently augmenting the pro-inflammatory environment. Our results also strengthen the notion that cellular senescence markers in macrophages are dynamic and may have more meaningful and diverse roles depending on the physiological status of macrophages such as prevalent stress. We also recommend that studies testing macrophages for cellular senescence should consider the functional aspects of macrophages such as those related to immunosurveillance of tissue senescent cells for a more meaningful interpretation and conclusion. Further validation of macrophage cellular senescence in vivo under inflammatory disease conditions (such as diabetes) is desirable to truly comprehend the senescence-associated dysfunctions in macrophages and its effects thereof.
Data availability
All data generated or analyzed during this study are included in this article.
References
Bertelli PM, Pedrini E, Hughes D, McDonnell S, Pathak V, Peixoto E, Guduric-Fuchs J, Stitt AW, Medina RJ (2022) Long term high glucose exposure induces premature senescence in retinal endothelial cells [Original Research]. Frontiers in Phys. https://doi.org/10.3389/fphys.2022.929118
Bylund J, MacDonald KL, Brown KL, Mydel P, Collins LV, Hancock RE, Speert DP (2007) Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-kappa B. Eur J Immunol 37(4):1087–1096. https://doi.org/10.1002/eji.200636651
Clark D, Brazina S, Yang F, Hu D, Hsieh CL, Niemi EC, Miclau T, Nakamura MC, Marcucio R (2020) Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell 19(3):e13112. https://doi.org/10.1111/acel.13112
Cudejko C, Wouters K, Fuentes L, Hannou SA, Paquet C, Bantubungi K, Bouchaert E, Vanhoutte J, Fleury S, Remy P, Tailleux A, Chinetti-Gbaguidi G, Dombrowicz D, Staels B, Paumelle R (2011) p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood 118(9):2556–2566. https://doi.org/10.1182/blood-2010-10-313106
Dash M, Palaniyandi K, Ramalingam S, Sahabudeen S, Raja NS (2021) Exosomes isolated from two different cell lines using three different isolation techniques show variation in physical and molecular characteristics. Biochim et Biophys Acta Biomembranes 1863(2):183490. https://doi.org/10.1016/j.bbamem.2020.183490
De Maeyer RPH, Chambers ES (2021) The impact of ageing on monocytes and macrophages. Immunol Lett 230:1–10. https://doi.org/10.1016/j.imlet.2020.12.003
de Mera-Rodríguez JA, Álvarez-Hernán G, Gañán Y, Martín-Partido G, Rodríguez-León J, Francisco-Morcillo J (2021) Is Senescence-associated β-galactosidase a reliable in vivo marker of cellular senescence during embryonic development? [Review]. Frontiers Cell Dev Biol. https://doi.org/10.3389/fcell.2021.623175
Diwan B, Sharma R (2022) Green tea EGCG effectively alleviates experimental colitis in middle-aged male mice by attenuating multiple aspects of oxi-inflammatory stress and cell cycle deregulation. Biogerontology 23(6):789–807. https://doi.org/10.1007/s10522-022-09976-9
Diwan B, Yadav R, Singh A, Kumar D, Sharma R (2023) Murine sterile fecal filtrate is a potent pharmacological agent that exerts age-independent immunomodulatory effects in RAW264.7 macrophages. BMC Complementary Med Therapies 23(1):362. https://doi.org/10.1186/s12906-023-04193-4
Duong L, Radley HG, Lee B, Dye DE, Pixley FJ, Grounds MD, Nelson DJ, Jackaman C (2021) Macrophage function in the elderly and impact on injury repair and cancer. Immunity Ageing 18(1):4. https://doi.org/10.1186/s12979-021-00215-2
Frescas D, Hall BM, Strom E, Virtuoso LP, Gupta M, Gleiberman AS, Rydkina E, Balan V, Vujcic S, Chernova OB, Gudkov AV (2017) Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle 16(16):1526–1533. https://doi.org/10.1080/15384101.2017.1339850
Friggeri A, Banerjee S, Biswas S, de Freitas A, Liu G, Bierhaus A, Abraham E (2011) Participation of the receptor for advanced glycation end products in efferocytosis. J Immunol 186(11):6191–6198. https://doi.org/10.4049/jimmunol.1004134
Giri B, Dey S, Das T, Sarkar M, Banerjee J, Dash SK (2018) Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed Pharm 107:306–328. https://doi.org/10.1016/j.biopha.2018.07.157
Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, Gudkov AV (2016) Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 8(7):1294–1315. https://doi.org/10.18632/aging.100991
Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin II, Leonova KI, Consiglio CR, Gollnick SO, Chernova OB, Gudkov AV (2017) p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 9(8):1867–1884. https://doi.org/10.18632/aging.101268
Hari P, Millar FR, Tarrats N, Birch J, Quintanilla A, Rink CJ, Fernández-Duran I, Muir M, Finch AJ, Brunton VG, Passos JF, Morton JP, Boulter L, Acosta JC (2019) The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci Adv. 5(6):eaaw0254. https://doi.org/10.1126/sciadv.aaw0254
Hattori H, Takaoka K, Ueta M, Oshitani M, Tamaoka J, Noguchi K, Kishimoto H (2021) Senescent RAW264.7 cells exhibit increased production of nitric oxide and release inducible nitric oxide synthase in exosomes. Mol Med Rep. https://doi.org/10.3892/mmr.2021.12320
Jiang Z, Jiang C, Jin L, Chen Z, Feng Z, Jiang X, Cao Y (2022) In vitro and in vivo effects of hyperglycemia and diabetes mellitus on nucleus pulposus cell senescence. J Orthopaedic Res 40(10):2350–2361. https://doi.org/10.1002/jor.25264
Jones AE, Divakaruni AS (2020) Macrophage activation as an archetype of mitochondrial repurposing. Mol Aspects Med 71:100838. https://doi.org/10.1016/j.mam.2019.100838
Kale A, Sharma A, Stolzing A, Desprez P-Y, Campisi J (2020) Role of immune cells in the removal of deleterious senescent cells. Immunity & Ageing 17(1):16. https://doi.org/10.1186/s12979-020-00187-9
Kim H, Xue X (2020) Detection of Total Reactive oxygen species in adherent cells by 2’,7’-dichlorodihydrofluorescein diacetate staining. J Vis Exp. https://doi.org/10.3791/60682
Kim K-H, Park B, Rhee D-K, Pyo S (2015) Acrylamide Induces senescence in macrophages through a Process Involving ATF3, ROS, p38/JNK, and a telomerase-independent pathway. Chem Res Toxicol 28(1):71–86. https://doi.org/10.1021/tx500341z
Kim A, Gwon M-H, Lee W, Moon H-R, Yun J-M (2022) Zerumbone suppresses high glucose and LPS-induced inflammation in THP-1-derived macrophages by inhibiting the NF-κB/TLR signaling pathway. Nutrition Res. https://doi.org/10.1016/j.nutres.2022.01.002
Kumar R, Sharma A, Kumari A, Gulati A, Padwad Y, Sharma R (2019) Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway. Biogerontology 20(2):171–189. https://doi.org/10.1007/s10522-018-9785-1
Kumar R, Sharma A, Padwad Y, Sharma R (2020) Preadipocyte secretory factors differentially modulate murine macrophage functions during aging which are reversed by the application of phytochemical EGCG. Biogerontology 21(3):325–343. https://doi.org/10.1007/s10522-020-09861-3
Li X, Yang K, Gao S, Zhao J, Liu G, Chen Y, Lin H, Zhao W, Hu Z, Xu N (2020) Carnosine stimulates macrophage-mediated clearance of senescent skin cells through activation of the AKT2 signaling pathway by CD36 and RAGE. Front Pharmacol 11:593832. https://doi.org/10.3389/fphar.2020.593832
Liu J-Y, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV, Sharpless NE (2019) Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence. Procee Natl Acad of Sci 116(7):2603–2611. https://doi.org/10.1073/pnas.1818313116
Lloberas J, Tur J, Vico T, Celada A (2019) Molecular and cellular aspects of macrophage aging. In: Fulop T, Franceschi C, Hirokawa K, Pawelec G (eds) Handbook of immunosenescence: basic understanding and clinical implications. Springer, Cham
Marino F, Scalise M, Salerno N, Salerno L, Molinaro C, Cappetta D, Torella M, Greco M, Foti D, Sasso FC, Mastroroberto P, De Angelis A, Ellison-Hughes GM, Sampaolesi M, Rota M, Rossi F, Urbanek K, Nadal-Ginard B, Torella D, Cianflone E (2022) Diabetes-induced cellular senescence and senescence-associated secretory phenotype impair cardiac regeneration and function independently of age. Diabetes 71(5):1081–1098. https://doi.org/10.2337/db21-0536
Matacchione G, Perugini J, Di Mercurio E, Sabbatinelli J, Prattichizzo F, Senzacqua M, Storci G, Dani C, Lezoche G, Guerrieri M, Giordano A, Bonafè M, Olivieri F (2022) Senescent macrophages in the human adipose tissue as a source of inflammaging. GeroSci 44(4):1941–1960. https://doi.org/10.1007/s11357-022-00536-0
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167(2):457-470.e413. https://doi.org/10.1016/j.cell.2016.08.064
