
Abstract
The skin coloration is one of the key characteristics of fish morphological diversity; understanding the molecular regulation mechanism of fish skin coloration has attracted a mounting number of attention and is also an important prerequisite for the future research on fish skin coloration genetics and skin coloration improvement breeding of fish. Pearlscale angelfish (Centropyge vrolikii) is a unique marine ornamental fish with tremendous market potential and is well favored by consumers in China, which has a wide variety of colorations and shapes. In this study, the distribution of pigment cells in the fins and scales was observed, and the pigment cells were mainly composed of melanocytes, xanthophore, iridophore, and erythrophore in C. vrolikii. The distribution of pigment cells in the tail scales is more concentrated than that in other parts. There are mainly melanocytes and yellow pigment cells in the anal fin, tail fin, and dorsal fin, and mainly xanthophore with a few melanocytes in the pectoral fin and abdominal fin. The tissues of the pectoral fin (PF), anal fin (AF), tail fin (TF), and eyeballs (EB) were sequenced by SOAPdenovo-Trans technology, the differentially expressed genes (DEGs) of skin coloration were screened, and the expression pattern of these genes in different tissues was analyzed. The results represented that a total of 159,213 all-unigene was assembled and gene annotation was carried out using COG, GO, KEGG, KOG, Pfam, Swissprot, eggNOG, and NR functional database. The Tyrp1, Slc45a2, Xdh, Mitf, Tyr, Hps5, Pomc, Pmel, and other genes related to the skin coloration were annotated. Through KEGG pathway enrichment analysis, it was found that tyrosine metabolism, MAPK, Wnt, and melanin production were involved in the regulation of skin coloration. At the same time, the DEGs related to melanin production (Mitf, Mc1r, Sox10, Tyr, Tyrp1, and Pomc) were screened to verify their expression in various tissues. The results also showed that Mitf and Tyr were highly expressed in eyeballs, Tyrp1 was highly expressed in skin, Sox10 and Pomc were highly expressed in the tail fin, and Mc1r was highly expressed in the heart and tail fin. The results suggested that these genes may play an important role in the process of skin coloration in C. vrolikii. This study will help to further understand the molecular regulatory mechanism of skin coloration and provide essential clues for accelerating skin coloration improvement in the future in C. vrolikii.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pigmentation of animal skin, eyes, and hair is mainly determined by the quality, quantity, and distribution of melanin. Melanosomes are located in melanocyte and are specifically responsible for the transfer of melanin to surrounding epidermal cells (also known as keratinocyte) (Ito and Wakamatsu 2008). The iridophore deposits guanine crystals which are assembled into multilayer reflective plates to produce the silver reflectivity. The xanthophore and erythrophore are similar in shape and size and have the same pteridine and carotenoid (Funt et al. 2017). The pigment cells of fish originate from neural crest cells and principally divided into melanocyte, iridophore, xanthophore, and erythrophore. The changes in skin coloration are jointly regulated by the quantity and quality of these pigment cells, which play an important role in communication, species formation, and ecological interaction in fish (Sefc et al. 2014). The skin coloration of fish is composed of one or more pigment cells. The different numbers and shapes of pigment cells determine the monotonous or colorful skin coloration of fish. For example, the skin coloration of large yellow croaker (Larimichthys crocea) is composed of melanocyte and xanthophore (Han et al. 2019; Luo et al. 2020). The pattern of alternating blue and golden stripes displayed by adult zebrafish is composed of three kinds of pigment cells: black melanophore, yellow xanthophore, and silvery-blue iridophore (Mahalwar et al. 2014; Parichy 2003). Generally, there are two ways in which pigment cells cause fish skin coloration. One is physiological changes, involving the diffusion or aggregation of pigment particles, as well as hormone and nerve regulation. The other is morphological changes, involving changes in the amount of pigment particles and pigment cells and the migration of pigment cells. No matter what kind of skin coloration changes, it is inseparable from the participation and regulation of a series of key genes, which is closely related to heredity.
In fish, humans, mice, and other vertebrates, melanocytes are the main or even the only pigment cells. Therefore, the research on the activities of melanocytes is extensive and in-depth. The melanocortin-1 receptor (Mc1r) is a dominant regulatory point of melanin synthesis to demonstrate the function of seven transmembrane G-protein coupled receptors (Guida et al. 2021; Rees 2000). Adrenocorticotropic hormone (ACTH), melanocortin peptides derived from the proopiomelanocortin (pomc) gene, α-melanocyte stimulating hormone (α-MSH), and the endogenous antagonist agouti signaling protein (Asip1) can regulate Mc1r to participate in the control of pigmentation (Cal et al. 2017). The adenylate cyclase is activated after α-MSH binded to Mc1r, and then the cAMP increases which can induce the expression of tyrosinase (Tyr), tyrosinase-related protein 1 (Tyrp1), and tyrosinase-related protein 2 (Tyrp2) (D’Mello et al. 2016; Qomaladewi et al. 2019; Xu et al. 2013). The tyrosinase-related family genes, Tyr, Tyrp1, and Tyrp2, are directly involved in the production and deposition of melanin (Zheng et al. 2010). Tyrp1 is an important downstream regulatory factor for body coloration formation in animals, and the expression of Tyrp1 will be reduced, and then the production of melanin will be inhibited after inhibiting the activity of melanocyte transcription factors such as microphthalmia-associated transcription factor (Mitf) (Fang et al. 2002; Wu et al. 2021a). Tyr enhancer was induced and activated by Mitf and sex-determining region Y-box protein 10 (Sox10) in mouse melanocyte, and then melanin synthesis was initiated (Murisier et al. 2007). Different from melanocyte, the pigment cells of frog liver can synthesize melanin through melanogenesis when Tyr-like (Tyrl) is activated (Cicero et al. 1989). Similarly, it has been confirmed that tyrosinase is one of the regulatory pathways involved in the regulation of cytochrome synthesis by Mitf in most animals. Mitf increases the expression of pre melanosome protein (Pmel) and then regulates the synthesis of melanin (Gutknecht et al. 2015; Zhang et al. 2013). In other ways, Mitf is regulated by the Gpnmb gene with the expression level of Mitf downregulated after overexpression of Gpnmb, and the expression level of Gpnmb is decreased after treatment of active small molecule inhibitors of Mitf (Gutknecht et al. 2015; Zhang et al. 2013). Therefore, Mitf is a key target site for the study of melanin synthesis in mammals. In addition, the development of melanocyte requires non autonomous or autonomous signal transduction through endothelin receptor type B (Ednrb) (Hou et al. 2004; Lee et al. 2003). As an upstream regulatory element of tyrosinase family genes, Ednrb is closely related to melanin synthesis and expressed in many cell types (Hou et al. 2004; Lee et al. 2003). In a word, a series of key skin coloration genes jointly regulates the development, variation, migration, and apoptosis of pigment cells which determine the skin coloration of fish single or colorful.
As a new transcriptome research technology, RNA-Seq technology has attracted more and more attention for its accuracy, efficiency, and speed. RNA-Seq technology can not only annotate and map the fish transcriptome, but also help to understand the biological information of fish, such as immunology, toxicology, physiology, evolutionary biology, and developmental biology (Qian et al. 2014). The DEGs in the skin tissues of seawater and freshwater sticklebacks (Gasterosteus aculeatus) and the light and black skin tissues of freshwater stickleback were screened by RNA-Seq technology, and then the differentially expressed genes (DEGs) were verified by qRT-PCR to clarify the mechanism of skin coloration variation (Greenwood et al. 2012). The transcriptome of the caudal fin of the wild-type and the albino-type of Aulonocara baenschi was sequenced by RNA-Seq to identify their pigment-related genes and analyze the expression patterns of the genes at the transcriptome level (Lee and Lee 2020). In the study of Yellow mutant rainbow trout (YR), YR skin transcriptomes were analyzed to explore temporal expression patterns of pigmentation-related genes in three different stages of skin color development (Wu et al. 2022). The key color variation-associated mRNAs and miRNAs between wild-type (WR) and yellow mutant rainbow trout (YR) were sequenced and identified, which also laid a solid foundation for genetic selection and breeding of fish skin coloration (Wu et al. 2021b). Comparing the fin transcriptome in the wild-type and albino-type of Pelteobagrus fulvidraco, it was found that the expressions of Tyr, Tyrp1, and Ednrbla were upregulated in albino types; this may result from the upregulation of cystine/glutamate transporter in albino types (Zou et al. 2015). By analyzing the transcriptome in the specific skin pigment pattern in trout (Salmo species) with the labyrinthine skin pattern and spotted skin pattern respectively, the DEGs possibly associated with skin pigment pattern were identified, and the expression of Gja5, Clcn2, Cdkn1a, and Tjp1 was detected (Djurdjevic et al. 2019). The application of transcriptome has greatly expanded the basic data of skin coloration research of fish, which is not only conducive to the screening of main genes and regulatory pathways, but also provides reliable auxiliary molecular markers for breeding.
Centropyge has a wide variety of colorations and shapes, which is favored by consumers. Pearlscale angelfish (Centropyge vrolikii), commonly known as the black-tail angelfish, is a unique marine ornamental fish with huge market potential in China and is mainly found near coral reefs in the Indian Ocean and the Pacific Ocean (Dibattista et al. 2016; Fernandez-Silva et al. 2018). In our previous research, the DEGs in gonads at different stages in C. vrolikii were screened using RNA-Seq technology for further analysis (Zhong et al. 2021), and the pluripotent transcription factor Nanog was cloned and analyzed (Zhong, et al. 2022a). Understanding the molecular regulation mechanism of skin coloration is an important prerequisite for the research of skin coloration inheritance and improvement in fish. However, little is known about its skin coloration at the molecular level. In the model organism, the core regulatory network of their skin coloration has been deeply studied, but the molecular mechanism and related technical means of the regulatory network in C. vrolikii have not been reported. In this study, the different colored tissues at different parts of C. vrolikii were sequenced by RNA-Seq technology, and the key genes affecting the development and variation of skin coloration were screened and then verified by qRT-PCR. The results will provide a new perspective on the mechanism of skin coloration in small sea angelfish and have important significance for the skin coloration breeding of C. vrolikii in the future.
Materials and methods
Sample collection
The tissue samples of C. vrolikii were obtained from the Aquatic Experimental Center of Fisheries College of Jimei University (Xiamen China). A total of 24 individual fish (more than 3.5 cm in length, 3 + years old) were caught in the Philippine Sea area, which was disinfected and fed temporarily with circulating seawater, and were taken and carefully dissected for the pectoral fin (PF), anal fin (AF), tail fin (TF), eyeballs (EB), and other tissues. Then the tissues were collected in RNAlater (Ambion) and stored at 4℃ for 24 h and then transferred to − 80℃ until RNA extraction. The squamae and fins were washed with normal saline and directly placed on the culture dish for observation by stereoscopic microscope.
RNA extraction and transcriptome sequencing
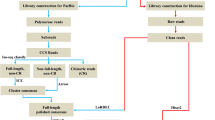
A total of 24 fish with a body length of more than 3.5 cm were selected, and the tissues of each 6 fish in the same part were taken as a sample for RNA extraction. RNA was extracted from PF, AF, TF, and EB using the Total RNA Kit II (OMEGA, USA). RNA concentration and purity were determined by the NanoDrop1000 Spectrophotometer (Thermo Scientific, MA, USA). Samples with A260/280 values of 1.8 ~ 2.0 could be utilized for downstream experiments. The integrity of the RNA was tested by 1.0% agarose gel electrophoresis. Subsequently, one part of RNA (PF, AF, TF, and EB) was performed reverse transcription immediately, and others were sent to Biomarker Technologies Co., Ltd. (Beijing, China) for quality testing and transcriptome sequencing.
Transcription sequence acquisition and gene annotation
Low-quality data were removed from raw reads, and clean reads were subjected to further analysis Trinity is an assembly software specially designed for high-throughput transcriptome sequencing. First, the sequenced reads are broken into shorter fragments (k-mer), then these small fragments were extended into longer fragments (contig), and the fragment set (component) was obtained by using the overlap among these fragments. Finally, transcripts were identified in each fragment set by using the de Bruijn diagram method and sequencing read information. The sequence of unigene was compared with NR, Swiss-Prot, COG, KOG, eggNOG4.5, and KEGG databases by using DIAMOND software, and the KEGG ontology results of unigene in KEGG were obtained by using KOBAS. The gene ontology (GO) results of new genes were analyzed by InterProScan (A software package that allows users to scan sequences against member database signatures) using the database integrated by InterPro, and the annotation information of unigene was obtained by comparing with the Pfam database in the HMMER software after the amino acid sequence of unigene was predicted. Bowtie was used to compare the sequenced reads with the UniGene library. According to the comparison results, the expression level was estimated in combination with RSEM (an accurate tool for quantifying transcript abundances from RNA-Seq data). The expression abundance of the corresponding unigene was indicated by the FPKM value. The Pearson’s correlation coefficient R was used as the evaluation index of correlation between samples.
Screening and enrichment of differential expression genes
To compare differences in the gene expression in TF, PF, AF, and EB groups of C. vrolikii, a method described by Benjamini–Hochberg was adopted to evaluate the tag frequency in the different RNA-Seq libraries. The threshold P value in multiple tests was determined by measuring the FDR (false discovery rate). In the process of screening DEGs, the FDR was less than 0.01, and the difference multiple FC (fold change) was greater than or equal to 2 as the screening standard. The DEGs were subjected to the enrichment analysis by GO (Gene Ontology) functional group, KEGG, COG (Cluster of Orthologous Groups of proteins) database, and EggNOG database. Moreover, the Volcano Plot can intuitively show the relationship between FDR and FC of all genes and quickly view the difference and statistical significance of gene expression level between the two groups of samples. The path enrichment analysis of DEGs can analyze whether the DEGs were over-presentation on a certain path. The enrichment degree of the pathway was analyzed by the enrichment factor, and the enrichment significance was calculated by the hypergeometric test method.
qRT-PCR verification of transcriptome sequencing results
In order to verify the accuracy of differential gene expression from transcriptome sequencing, 13 genes were screened for qRT-PCR and primers as shown in Table 1. At the same time, the expression levels of 6 genes related to pigment synthesis were verified in various tissues. And the STRING is a database of protein–protein interactions predicted and experimentally verified by multiple species, including direct physical interactions and indirect functional correlations. Combined with the results of DEGs, qRT-PCR analysis, and the interaction pairs recorded in the database, the interaction network of DEGs was constructed. Using BLAST software, the protein sequence of the target genes (the skin coloration–related genes of C. vrolikii) was compared in the database to find the homologous proteins for the construction of a protein interaction network.