Moganti K, Li F, Schmuttermaier C, Riemann S, Klüter H, Gratchev A, Harmsen MC, Kzhyshkowska J (2017) Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology 222(10):952–959. https://doi.org/10.1016/j.imbio.2016.07.006
Moghadam ZM, Henneke P, Kolter J (2021) From flies to men: ROS and the NADPH oxidase in phagocytes [Review]. Frontiers Cell Dev Biol. https://doi.org/10.3389/fcell.2021.628991
Murakami Y, Mizoguchi F, Saito T, Miyasaka N, Kohsaka H (2012) p16(INK4a) exerts an anti-inflammatory effect through accelerated IRAK1 degradation in macrophages. J Immunol 189(10):5066–5072. https://doi.org/10.4049/jimmunol.1103156
Mytych J, Romerowicz-Misielak M, Koziorowski M (2017) Long-term culture with lipopolysaccharide induces dose-dependent cytostatic and cytotoxic effects in THP-1 monocytes. Toxicol Vitro 42:1–9. https://doi.org/10.1016/j.tiv.2017.03.009
Oh C, Koh D, Jeon HB, Kim KM (2022) The role of extracellular vesicles in senescence. Mol Cells 45(9):603–609. https://doi.org/10.14348/molcells.2022.0056
Oishi Y, Manabe I (2016) Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech Dis 2:16018. https://doi.org/10.1038/npjamd.2016.18
Ortiz-Montero P, Londoño-Vallejo A, Vernot J-P (2017) Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun and Signaling 15(1):17. https://doi.org/10.1186/s12964-017-0172-3
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A, Windheim J, Tsoory M, Schirmbeck R, Amit I, Geiger H, Krizhanovsky V (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9(1):5435. https://doi.org/10.1038/s41467-018-07825-3
Park J, Lee J, Choi C (2011) Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE 6(8):e23211. https://doi.org/10.1371/journal.pone.0023211
Parvizi M, Ryan ZC, Ebtehaj S, Arendt BK, Lanza IR (2021) The secretome of senescent preadipocytes influences the phenotype and function of cells of the vascular wall. Biochim Biophys Acta Mol Basis Dis 1867(1):165983. https://doi.org/10.1016/j.bbadis.2020.165983
Pavlou S, Lindsay J, Ingram R, Xu H, Chen M (2018) Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol 19(1):24. https://doi.org/10.1186/s12865-018-0261-0
Pignolo RJ, Passos JF, Khosla S, Tchkonia T, Kirkland JL (2020) Reducing senescent cell burden in aging and disease. Trends Mol Med 26(7):630–638. https://doi.org/10.1016/j.molmed.2020.03.005
Prata L, Ovsyannikova IG, Tchkonia T, Kirkland JL (2018) Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin Immunol 40:101275. https://doi.org/10.1016/j.smim.2019.04.003
Sebastián C, Herrero C, Serra M, Lloberas J, Blasco M, a. A., & Celada, A. (2009) Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation1. J Immunol 183(4):2356–2364. https://doi.org/10.4049/jimmunol.0901131
Sharma R (2021) Perspectives on the dynamic implications of cellular senescence and immunosenescence on macrophage aging biology. Biogerontology 22(6):571–587. https://doi.org/10.1007/s10522-021-09936-9
Sharma R, Diwan B, Sharma A, Witkowski JM (2022a) Emerging cellular senescence-centric understanding of immunological aging and its potential modulation through dietary bioactive components. Biogerontology 23(6):699–729. https://doi.org/10.1007/s10522-022-09995-6
Sharma R, Kumar R, Sharma A, Goel A, Padwad Y (2022) Long-term consumption of green tea EGCG enhances murine health span by mitigating multiple aspects of cellular senescence in mitotic and post-mitotic tissues, gut dysbiosis, and immunosenescence. J Nutr Biochem. https://doi.org/10.1016/j.jnutbio.2022.109068
Sousa ESA, Queiroz LAD, Guimarães JPT, Pantoja KC, Barros RS, Epiphanio S, Martins JO (2023) The influence of high glucose conditions on macrophages and its effect on the autophagy pathway [Original Research]. Frontiers in Immunol. https://doi.org/10.3389/fimmu.2023.1130662
Suzuki T, Yamashita S, Hattori K, Matsuda N, Hattori Y (2021) Impact of a long-term high-glucose environment on pro-inflammatory responses in macrophages stimulated with lipopolysaccharide. Naunyn Schmiedebergs Arch Pharmacol 394(10):2129–2139. https://doi.org/10.1007/s00210-021-02137-8
Talma N, Gerrits E, Wang B, Eggen BJL, Demaria M (2021) Identification of distinct and age-dependent p16High microglia subtypes. Aging Cell 20(10):e13450. https://doi.org/10.1111/acel.13450
Théry, C., Amigorena, S., Raposo, G., & Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol, Chapter 3, Unit 3.22.