The first strand of cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, MA, USA) from 1.0 μg total RNA samples. The product of reverse transcription was diluted 100 times with sterile water and then stored at − 20℃ as a qRT-PCR template. The qRT-PCR was based on the QuantiFast SYBR Green PCR Kit (Qiagen, Nangjing, China) on 384-well PCR plates. The 10 μL of reaction system was adopted: SYBR Green Realtime PCR Mster Mix was 5 μL, the positive and negative primers were 0.25 μL (10 μmoL/L), the cDNA template was 4.5 μL, and the reaction was conducted at 95℃ for 5 min, circulated for 45 times as follows: denaturation at 95℃ for 10 s, annealing at 59℃ for 10 s, extension at 72℃ for 10 s. In this experiment, there were 6 biological replicates for each tissue and 3 technical replicates for each sample. Rps29 was used as a reference gene. The relative fold change of the gene expression level was calculated by the 2−ΔΔCt method (Zhong, et al. 2022b).
Statistical analysis
All statistical analyses were performed using SPSS20.0 software, and one-way ANOVA was used to compare the relative expression levels. The level of significance (P) was less than 0.05.
Result
Pigment distribution
The skin coloration of C. vrolikii was primarily grayish black over the entire. The pectoral fin and ventral fin were both bright yellow color, and the anal fin and tail fin were both entirely black, while the dorsal fin was solid black in the back half and golden brown in the front half (Fig. 1A). Similar to the anal fin, the dorsal fin were primarily made up of melanocytes with a few xanthophore scattered across the base of the fin (Fig. 1B and C). Melanocytes predominated over the whole of the tail fin, and a circle of iridophore was distributed around its margin with blue patches (Fig. 1D and D-1). The morphology of pectoral fin and ventral fin was comparable, and the majority of the pigment cells were xanthophore, which were dispersed in the fin rays and uniformly distributed in the area between the fins and spines (Fig. 1E, E-1, and E-2, and F, F-1, and F-2).

The pigment cell distribution in the fins. Note: A the biological form of fish; B dorsal fin; C anal fin; D and D-1 tail fin; E and E-1 ventral fin; F and F-1 pectoral fin; X, xanthophore; M, melanocyte; I, iridophore
The tail part of C. vrolikii was dark and black in color caused by its scales. The melanocytes and xanthophore were visible as the pigment cells of scales in the tail, which were mostly gathered at the base region of scales (Fig. 2 to A-4). There were two types of melanocytes: type I cells were big, scattered, diffuse, or dendritic; type II cells were smaller than type I cells in size, absence of dendritic structure, and have high-density clumps and granules (Fig. 2).

The pigment cell distribution in the squamae. Note: A and A-1 the squamae of the posterior ocular head; B and B-1 the squamae in the middle of the fish body; C and C-1 the squamae at the front end of the tail; D and D-1 the squamae of the tail. X, xanthophore; M, melanocyte
The color of the abdomen was gray. And the melanocytes, xanthophore, and iridophore made up the majority of pigment cells of scales in C. vrolikii. On the comb side, the pigment cells were uniformly dispersed (Fig. 2 to B-4). The iridophore was substantially larger than other pigment cells, primarily consisted of long oval or polygonal short rods, and exhibited reflecting qualities with transparency under light (Fig. 2 to B-4). The pigment cells of the base of the tail did not seem rich (Fig. 2 to B-4).
The coloration of the pectoral had light yellow sections on the front and back of C. vrolikii. The concentration of pigment granules of scales in the base of the front and back of the pectoral was the most dispersed relative to the tail and abdominal, and there was no pigment cell granular zone at the end of the basal area of pectoral (Fig. 2 to C-4). The red and yellow masses of scales were dispersed in the base region, and there were just a few sporadic pigment cells in the top region of the pectoral (Fig. 2 to C-4). The structure of erythrophore was thicker than melanocytes without evident branching (Fig. 2).
Assembly of transcriptome sequencing data
The RNA with good quality of each sample was used for downstream experiments. Illumina HiSeq platform was used for transcriptome sequencing of PF, AF, TF, and EB from C. vrolikii. The samples were divided into four groups with three parallels: PF group (PF-1, PF-2, PF-3), AF group (AF-1, AF-2, AF-3), TF group (TF-1, TF-2, TF-3), and EB group (EB-1, EB-2, EB-3). After the quality control, a total of 76.27 GB of clean data was obtained, and the Q30 base percentage of each sample was not less than 93.30%. After the removal of low-quality and sub sequences, the clean reads were further analyzed and mapped to the reference genome sequence obtained, and the GC content of all samples in these four groups is shown in Table S2. The clean data of each sample was sequenced with the assembled transcript or unigene library, and the alignment results were compared as shown in Table S2. The reads written to the transcript or unigene were described as mapped reads shown in Table 1 and will be used for subsequent analysis. The total number of all-unigene is 159,213, the total length was 258,944,660, the mean length is 1626.40, and the N50 length (the length of unigene’s N50) is 2928.
Function annotation of all-unigene
A total of 159,213 all-unigene samples were assembled from the transcriptome of PF, AF, TF, and EB in C. vrolikii and compared to the functional database of COG, GO, KEGG, KOG, Pfam, Swissprot, eggNOG, and NR, respectively (Table 2). The annotation results of unigenes by the four databases (KOG, Pfam, SwissProt, and NR) were depicted by an E-value threshold of 1e-5 (1 × 10−5) in the Venn diagram (Fig. 3). The results of all-unigene annotated by NR can be further transferred to the GO database which includes three levels: cellular component, molecular function, and biological process (Fig. 4A). The cellular component contains intracellular, protein-containing complex, cellular anatomical entity, etc.; the molecular function contains the catalytic activity, the structural molecular activity, the transporter activity, the binding, etc.; the biological process contains reproduction, immune system process, behavior, metabolic process, cellular process, etc. (Fig. 4A). The COG database was used for functional annotation of unigene, and 26 COG functional classification information of gene homologous was obtained (Fig. 4B); among them, the top three were the general function prediction only, the carbohydrate transport and metabolism, and the signal transduction mechanisms. The result of BLAST top-hit species distributions for matches between the assembled unigenes of C. vrolikii and genes in the NCBINR database showed that 5046 unigenes of C. vrolikii were significantly similar to Morone saxatilis (17.20%), followed by L. crocea (13.16%), Sparus aurata (9.26%), and Lates calcarifer (5.70%) (Fig. 4C).

Venn diagram of annotation results of PF, AF, TF, and EB in C. vrolikii against four protein databases. Note: The number in each colored block indicates the number of unigenes that can be annotated by single or multiple databases (color Fig. online)

The functional distribution statistics of the transcriptome of C. vrolikii. Note: A: all-unigene GO functional distribution statistics of the transcriptome of C. vrolikii; B: all-unigene COG functional distribution statistics of transcriptome of C. vrolikii; C: Top-hit species distributions for sequences from C. vrolikii submitted to BLASTX against the NCBI-NR database
Analysis of gene expression
The gene expression was detected by the transcriptome data with high sensitivity. The FPKM value of protein-coding gene expression level can be sequenced ranging from 10−2 to 104, the expression levels were similar between different samples (Fig. 5A), and the overall discrete angle of gene expression level distribution of a single sample is not far different (Fig. 5B), which can measure the expression level of each sample from the perspective of the overall dispersion of expression. The closer R2 is to 1, the stronger the correlation between the two samples. The correlation between the gene expression of each sample is illustrated in Fig. 5C. There was no significant difference between the samples of fin rays in each group, but there was a great difference among the samples of fin rays and eyeballs.

The relationship between samples and SNP site prediction. Note: A the FPKM value of protein-coding gene expression level; B the FPKM distribution box diagram of each sample; C the correlation of gene expression of each group of samples
Analysis of differential expression genes
By comparing the transcriptome data of PF, AF, TF, and EB in C. vrolikii, the genes with a difference multiple greater than two and a Q-value < 0.001 were identified as significantly differential expression genes (DEGs). The transcriptome data of PF, AF, TF, and EB were compared in pairs, and the number of DEGs was obtained (Fig. 6; Table 3). The classification of GO functional annotation of DEGs was conducted (see Fig. S1 in Supplementary). In organisms, the different gene products coordinate with each other to perform the biological functions. The analysis of pathway annotation of DEGs is helpful to further clarify the gene functions. The DEGs were classified according to KEGG Pathway annotations (see Fig. S2 in Supplementary), and the first 20 KEGG pathways with the smallest Q-value were made into enrichment bubble diagrams (see Fig. S3 in Supplementary). According to the pathway enrichment analyses, the DEGs identified from a number of KEGG pathways such as “Melanogenesis,” “Tyrosine metabolism,” “Glycosphingolipid biosynthesis-ganglio series,” “Axon guidance,” “Cell adhesion molecules,” “Osteoclast differentiation,” “Nitrogen metabolism,” “Wnt signaling pathway,” “Metabolism of xenobiotics by cytochrome P450,” and “Drug metabolism-cytochrome P450.”

Volcano map of DEGs. Note: A to F volcano map of DEGs in PF vs AF, PF vs TF, PF vs EB, AF vs TF, AF vs EB, TF vs EB, respectively
Expression of DEGs and identification of protein interaction
The DEGs in different tissues were screened by transcriptome sequencing, and some potential genes regulating skin coloration were obtained, including Slc45a2, Slc24a5, Tryp1, Xdh, Tryp1a, Hps5, Pomc, Pmel, Sox10, Mitf, and Tyr (Table 4). Among them, Slc24a5, Mitf, Hes1-b, and Tyr were highly expressed in EB, Hps5, Alk, and Map2k4a were highly expressed in AF; Xdh, Tryp1, Tryp1a, Pomc, Pmel, and Sox10 were highly expressed in PF, while Flt1 and Pdgfrl were highly expressed in TF (Table 4) . Based on the results of differential expression analysis and the interaction pairs included in the database, the differential expression protein interaction network was constructed. During the analysis, the differentially expressed proteins were mapped into the STRING database of version 11.0 to obtain the interaction relationship information of differentially expressed proteins (Fig. 7). The interaction of Tyr, Mc1r, Mitf, and Sox10 jointly regulate the production and accumulation of melanin. In addition, Tyr, one of the differentially expressed genes, may be a key factor in the core regulation of melanin production, and its dense interaction network with a large number of proteins in the biological system indicates that Tyr actively participates in the pathway of melanin production.

The protein–protein interaction by STRING
qRT-PCR verification
The expression of thirteen DEGs from the transcriptome data of C. vrolikii was verified by qRT-PCR using Rps29 as a reference gene. Among them, in PF vs AF, the mRNA expression levels of Slc24a5, Tryp1, Pomc, Pmel, Sox10, Flt4, and Tyr were increased, while those of Hps5, Alk, and Mib1 were reduced, and those of Xdh, Mitf, and Trpm7 were basically unchanged (Fig. 8A); in PF vs TF, the mRNA expression levels of Slc24a5, Tryp1, Pomc, Pmel, Sox10, Mitf, Tyr, and Flt4 were increased, and other genes were reduced (Fig. 8B); in PF vs EB, the mRNA expression levels of Slc24a5, Tryp1, Pomc, Pmel, Mitf, and Tyr were increased, while other genes were reduced (Fig. 8C); in AF vs TF, the mRNA expression levels of all thirteen DEGs were reduced (Fig. 8D); in AF vs EB, the mRNA expression levels of Slc24a5, Tryp1, Mitf, and Tyr were increased, and other genes were reduced (Fig. 8E); in TF vs EB, the mRNA expression levels of Slc24a5, Tryp1, Hps5, Mitf, and Tyr were increased, while other genes were reduced (Fig. 8F). The above results showed similar patterns of mRNA abundance in RNA-Seq and qRT-PCR analysis, and the R square of the correlation between RNA-seq and qRTPCR was 0.95 (see Fig. S4 in Supplementary), indicating a strong correlation between RNA-seq and qRT-PCR, demonstrating the reliability of the RNA-Seq results in C. vrolikii.

DEG verification by qRT-PCR and comparison between RNA-Seq results and qRT-PCR. Note: The abscissa represents the validated genes, and the ordinate represents the log2 ratios of expression in PF vs AF (A), PF vs TF (B), PF vs EB (C), AF vs TF (D), AF vs EB (E) and TF vs EB (F), respectively
Six color genes, namely Mitf, Mc1r, Sox10, Tyr, Tyrp1, and Pomc, were identified as upregulated candidates in black fins or eyeballs. Based on the transcriptome data of different parts of fins and tissues, the DEGs related to melanin production (Mitf, Mc1r, Sox10, Tyr, Tyrp1, and Pomc) were screened to verify their expression in various tissues (Fig. 9). The qRT-PCR results showed that Mitf and Tyr were highly expressed in eyeballs, and from then on, the expression levels of Mitf and Tyr were also relatively high in the dark fin (tail fin, anal fin) and skin; Tyrp1 was highly expressed in the skin; Sox10 and Pomc were highly expressed in tail fin; Mc1r was highly expressed in the heart and tail fin. And the expression levels of these genes in the dark tissues or black fins were higher than that in the light-colored tissue or yellow fins.

The expression of the DEGs related to melanin production in various tissues by qRT-PCR. Note: “Adjacent letters (a, b, b, c or c, d)” means 0.01 < p < 0.05, “Alternating letters (a, c or b, d)” means 0.001 < p < 0.01, “separated by 2 letters (a, d)” represents p < 0.001
Discussion
In fish, skin pigmentation is associated with several biological processes, including camouflage, UV protection, and nutritional (Colihueque 2010). The skin colorations and patterns displayed by many kinds of fish depend on the presence of several pigment cells, such as melanophore and xanthophore (Bar et al. 2013). The pigment cells of the squamae and fins of C. vrolikii include melanocyte, xanthophore, and iridophore (Figs. 1 and 2). In fish, the skin coloration is an important commercial trait, which is determined by a single gene or multiple genes. Therefore, controlling melanin production is intensely important for regulating skin pigmentation. Melanin production was a biosynthetic pathway in which melanocyte synthesizes melanin through a series of complex chemical and enzymatic catalytic reactions (Pillaiyar et al. 2017). The cAMP/PKA pathway was an important pathway to regulate melanin synthesis by stabilizing and activating the MAPK, Wnt, and p53 signal pathway (Kadekaro et al. 2012; Kingo et al. 2008).