van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM (2019) Metabolic alterations in aging macrophages: ingredients for inflammaging? Trends Immunol 40(2):113–127. https://doi.org/10.1016/j.it.2018.12.007
Virág L, Jaén RI, Regdon Z, Boscá L, Prieto P (2019) Self-defense of macrophages against oxidative injury: fighting for their own survival. Redox Biol 26:101261. https://doi.org/10.1016/j.redox.2019.101261
Wan Y, Liu Z, Wu A, Khan AH, Zhu Y, Ding S, Li X, Zhao Y, Dai X, Zhou J, Liu J, Li Y, Gong X, Liu M, Tian XL (2022) Hyperglycemia promotes endothelial cell senescence through AQR/PLAU signaling axis. Int J Mol Sci. https://doi.org/10.3390/ijms23052879
Wang H, Fu H, Zhu R, Wu X, Ji X, Li X, Jiang H, Lin Z, Tang X, Sun S, Chen J, Wang X, Li Q, Ji Y, Chen H (2020) BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging (Albany NY) 12(10):9240–9259. https://doi.org/10.18632/aging.103200
Xu M, Sun S, Ge J, Shen Y, Li T, Sun X (2020) Bupleurum chinense Polysaccharide Improves LPS-Induced Senescence of RAW264.7 Cells by Regulating the NF-κB Signaling Pathway. Evid Based Complement Alternat Med. https://doi.org/10.1155/2020/7060812
Yin M, Zhang Y, Yu H, Li X (2021) Role of hyperglycemia in the senescence of mesenchymal stem cells. Front Cell Dev Biol 9:665412. https://doi.org/10.3389/fcell.2021.665412
Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, Cui Y, Angelini L, Lee K-A, McGowan SJ, Burrack AL, Wang D, Dong Q, Lu A, Sano T, O’Kelly RD, McGuckian CA, Kato JI, Bank MP, Wade EA, Pillai SPS, Klug J, Ladiges WC, Burd CE, Lewis SE, LaRusso NF, Vo NV, Wang Y, Kelley EE, Huard J, Stromnes IM, Robbins PD, Niedernhofer LJ (2021) An aged immune system drives senescence and ageing of solid organs. Nature 594(7861):100–105. https://doi.org/10.1038/s41586-021-03547-7
Yu X, Guo C, Fisher PB, Subjeck JR, Wang X-Y (2015) Chapter Nine - Scavenger Receptors: Emerging Roles in Cancer Biology and Immunology. In: Wang X-Y, Fisher PB (eds) Advances in cancer research. academic press, Cham
Zhang P, Wang Q, Nie L, Zhu R, Zhou X, Zhao P, Ji N, Liang X, Ding Y, Yuan Q, Wang Q (2019) Hyperglycemia-induced inflamm-aging accelerates gingival senescence via NLRC4 phosphorylation. J Biol Chem 294(49):18807–18819. https://doi.org/10.1074/jbc.RA119.010648
Zhang B, Yang Y, Yi J, Zhao Z, Ye R (2021) Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res 56(5):991–1005. https://doi.org/10.1111/jre.12912
Zhang Y-X, Ou M-Y, Yang Z-H, Sun Y, Li Q-F, Zhou S-B (2023) Adipose tissue aging is regulated by an altered immune system [Review]. Frontiers in Immunol. https://doi.org/10.3389/fimmu.2023.1125395
Acknowledgements
The authors acknowledge infrastructure support from Shoolini University, Solan for this research work.
Funding
Funding support by the Science and Engineering Research Board, Department of Science and Technology, Government of India, under the SERB-Research Scientist Scheme (SB/SRS/2022–23/75/LS) is thankfully acknowledged for this manuscript.
Author information
Authors and Affiliations
Contributions
RS conceived and designed the study. BD and RY performed experimental procedures. RG provided laboratory support. RS analyzed the data and wrote the manuscript. All authors reviewed the manuscript before final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Diwan, B., Yadav, R., Goyal, R. et al. Sustained exposure to high glucose induces differential expression of cellular senescence markers in murine macrophages but impairs immunosurveillance response to senescent cells secretome. Biogerontology 25, 627–647 (2024). https://doi.org/10.1007/s10522-024-10092-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-024-10092-z