In recent years, with the continuous high-quality development of molecular biology technology, RNA-Seq technology, as a new generation of transcriptome sequencing technology, has been gradually applied to the study of the molecular mechanism of fish coloration development, such as carp (Zhang et al. 2017b) and catfish (Zhang et al. 2019b). In this study, RNA-seq technology was used for the transcriptome analysis, and many genes related to skin coloration development and variation were identified and screened in C. vrolikii for understanding their corresponding functions from a biological perspective. After sequencing, a total of 159,213 unigenes were assembled in our study, and the N50 value of the assembled unigenes was 2263 bp (Table 1). The results of all-unigene function annotation showed that 17.20% of unigenes had significant similarity to M. saxatilis (5046) (Fig. 4C), which indicated that the gene homology was conserved between C. vrolikii and M. saxatilis, and C. vrolikii was more closely related to M. saxatilis. Consistent with this, both of them belong to the order Perciformes in the natural classification system. Through the analysis of the KEGG pathway in the transcriptome of skin in red and white crucian carp, the DEGs are mainly enriched in the Wnt signal pathway, tyrosine metabolism pathway, MAPK signal pathway, and melanogenesis pathway (Zhang et al. 2017b). In the transcriptome data of skin in red and yellow river carp, the skin coloration was regulated by the melanin production pathway, MAPK signal pathway, and Wnt signal pathway (Zhang et al. 2019a). Similarly, the KEGG pathway analysis in the transcriptome of C. vrolikii showed that the DEGs were mainly concentrated in the melanogenesis pathway, tyrosine metabolism, pyruvate metabolism, MAPK signal pathway, Wnt signal pathway, and melanin production pathway (Fig. S3 in Supplementary). The results showed that the tyrosine metabolism pathway, melanogenesis pathway, MAPK signaling pathway, and Wnt signaling pathway were closely related to the variation of skin coloration in fish.
Skin is an important barrier for higher animals to isolate from the external environment. The melanocytes dominate the coloration of skin to some extent. The function of melanocytes was regulated by a series of genes in the heredity. The genes related to melanin production regulate the growth and development of melanocytes and result in the blackening of skin coloration (D’Mello et al. 2016). The transcription of Tyr and Tyrp1 is stimulated by Mitf, a key target of the melanin synthesis pathway, which can lead to the production of melanin (Liu and Fisher 2010). The A5 member of the solute carrier 24 family (Slc24a5) is distributed in the inner membrane of cells, whose golden mutation site may lead to the mutation of skin coloration to gold in zebrafish (Lamason et al. 2005). The transcriptome of the squamae by RNA-Seq in three different skin colorations of orange double crown cichlid (Midas cichlids) showed that Slc24a5 was significantly expressed in gray and golden squamae, which may play a regulatory role in the transformation of skin coloration from gray to gold (Henning et al. 2013). In Red tilmpa, the mRNA expression level of Slc24a5 was upregulated in pink skin with black spots compared with all pink skin (Zhu et al. 2016). Similar to these results, the expression level of Slc24a5 in the transcriptome and qRT-PCR of C. vrolikii was significantly higher in the black fins and tissues than that in the yellow fins or light tissues (Fig. 8 and Table 4). The results suggested that Slc24a5 may regulate the blackening of the skin coloration in fish. In addition, Xdh plays the important role in pteridine coloration, and the expression level of Xdh in the red tail fin was significantly higher than that in the yellow tail fin of Nothobranchius furzeri (Ng’oma et al. 2014). The transcriptome data of C. vrolikii showed that the expression levels of Xdh and Pomc were upregulated in the process of skin coloration variation from light gray or yellow to black (Fig. 8 and Table 4). It is speculated that these genes may be involved in the synthesis of melanin.
In order to further study the molecular mechanism of skin coloration of fish, the expression levels of genes related to melanin production in various tissues of fish were analyzed by qRT-PCR. The mRNA expression level of Sox10 in light and black fins was not significantly different, while the mRNA expression levels of Tyr, Mc1r, Mitf, Pomc, and Tyrp1 in black fins or tissues were significantly higher than those in light fins or tissues (Fig. 9). Studies on the expression of Mitf in Oryzias latipes (Li et al. 2013a), Cyprinus carpio and Carassius auratus (Xu et al. 2014 and Liu et al. 2015), and zebrafish (Elworthy et al. 2003) have also been carried out, and the role of Mitf in fish skin coloration formation was preliminarily explored. The skin coloration–related genes, such as Sox10, Tyrp1, Tyr, and Mc1r, have also been reported in Cyprinus carpio var. color (Gu 2016; Li et al. 2013b and Li 2014), Cyprinus carpio (Bar et al. 2013), and other species with similar evolutionary relationship (Selz et al. 2007; Gross et al. 2009; Kobayashi et al. 2016; Sánchez et al. 2010). In melanocyte, Sox10 directly activated Mitf to induce Tyr and Tyrp2 to produce pigmentation (Hou et al. 2008), and Wnt3a promoted melanin production by upregulating the expression of Mitf, Tyrp1, and Tyr (Guo et al. 2012). Tyr and Tyrp1 are members of the tyrosine gene family, encoding proteins related to melanin production (Camacho-Hubner et al. 2002). Tyr mRNA was expressed in the liver, cecum, gill, muscle, and skin of adult Japanese amphioxus (Branchiostoma japonicum), but not in the intestine, neural tube, and testis (Pang et al. 2013), which is similar to the expression level of Tyr in our study (Fig. 9 B). Through transcriptome analysis of catfish skin of albino and wild-type, Tyrl and Tyrp1 were significantly upregulated in albino (Zhang et al. 2019a). The mRNA expression level of Tyrp1 in gray or black skin was upregulated than that in red skin of red crucian carp (Carassius auratus) and red fin snapper (Lutjanus erythropterus), respectively (Zhang et al. 2015 and 2017). In C. vrolikii, the mRNA expression levels of Tyr, Mc1r, and Mitf in black fin were significantly higher than that in yellow fin, which may be one of the reasons for its regulation of melanin and blackening (Fig. 9). It can be seen that the mRNA expression level of Tyrp1 in gray black skin of fish is higher than that in light coloration skin. Similarly, the mRNA expression levels of Tyr and Tyrp1 in black fins or dark tissues were significantly upregulated compared with yellow fins or light tissues of C. vrolikii. All these showed that Tyr and Tyrp1 were significantly expressed in the process of light coloration blackening, indicating they may play a key role in the process of skin coloration blackening.
Interestingly, these six genes showed high expression level in spleen tissue and some in heart tissue and low expression level in liver tissue. Research has shown that Sox10 has been shown to be extensively expressed in adult tissues such as the brain and heart, with little or no expression in the prostate, bladder, pancreas, liver, and stomach of mammals (Volff and Schartl 2003). In Cynoglossus semilaevis, in addition to a large expression in the brain, gills, skin, and eyes, there is also a high expression in the spleen, heart, head, and muscles, while there is a small expression in the kidneys and intestines, but not in the liver and gonads (Potterf et al. 2001). This study found that Sox10 was mostly in the anal fin, caudal fin, and skin, followed by the kidneys, pectoral fins, heart, eyes, and spleen, with little or no expression in the ovaries, intestines, and testes. Mitf regulates the transcription of tyrosinase (Tyr), and the degree of Mitf expression directly influences melanin production (Curran et al. 2010). In Yellow River carp, it is abundantly expressed in the intestines, brain, gills, liver, and pancreas, as well as the skin, buttocks, and tail (Zhang et al. 2017a); they believe Mitf is engaged in a variety of cellular processes and has several impacts. In this study, Mitf was shown to be expressed to diverse degrees in many tissues, with comparatively high levels of expression in the eyes, kidneys, tail fin, and anal fin, this supports the notion that Mitf participates in a variety of cellular processes and has a wide range of biological roles and consequences. Compared to muscle and liver, the zebrafish’s Mc1r is significantly expressed in the brain, eyes, gills, skin, and testes (Selz et al. 2007). This study demonstrates that there are variations in the expression of Mc1r in various fish tissues, with the heart and spleen exhibiting the highest levels of expression. In contrast to other tissues, where it is little expressed, the gills, skin, and pectoral fin do exhibit some expression. The expression of Mc1r varies from that of other fish, demonstrating the species-specific nature of gene expression. Tyr, Tyrp1, and Pomc were widely expressed in a variety of fish tissues, which implies that they may be involved in a number of cellular processes. Further evidence that these genes may be involved in the growth and differentiation of melanocytes comes from the high expression levels of these genes in the skin, anal fin, and tail fin. The function of these genes in the development of melanocytes and the production of fish body color requires more study.
Conclusion
The differential expression genes (DEGs) related to the skin coloration in the transcriptome data of the pectoral fin (PF), anal fin (AF), tail fin (TF), and eyeballs (EB) of C. vrolikii have been screened and analyzed, respectively. The variation trend of qRT-PCR was consistent with that of transcriptome data. Among the results, some genes related to the skin coloration variation were screened and enriched through the KEGG pathway. It was found that they were involved in the regulation of melanogenesis pathway, tyrosine metabolism, pyruvate metabolism, MAPK signal pathway, and Wnt signal pathway and then affected the skin coloration of C. vrolikii. The information on the interaction between the fish skin coloration and the regulatory pathways is worthy of further investigation. The expression levels of the DEGs related to melanin production (Mitf, Mc1r, Sox10, Tyr, Tyrp1, and Pomc) in various tissues of C. vrolikii were screened and validated. The interaction of these genes may affect the skin coloration under the influence of abiotic factor such as environments during the fish growth. These results are helpful to understand the molecular regulatory mechanism of skin coloration, also present basic support for skin coloration control of C. vrolikii in the aquaculture industry, and provide a theoretical basis and a data source for fish developmental biology.
Data availability
Not applicable.
Code availability
Not applicable.
References
Bar I, Kaddar E, Velan A, David L (2013) Melanocortin receptor 1 and black pigmentation in the Japanese ornamental carp (Cyprinus carpio var. Koi). Front Genet. 4: 6. https://doi.org/10.3389/fgene.2013.00006
Cal L, Suarez-Bregua P, Cerda-Reverter JM, Braasch I, Rotllant J (2017) Fish pigmentation and the melanocortin system. Comp Biochem Phys a 211:26–33. https://doi.org/10.1016/j.cbpa.2017.06.001
Camacho-Hubner A, Richard C, Beermann F (2002) Genomic structure and evolutionary conservation of the tyrosinase gene family from Fugu. Gene 285(1–2):59–68. https://doi.org/10.1016/S0378-1119(02)00411-0
Cicero R, Mallardi A, Maida I, Gallone A, Pintucci G (1989) Melanogenesis in the pigment cells of rana esculenta l. liver: evidence for tyrosinase-like activity in the melanosome protein fraction. Pigm Cell R. 2(2):100–108. https://doi.org/10.1111/j.1600-0749.1989.tb00169.x
Colihueque N (2010) Genetics of salmonid skin pigmentation: clues and prospects for improving the external appearance of farmed salmonids. Rev Fish Biol Fisher 20(1):71–86. https://doi.org/10.1007/s11160-009-9121-6
Curran K, Lister JA, Kunkel GR, Prendergast A, Parichy DM, Raible DW (2010) Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Dev Biol 344(1):107–118. https://doi.org/10.1016/j.ydbio.2010.04.023
Dibattista JD, Gaither MR, Hobbs JPA, Rocha LA, Bowen BW (2016) Angelfishes, Paper tigers, and the Devilish taxonomy of the Centropyge flavissima complex. J Hered 107(7):647–653. https://doi.org/10.1093/jhered/esw062
Djurdjevic I, Furmanek T, Miyazawa S, Bajec SS (2019) Comparative transcriptome analysis of trout skin pigment cells. BMC Genomics 20:359. https://doi.org/10.1186/s12864-019-5714-1
D’Mello SAN, Finlay GJ, Baguley BC, Askarian-Amiri ME (2016) Signaling pathways in melanogenesis. Int J Mol Sci 17(7):1144. https://doi.org/10.3390/ijms17071144
Elworthy S, Lister JA, Carney TJ, Raible DW, Kelsh RN (2003) Transcriptional regulation of mitfa accounts for the sox10 requirement in zebrafish melanophore development. Development 130(12):2809–2818. https://doi.org/10.1242/dev.00461
Fang D, Tsuji Y, Setaluri V (2002) Selective down-regulation of tyrosinase family gene Tyrp1 by inhibition of the activity of melanocyte transcription factor. Mitf Nucleic Acids Res 30(14):3096–3106. https://doi.org/10.1093/nar/gkf424
Fernandez-Silva I, Henderson JB, Rocha LA, Simison WB (2018) Whole-genome assembly of the coral reef Pearlscale Pygmy Angelfish (Centropyge vrolikii). Sci Rep 8(1):1489. https://doi.org/10.1038/s41598-018-19430-x
Funt N, Palmer BA, Weiner S, Addadi L (2017) Koi fish-scale iridophore cells orient guanine crystals to maximize light reflection. Chem Plus Chem 82(6):914–923. https://doi.org/10.1002/cplu.201700151
Greenwood AK, Cech JN, Peichel CL (2012) Molecular and developmental contributions to divergent pigment patterns in marine and freshwater sticklebacks. Evol Dev 14(4):351–362. https://doi.org/10.1111/j.1525-142X.2012.00553.x
Gross JB, Borowsky R, Tabin CJ (2009) A novel role for Mc1r in the parallel evolution of depigmentation in independent populations of the cavefish Astyanax mexicanus. Plos Genet 5(1):1000326. https://doi.org/10.1371/journal.pgen.1000326
Gu ZZ (2016) Study on selection pressure of four color related genes and gowth combining effects of the two patterns in Oujiang color common carp. Shanghai Ocean University (In Chinese), Color
Guida S, Ciardo S, De-Pace B, De-Carvalho N, Farnetani F, Pezzini C, Chester J, Shaniko K, Manganelli M, Guida G, Pellacani G (2021) Atrophic and hypertrophic skin photoaging and melanocortin-1 receptor (Mc1r): the missing link. J Am Acad Dermatol 84(1):187–190. https://doi.org/10.1016/j.jaad.2020.04.075
Guo HY, Yang K, Deng F, Ye JX, Xing YZ, Li YH, Lian XH, Yang T (2012) Wnt3a promotes melanin synthesis of mouse hair follicle melanocytes. Biochem Bioph Res Co 420(4):799–804. https://doi.org/10.1016/j.bbrc.2012.03.077
Gutknecht M, Geiger J, Joas S, Dorfel D, Salih HR, Muller MR, Grunebach F, Rittig SM (2015) The transcription factor Mitf is a critical regulator of Gpnmb expression in dendritic cells. Cell Commun Signal 13:19. https://doi.org/10.1186/s12964-015-0099-5
Han J, Hong WS, Wang Q, Zhang TT, Chen SX (2019) The regulation of melanocyte-stimulating hormone on the pigment granule dispersion in the xanthophores and melanophores of the large yellow croaker (Larimichthys crocea). Aquaculture 507:7–20. https://doi.org/10.1016/j.aquaculture.2019.03.074
Henning F, Jones JC, Franchini P, Meyer A (2013) Transcriptomics of morphological color change in polychromatic Midas cichlids. BMC Genomics 14:171. https://doi.org/10.1186/1471-2164-14-171
Hou L, Pavan WJ (2008) Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res 18(12):1163–1176. https://doi.org/10.1038/cr.2008.303
Hou L, Pavan WJ, Shin MK, Arnheiter H (2004) Cell-autonomous and cell non-autonomous signaling through endothelin receptor B during melanocyte development. Development 131(14):3239–3247. https://doi.org/10.1242/dev.01193
Ito S, Wakamatsu K (2008) Chemistry of mixed melanogenesis - pivotal roles of dopaquinone. Photochem Photobiol 84(3):582–592. https://doi.org/10.1111/j.1751-1097.2007.00238.x
Kadekaro AL, Chen JP, Yang J, Chen SN, Jameson J, Swope VB, Cheng T, Kadakia M (2012) Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol Cancer Res 10(6):778–786. https://doi.org/10.1158/1541-7786.MCR-11-0436
Kingo K, Aunin E, Karelson M, Ratsep R, Silm H, Vasar E, Koks S (2008) Expressional changes in the intracellular melanogenesis pathways and their possible role the pathogenesis of vitiligo. J Dermatol Sci 52(1):39–46. https://doi.org/10.1016/j.jdermsci.2008.03.013
Kobayashi Y, Hamamoto A, Takahashi A, Saito Y (2016) (2016) Dimerization of melanocortin receptor 1 (MC1R) and MC5R creates a ligand-dependent signal modulation: potential participation in physiological color change in the flounder. Gen Comp Endocr 230:103–109. https://doi.org/10.1016/j.ygcen.2016.04.008
Lamason RL, Mohideen MAPK, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao XY, Humphreville VR, Humbert JE, Sinha S, Moore JL, Jagadeeswaran P, Zhao W, Ning G, Makalowska I, McKeigue PM, O’Donnell D, Kittles R, Parra EJ, Mangini NJ, Grunwald DJ, Shriver MD, Canfield VA, Cheng KC (2005) Slc24a5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310(5755):1782–1786. https://doi.org/10.1126/science.1116238
Lee SY, Lee HJ (2020) Comprehensive RNA-seq analysis to evaluate the pigmentation-related genes involved in albinism of cichlid fish. Aulonocara Baenschi Front Mar Sci 7:723. https://doi.org/10.3389/fmars.2020.00723
Lee HO, Levorse JM, Shin MK (2003) The endothelin receptor-B is required for the migration of neural crest-derived melanocyte and enteric neuron precursors. Dev Biol 259(1):162–175. https://doi.org/10.1016/S0012-1606(03)00160-X
Li KL (2014) Molecular cloning and expression analysis of pigmentation-related genes, Sox10, Agouti, Tyrp1 and Dct, in Oujiang color common carp. Color, Shanghai Ocean University (In Chinese), Cyprinus carpio var
Li M, Yuan Y, Hong Y (2013a) (2013a) Identification of the RNAs for transcription factor Mitf as a component of the Balbiani body. J Genet Genomics 40(002):75–81. https://doi.org/10.1016/j.jgg.2012.12.006
Li KL, Hu JZ, Yan B, Wang CH (2013b) Isolation and expression analysis of Sox10 gene relating body color variation in Oujiang color common carp. J Fish Sci China 20(6):1139–1147. https://doi.org/10.3724/SP.J.1118.2013.01139
Liu JJ, Fisher DE (2010) Lighting a path to pigmentation: mechanisms of Mitf induction by UV. Pigm Cell Melanoma r 23(6):741–745. https://doi.org/10.1111/j.1755-148X.2010.00775.x
Liu JH, Wen S, Luo C, Zhang YQ, Tao M, Wang DW, Deng SM, Xiao YM (2015) 2015) Involvement of the mitfa gene in the development of pigment cell in Japanese ornamental (Koi) carp (Cyprinus carpio L. Genet Mol Res 14(1):2775–2784. https://doi.org/10.4238/2015.March.31.7
Luo K, Li J, Chen J, Pan Y, Zhang YJ, Zhou HH, Zhang WB, Mai KS (2020) Proteomics analysis of skin of large yellow croaker Larimichthys crocea fed different dietary carotenoids. Aquacult Nutr 26(6):1981–1993. https://doi.org/10.1111/anu.13140
Mahalwar P, Walderich B, Singh AP, Nusslein-Volhard C (2014) Local reorganization of xanthophores fine-tunes and colors the striped pattern of zebrafish. Science 345(6202):1362–1364. https://doi.org/10.1126/science.1254837
Murisier F, Guichard S, Beermann F (2007) The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigm Cell r 20(3):173–184. https://doi.org/10.1111/j.1600-0749.2007.00368.x
Ng’oma E, Groth M, Ripa R, Platzer M, Cellerino A (2014) Transcriptome profiling of natural dichromatism in the annual fishes Nothobranchius furzeri and Nothobranchius kadleci. BMC Genomics 15:754. https://doi.org/10.1186/1471-2164-15-754
Pang QX, Liu XM, Sun HH, Zhang SC, Song XH, Zhang XF, Zhang M, Bai Y, Gao L, Zhao BS (2013) Cloning, characterization and expression of tyrosinase-like gene in amphioxus Branchiostoma japonicum. Fish Shellfish Immun 34(1):356–364. https://doi.org/10.1016/j.fsi.2012.11.013
Parichy DM (2003) Pigment patterns: fish in stripes and spots. Curr Biol 13(24):R947–R950. https://doi.org/10.1016/j.cub.2003.11.038
Pillaiyar T, Manickam M, Jung SH (2017) Recent development of signaling pathways inhibitors of melanogenesis. Cell Signal 40:99–115. https://doi.org/10.1016/j.cellsig.2017.09.004
Potterf SB, Mollaaghababa R, Hou L, Southard-Smith EM, Hornyak TJ, Arnheiter H, Pavan WJ (2001) Analysis of SOX10 function in neural crest-derived melanocyte development: SOX10-dependent transcriptional control of dopachrome tautomerase. Dev Biol 237(2):245–257. https://doi.org/10.1006/dbio.2001.0372
Qian X, Ba Y, Zhuang QF, Zhong GF (2014) RNA-Seq technology and its application in fish transcriptomics. OMICS 18(2):98–110. https://doi.org/10.1089/omi.2013.0110
Qomaladewi NP, Kim MY, Cho JY (2019) Rottlerin reduces cAMP/CREB-mediated melanogenesis via regulation of autophagy. Int J Mol Sci 20(9):2081. https://doi.org/10.3390/ijms20092081
Rees JL (2000) The melanocortin 1 receptor (Mc1r): more than just red hair. Pigm Cell R 13(3):135–140. https://doi.org/10.1034/j.1600-0749.2000.130303.x
Sánchez E, Rubio VC, Cerdá-Reverter JM (2010) Molecular and pharmacological characterization of the melanocortin type 1 receptor in the sea bass. Gen Comp Endocr 165(1):163–169. https://doi.org/10.1016/j.ygcen.2009.06.008
Sefc KM, Brown AC, Clotfelter ED (2014) Carotenoid-based in cichlid fishes. Comp Biochem Phys a 173:42–51. https://doi.org/10.1016/j.cbpa.2014.03.006
Selz Y, Braasch I, Hoffmann C, Schmidt C, Schultheis C, Schartl M, Volff JN (2007) Evolution of melanocortin receptors in teleost fish: the melanocortin type 1 receptor. Gene 401(1/2):114–122. https://doi.org/10.1016/j.gene.2007.07.005
Volff JN, Schartl M (2003) Evolution of signal transduction by gene and genome duplication in fish. J Struct Funct Genomics 3:139–150. https://doi.org/10.1023/A:1022678305005
Wu SJ, Huang JQ, Li YJ, Liu Z, Zhang Q, Pan YC, Wang XL (2021) Cloning, sequence analysis, and expression of tyrp1a and tyrp2 genes related to body colour in different developmental stages and tissues of rainbow trout Oncorhynchus mykiss. Aquacult Int 29(3):941–961. https://doi.org/10.1007/s10499-021-00666-w
Wu SJ, Huang JQ, Li YJ, Zhao L, Liu Z, Kang YJ, Wang JF (2021) Integrative mRNA-miRNA interaction analysis reveals the molecular mechanism of skin color variation between wild-type and yellow mutant rainbow trout (Oncorhynchus mykiss). Comp Biochem Phys D. 40:100914. https://doi.org/10.1016/j.cbd.2021.100914
Wu SJ, Huang JQ, Li YJ, Zhao L, Liu Z (2022) Analysis of yellow mutant rainbow trout transcriptomes at different developmental stages reveals dynamic regulation of skin pigmentation genes. Sci Rep 12(1):256. https://doi.org/10.1038/s41598-021-04255-y
Xu Y, Zhang XH, Pang YZ (2013) Association of tyrosinase (Tyr) and tyrosinase-related protein 1 (Tyrp1) with melanic plumage color in Korean quails (Coturnix coturnix). Asian Austral J Anim 26(11):1518–1522. https://doi.org/10.5713/ajas.2013.13162
Xu P, Zhang X, Wang X, Li J, Liu G, Kuang Y, Xu J, Zheng X, Ren L, Wang G, Zhang Y, Huo L, Zhao Z, Cao D, Lu C, Li C, Zhou Y, Liu Z, Fan Z, Shan G, Li X, Wu S, Song L, Hou G, Jiang Y, Jeney Z, Yu D, Wang L, Shao C, Song L, Sun J, Ji P, Wang J, Li Q, Xu L, Sun F, Feng J, Wang C, Wang S, Wang B, Li Y, Zhu Y, Xue W, Zhao L, Wang J, Gu Y, Lv W, Wu K, Xiao J, Wu J, Zhang Z, Yu J, Sun X (2014) Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet 46:1212–1219. https://doi.org/10.1038/ng.3098
Zhang P, Liu W, Yuan XY, Li DG, Gu WJ, Gao TW (2013) Endothelin-1 enhances the melanogenesis via Mitf-Gpnmb pathway. BMB Rep 46(7):364–369. https://doi.org/10.5483/BMBRep.2013.46.7.250
Zhang YP, Wang ZD, Guo YS, Liu L, Yu J, Zhang S, Liu SJ, Liu CW (2015) Morphological characters and transcriptome profiles associated with black skin and red skin in Crimson snapper (Lutjanus erythropterus). Int J Mol Sci 16(11):26991–27004. https://doi.org/10.3390/ijms161126005
Zhang YQ, Liu JH, Fu W, Xu WT, Zhang HQ, Chen SJ, Liu WB, Peng LY, Xiao YM (2017) Comparative transcriptome and DNA methylation analyses of the molecular mechanisms underlying skin color variations in Crucian carp (Carassius carassius L.). Bmc Genetics 18:95. https://doi.org/10.1186/s12863-017-0564-9
Zhang YQ, Liu JH, Peng LY, Ren L, Zhang HQ, Zou LJ, Liu WB, Xiao YM (2017) Comparative transcriptome analysis of molecular mechanism underlying gray-to-red body color formation in red crucian carp (Carassius auratus, red var.). Fish Physiol Biochem 43(5):1387–1398. https://doi.org/10.1007/s10695-017-0379-7
Zhang SH, Gong YW, Xu J, Hu M, Xu P, Jiang YL (2019) The genetic basis of skin color and body shape of domesticated purse red carp revealed by comparative transcriptome analysis. Pak J Zool 51(5):1675–1684. https://doi.org/10.17582/journal.pjz/2019.51.5.1675.1684
Zhang SY, Li X, Pan JL, Wang MH, Zhong LQ, Wang J, Qin Q, Liu HY, Shao JJ, Chen XH, Bian WJ (2019b) Use of comparative transcriptome analysis to identify candidate genes related to albinism in channel catfish (Ictalurus punctatus). Aquaculture 500:75–81. https://doi.org/10.1016/j.aquaculture.2018.09.055
Zheng HQ, Li XL, Zhou RY, Li LH, Guo XL, Kang JF, Li DF (2010) Bioinformatics analysis of tyrosinase-related protein 1 gene (Tyrp1) from different species. Front Agric China 4:109–115. https://doi.org/10.1007/s11703-009-0081-3
Zhong ZW, Ao LL, Wang YL, Wang SH, Zhao LP, Ma SW, Jiang YH (2021) Comparison of differential expression genes in ovaries and testes of Pearlscale angelfish Centropyge vrolikii based on RNA-seq analysis. Fish Physiol Biochem 47:00997. https://doi.org/10.1007/s10695-021-00977-y
Zhong ZW, Xu Y, Feng Y, Ao LL, Wang YL, Jiang YH (2022a) Characterization of Nanog gene involved in the gonad development in pearlscale angelfish (Centropyge vrolikii). Fish Physiol Biochem 48(2):303–319. https://doi.org/10.1007/s10695-022-01054-8
Zhong ZW, Ao LL, Zhao LP, Zhang ZY, Jiang YH (2022b) Screening and validation of reference genes for qPCR analysis in gonads and embryos of Takifugu bimaculatus. Aquac Fish 7(3):278–286. https://doi.org/10.1016/j.aaf.2020.10.002
Zhu WB, Wang LM, Dong ZJ, Chen XT, Song FB, Liu N, Yang H, Fu JJ (2016) Comparative transcriptome analysis identifies candidate genes related to skin color differentiation in red tilapia. Sci Rep 6:31347. https://doi.org/10.1038/srep31347
Zou M, Zhang XT, Shi ZC, Lin L, Ouyang G, Zhang GR, Zheng H, Wei KJ, Ji W (2015) A comparative transcriptome analysis between wild and albino yellow catfish (Pelteobagrus fulvidraco). PLoS One. 10(6):e0131504. https://doi.org/10.1371/journal.pone.0131504
Acknowledgements
Thanks to Prof. Shu-hong Wang, Fisheries College, Jimei University (Xiamen), for providing the fish for this study.
Funding
This research was supported by the Natural Science Foundation of Fujian Province (2021J01824) and the Ornamental Aquarium Engineering Research Centre at the University of Fujian province; Fujian Provincial Department of Education (JAT190350); Innovation Training program for College students of Fujian province (202110390039; 202110390043).
Author information
Authors and Affiliations
Contributions
Li-jun He, Zhao-Wei Zhong, and Zhen Yang are responsible for the sampling, proposal of research scheme, carrying out the experiments, bioinformatics analysis, and manuscript writing. Yan Xu, Yan Feng, and Zhen Ding are responsible for the sampling, cell interference experiment, and quantitative analysis. Yong-Hua Jiang and Hong-Wei Liu are responsible for the task proposal, funding and technology, the research proposal, and implementation support and critically edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
The sample collection and experimental protocols were approved by the Animal Care and Use Committee of the Fisheries College of Jimei University (Animal Ethics No. 2021–4). All animal handling and methods were performed according to the relevant guidelines.
Consent to participate
All authors have discussed the study procedures and have been satisfied with the relevant questions, and all have agreed to participate in the study.
Consent for publication
All authors read and approved the final manuscript for publication.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Pierre Boudry
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, Lj., Yang, Z., Zhong, Zw. et al. Screening and expression analysis of genes related to skin coloration in pearlscale angelfish (Centropyge vrolikii). Aquacult Int 32, 137–159 (2024). https://doi.org/10.1007/s10499-023-01145-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-023-01145-0